
Abstract
Genes differentially expressed in different tissues, during development, or during specific pathologies are of foremost interest to both basic and pharmaceutical research. “Transcript profiles” or “digital Northerns” are generated routinely by partially sequencing thousands of randomly selected clones from relevant cDNA libraries. Differentially expressed genes can then be detected from variations in the counts of their cognate sequence tags. Here we present the first systematic study on the influence of random fluctuations and sampling size on the reliability of this kind of data. We establish a rigorous significance test and demonstrate its use on publicly available transcript profiles. The theory links the threshold of selection of putatively regulated genes (e.g., the number of pharmaceutical leads) to the fraction of false positive clones one is willing to risk. Our results delineate more precisely and extend the limits within which digital Northern data can be used.
Very large-scale, single-pass partial sequencing of cDNA clones from a large number of libraries has led to the identification of ∼50,000 human genes (Adams et al. 1995; Aaronson et al. 1996;Hillier et al. 1996). However, a precise function or a complete transcript sequence are known for <5000 of these (Adams et al. 1995; Boguski and Schuler 1995). In the absence of functional clues for most of the newly identified genes, evidence of differential expression is the most important criteria to prioritize the exploitation of anonymous sequence data in both basic and pharmaceutical (Nowak 1995; Adams 1996; Bains 1996; Editorial 1996) research. For example, the study of expression profiles in various tumors is central to the new Cancer Genome Anatomy project (Kuska 1996;O’Brien 1997). In contrast to functional assays, the quantitative analysis of gene expression level lends itself to large-scale implementation. Two main approaches have been proposed (1) “analog” methods based on hybridization to arrayed cDNA libraries (Lennon and Lehrach 1991; Gress et al. 1992; Nguyen et al. 1995; Schena et al. 1995; Zhao et al. 1995) or oligonucleotide “chips” (Fodor et al. 1991; Southern et al. 1992; Guo et al. 1994; Matson et al. 1995); and (2) “digital” methods, based on the generation of sequence tags. This paper focuses on the latter. The sequence tag-based method (Okubo et al. 1992; Matsubara and Okubo 1994) consists of generating a large number (thousands) of expressed sequence tags (ESTs) (Adams et al. 1991; Wilcox et al. 1991; Adams et al. 1992; Khan et al. 1992) from 3′-directed regional non-normalized cDNA libraries. Recently, Velculescu et al. (1995) have introduced the serial analysis of gene expression (SAGE). Although tags are 100–300 nucleotides in length in the original EST approach, the SAGE method only requires nine nucleotides, therefore allowing a larger throughput. In both protocols, the number of tags is reported to be proportional to the abundance of cognate transcripts in the tissue or cell type used to make the cDNA library. The variation in the relative frequency of those tags, stored in computer databases, is then used to point out the differential expression of the corresponding genes: This is the concept of a “digital Northern” comparison. In the absence of a sound theoretical framework, the validity of the method has only been verified for a handful of genes in the context of two cellular differentiation systems (Lee et al. 1995; Okubo et al. 1995) inducible in vitro. Yet, with a total number of human genes of ∼80,000 or more, it is not intuitive that sequencing a mere few thousand tags (a typical experiment) from highly redundant non-normalized cDNA libraries can produce a useful picture, or realistic “transcript profile,” of a given tissue, development stage, or cell type. What variations in tag numbers allow for a reliable inference about differential expression? How many tags should be generated? Here we present the statistical framework required to answer those questions and analyze transcript profiles in a quantitative manner.
RESULTS
In Methods we establish the probability distribution governing the occurrence of the same rare event in duplicate experiments. This probability distribution is a general result applicable to a wide variety of experimental situations, although this paper focuses on its use to analyze digital gene expression patterns. The main and only mathematical assumption behind the derivation is that the observed events are rare and part of a large population of possible outcomes (the distribution of which is not specified). In the context of a digital Northern, one event is the observation of a given cDNA sequence tag, and the experiment consists of the random picking and partial sequencing of a number N of cDNA clones. Given the usual complexity (i.e., the number of different genes expressed) of cDNA libraries, observing a given cDNA qualifies as a rare event, as the abundance of most individual messages is of the order of a few percents or less.
Random Fluctuation vs. Significant Change in Tag Number: When to Infer Differential Expression
Let us randomly pick N = 1000 clones from a cDNA library and generate the corresponding sequence tags; a given message (e.g., interleukin-2) will be picked x (e.g., two) times, withx in a typical (0–10) range. If we now redo this experiment, that is, again pick 1000 clones and generate the tags, the same message will now be picked y (e.g., 3) times. If the experiments have been duplicated correctly and the clones selected at random, we expectx and y to be close, albeit often different because of random fluctuations. In the Methods section, we show that the expected probability of observing y occurrences of a clone already observed x times is given by the simple formula:
Confidence Intervals in Function of the Value ofx

|
[i] The value of x (first column), one of the occurrence numbers. The intervals are given for the 95% (2 ɛ = 0.05) and 99% (2ɛ = 0.01) confidence levels. Up to x = 20, the exact boundaries, immediately outside the confidence interval (first significantly different values) are indicated. A star is used when none are possible. For larger values, the boundaries are given as percentages to be subtracted or added to x. Ricker’s confidence interval characterizes the value of λ, not y(see Methods). The use of a flat p (λ) prior distribution results in the most stringent test, as expected. Although the number (N) of clones sampled does not appear in the expression ofp(y|x) (Equation { label needed for disp-formula[@id='E1'] }), its influence shows in the fact that the confidence interval becomes proportionally smaller asx (and y) increases (e.g., 1 ➝ 7 has the same statistical significance as 40 ➝ 60). For the same expression level, larger N will result in larger absolute values forx and y, making the detection of significant differential expression more sensitive.
The same confidence intervals listed in Table 1 can in fact be used to analyze the results of sampling N clones from two different libraries. Provided all experimental factors are well replicated, significant discrepancies between x (from one library) andy (from the other) will now characterize differentially expressed genes, for example, the relative abundance of which is unlikely to be the same in the two libraries. Simply reading Table 1, we see that variations in counts such as 7 → 0, or 2 → 12 are significant (P < 0.01) evidence of regulated gene expression, whereas variations such as 3 → 0 or 8 → 16 are not (P > 0.05). However, we do not advocate the use of rigid significance thresholds to analyze digital transcript profiles, as discussed below.
Influence of the Sampling Size
Surprisingly at first, p(y | x) in Equation { label needed for disp-formula[@id='E1'] } does not involve the sampling size N, that is, the total number of picked clones. The fluctuation probabilities, and confidence intervals, depend only on the values of the observed counts. To understand why, we must remember that Equation { label needed for disp-formula[@id='E1'] } governs the results of strictly duplicated experiments. Given N clones are sampled, the most likely tags to be picked up are, intuitively, those corresponding to cDNA, the abundance of which is of the order of 1/N, or larger (according to Equation { label needed for disp-formula[@id='E3'] }, the probability of finding a given cDNA with 1/N abundance while picking upN clones is 0.63, see also Equation { label needed for disp-formula[@id='E13'] }). Choosing a sampling size therefore corresponds to targeting a given subset of genes, the level of expression of which allows their tags to occur at reasonable frequencies.
As expected, more reliable inferences can be made on clones corresponding to larger absolute frequencies (i.e., the ones more often picked up). For example (see Table 1), a variation in counts from 1–3 (threefold increase) is not indicative of a significant (P < 0.05) increase, whereas a variation from 4–12 is significant at P < 0.05, and a variation from 7–21 is significant at P < 0.01. For a gene expressed at a given rate, increasing the sampling size N leads to higher tag counts, and allows more stringent statistical inference to be made, for the same proportional variation.
Most often in practice one wishes to compare digital Northerns or gene profiles that have been computed from the random picking of different numbers of clones, N 1 and N 2. The mathematical problem is now to establish the probability for a given cDNA (e.g., interleukin-2) to be picked up x times when the sampling size was N 1 and y times when the sampling size was N 2. Equation { label needed for disp-formula[@id='E1'] } then becomes (see Methods):
Comparison with Fisher’s (2 × 2) Exact Test
The (2 × 2) contingency tables arising from treatment versus control experiments are traditionally analyzed with Fisher’s exact test (Siegel 1956; Agresti 1996). Differential EST count data can be presented in a tabulated form so as to suggest the use of this test, as follows:
The statistical significance according to Fisher’s exact test for such a result is 4.6% (two-tail P-value, i.e., the probability for such a table to occur in the hypothesis that actin EST frequencies are independent of the cDNA libraries). In comparison, theP-value computed from the cumulative form (Equation 9, see Methods) of Equation { label needed for disp-formula[@id='E2'] } (i.e., for the relative frequency of actin ESTs to be the same in both libraries, given that at least 11 cognate ESTs are observed in the liver library after two were observed in the brain library) is 1.6%. Fisher’s (2 × 2) exact test is always more conservative than our test (e.g., Fisher’s P-value of 1.6% requires a 2 → 13 EST count transition in the above setting). Besides being too conservative, there is a more fundamental difficulty in using this test to analyze EST count data. The sampling scheme assumed by Fisher’s exact test in principle requires the total number of data values in the contingency table to be fixed, as well as both the row marginal total and the column marginal totals. In our prospective experimental situation, only the column marginals (i.e., the numbers of clones sampled from each library) are fixed. The extension of Fisher’s exact test to cases where only one set of marginal totals is fixed (Tocher 1950) is still controversial. In the context of the above EST counting results, there is an additional problem with the lack of homogeneity in the definition of the “other EST” category. This category represents different subsets of transcripts for different libraries.
The use of Fisher’s (2 × 2) exact test is more natural for a different type of EST data analysis: the study of library-dependent alternative transcripts of the same gene (i.e., splice or polyadenylation variants) (D. Gautheret, O. Poirot, F. Lopez, S. Audic, and J.-M. Claverie, in prep.). Here, the results for an hypothetical gene G1 may look as follows:
where the alternative categories are unambiguously defined and refer to the same objects. For example, the above results constitute good evidence that G1 is expressed in different forms in those tissues (Fisher’s exact test two-tailP-value = 1.2%).
False Leads in the Selection of Candidate Genes
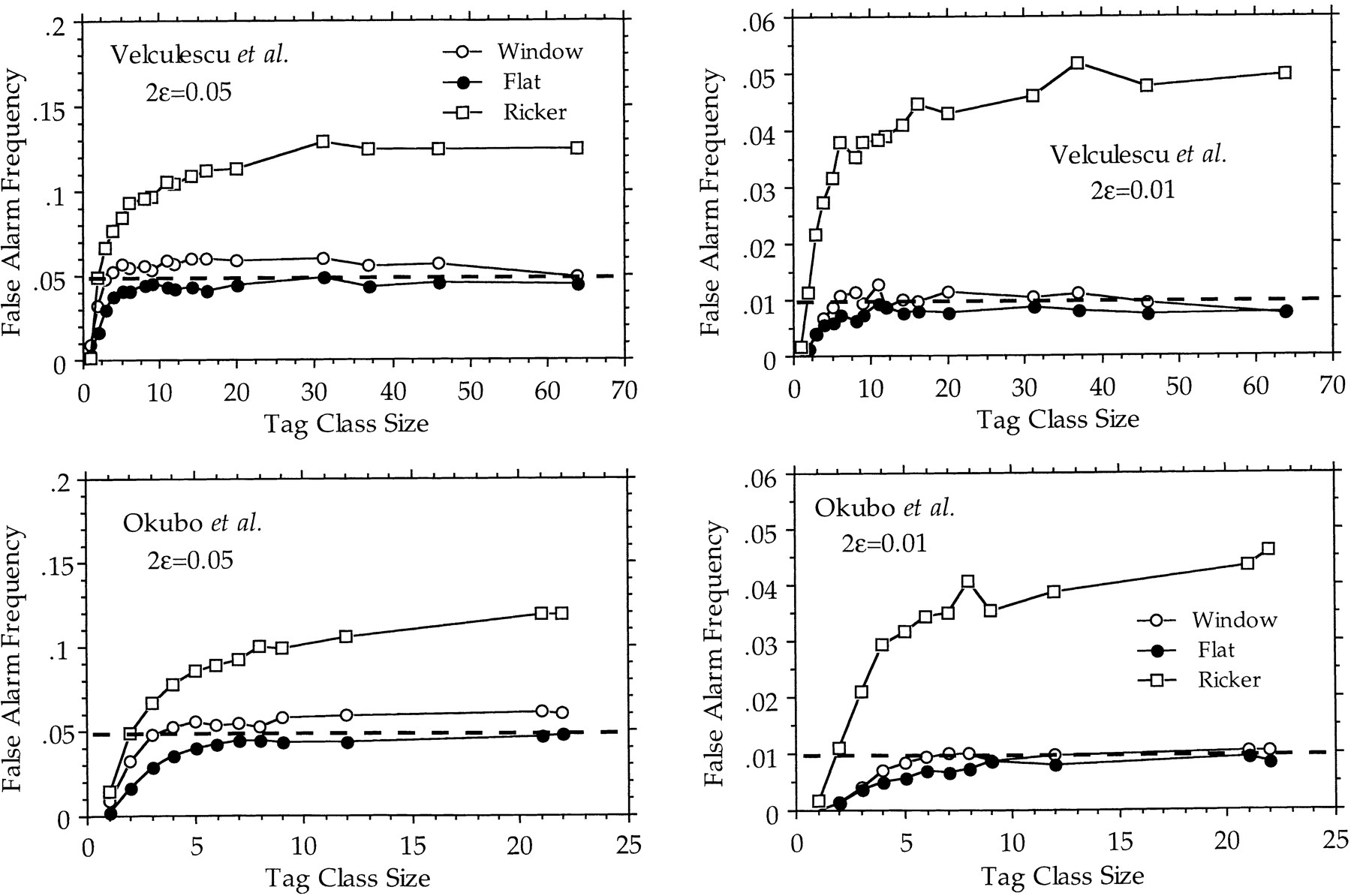
A crucial measure of the power of statistical significance tests is their rate of false alarm, that is, how often random fluctuations are expected to be mistaken for significant differences in the results. When analyzing the transcript profiles from two different libraries, a false alarm would cause a gene to be deemed differentially transcribed, whereas in fact it is not. The rate of false alarm is therefore a direct estimate of the fraction of false leads, when searching for differentially expressed genes on the basis of differences in tag counts. The rates of false alarm associated with theP < 0.01 and P < 0.05 confidence intervals listed in Table 1 have been computed by Monte-Carlo simulation on the basis of two experimental sequence tag distributions (Table2; Fig. 1). The rate of false alarms associated with the use of Equation { label needed for disp-formula[@id='E1'] } (in fact, its cumulative form Equation 9, see Methods) is very small for genes represented by small tag counts and slowly increases for higher tag counts, without ever exceeding the selected significance level. Such good behavior validates the use of the confidence intervals (Table 1) computed from Equation { label needed for disp-formula[@id='E1'] } and Equation 9 to assess the statistical significance of variations in digital Northern data. The curves labeled “window” characterize the very similar behavior of a slightly less conservative derivation of the same test (see Methods, Equation { label needed for disp-formula[@id='E15'] }). For comparison, Figure 1 also presents the behavior of another test, based on an inappropriate application of Ricker’s confidence intervals (Ricker 1937) (see Methods).
Publicly Available Distributions of Sequence Tags

|
[i] (Left) Data from Velculescu et al. (1995): Frequency of occurrence of each of the 428 transcript species represented in 840 SAGE tags randomly generated from a 3′-directed cDNA library from human pancreas. (Right) Data from Okubo et al. (1992): Frequency of occurrence of each of 641 transcript species represented in 982 randomly sequenced clones from a 3′-directed cDNA library from human liver cell line HepG2.
Rate of false alarm computed according to the confidence intervals listed in Table 1. (Top) Monte-Carlo simulation of the random sampling of 840 tags distributed according to the data from Velculescu et al. (1995; see Table 2). (Bottom) Monte-Carlo simulation of the random sampling of 982 ESTs distributed according to the data fromOkubo et al. (1992; see Table 2). The frequency of false alarm was computed for two significance levels (2ε = 5%, leftand 2ε = 1%, right) and plotted in function of the tag class size (from 1–64 for Velculescu et al., from 1–22 for Okubo et al.). In all cases, the rate of false alarm increases up to a plateau for larger class sizes. The test (cumulative form of Equation{ label needed for disp-formula[@id='E1'] }) derived from the flat p(λ) prior shows perfect behavior with a maximal rate of false alarm always less than the significance levels (broken lines). The test (cumulative form of Equation { label needed for disp-formula[@id='E15'] }) derived from the window p(λ) prior exhibits a slightly higher rate of false alarms. Both versions of the test exhibit conservative behaviors for class size <5, with a false alarm rate even less than expected. In contrast, Ricker’s confidence intervals (Equation 12) are grossly inadequate and lead to false alarm rates up to four times the significance level. Graphs are computed from the analysis of 1000 repetitions of each experiment.
DISCUSSION
An appropriate statistical test is now at our disposal to begin analyzing digital gene expression profiles in a more quantitative way. For example, the test can be used to determine how many genes appear regulated at various confidence levels using the data from a typical experiment (e.g., sampling a thousand clones). We analyzed the data gathered by Okubo et al. (1995) on the human promyelocytic leukemia cell line HL60 induced by dimethylsulfoxide (DMSO) or tetradecanoylphorbolacetate (TPA). Table 3 shows the 21 EST classes the occurrences of which exhibit significant variations at the 1% level. Most of the corresponding genes make biological sense in term of differentiation along the granulocyte or monocyte pathways.
List of ESTs Exhibiting Significant (P < 0.01) Differences in Abundance in the HL60 Cell Line Induced by DMSO or TPA
| EST ID | HL60 | HL60 + TPA | HL60 + DMSO | Significance |
| 418 | 22 | 10 | 1 | 3 × 10−7 |
| 211 | 24 | 10 | 2 | 4 × 10−7 |
| 19 | 8 | 23 | 2 | 8 × 10−7 |
| 356 | 16 | 2 | 0 | 3 × 10−6 |
| 380 | 12 | 1 | 0 | 6 × 10−5 |
| 135 | 4 | 12 | 0 | 6 × 10−5 |
| 285 | 14 | 8 | 1 | 1 × 10−4 |
| 2015 | 0 | 11 | 0 | 2 × 10−4 |
| 244 | 0 | 1 | 14 | 3 × 10−4 |
| 293 | 13 | 6 | 1 | 3 × 10−4 |
| 292 | 11 | 0 | 1 | 5 × 10−4 |
| 650 | 14 | 5 | 2 | 5 × 10−4 |
| 335 | 15 | 3 | 3 | 9 × 10−4 |
| 444 | 10 | 4 | 1 | 2 × 10−3 |
| 1674 | 0 | 8 | 1 | 4 × 10−3 |
| 155 | 0 | 8 | 3 | 4 × 10−3 |
| 861 | 6 | 1 | 0 | 7 × 10−3 |
| 305 | 6 | 2 | 0 | 7 × 10−3 |
| 1806 | 0 | 6 | 0 | 7 × 10−3 |
| 1808 | 0 | 6 | 0 | 7 × 10−3 |
| 1766 | 0 | 6 | 0 | 7 × 10−3 |
[i] Only the probability (computed according to Equations { label needed for disp-formula[@id='E7'] } and { label needed for disp-formula[@id='E8'] }) corresponding to the most significant transition (numbers in bold) is listed (Okubo et al. 1995). The total EST numbers sampled from the HL60, HL60 + TPA and HL60 + DMSO cDNA libraries are 845, 845, and 1058, respectively. ESTs 418, 211, 356, 285, 293, 292, 650, 335, 444, 861, 305 corresponding to ribosomal proteins, and EST 380, a tag to an unkown gene, exhibit a marked reduction of expression level in the DMSO- and/or TPA-induced differentiated states. In constrast, ESTs 135 (ferritin), 2015 (LD78/macrophage inflammatory protein), 1674 (methionine adenosyltransferase), 155 (thymosin β-4), 1806 (lipocortin), 1808 (thymosin β-10), and 1766 (a metallothionein) appear more abundant in the TPA-induced state, also highly enriched in EST 19 (the ubiquitous elongation factor 1-α). β-Actin (EST 244), is the only markedly increased tag in the DMSO-induced state. EST numbers, abundance data, and protein assignments are from the “body map” public expression data repository athttp://www.imcb.osaka-u.ac.jp (K. Okubo and K. Matsubara).
This example serves to discuss a subtle point in the interpretation of the P values computed from Equation { label needed for disp-formula[@id='E1'] }, 2, and 9. Rigorously, these equations apply to the case where a given gene (e.g., lipocortin) would have been selected for scrutiny before looking at the differences in cognate tag counts between libraries. When comparing two libraries without specifying in advance the transcripts we want to follow, and then focusing a posteriori on any of those exhibiting significant variations, the average number of expected false positiveN false isN false = PN species, whereN species is the number of different transcript species encountered and p is a given significance level. For instance, in the experiment analyzed in Table 3,N species is of the order of 600 (Okubo et al. 1995). It is therefore possible that up to four (600 × 7 × 10−3) out of the 21 transcript species listed in Table 3 are not truly differentially expressed.
Therefore, when two libraries are compared without prior gene selection, the use of a predetermined significance threshold is not advisable. The P values computed from Equation { label needed for disp-formula[@id='E1'] }, 2, and 9 should simply be used to rank all observed variations by order of decreasing statistical significance (analogous to how “similarity hits” are listed after database searches). The end-users can then make their own choice about the number of candidate target genes to be retained from the top of the list, bearing in mind the corresponding number of expected false positives.
Although the present interpretation of a digital Northern focuses on the genes exhibiting the most spectacular differential expressions, there is already ample evidence that small changes can cause drastic effects. Disease states caused by haploinsufficiency and trisomy suggest that 2 → 1 or 2 → 3 proportional changes in expression level may be of biological significance. Table 1 shows that there is no theoretical limit to the detection of such small variations from the comparison of digital expression patterns. Simply, the sampling size has to be increased enough for the required numbers of cDNA tags to reach a significance threshold (for instance 40 → 60, for a confidence level of 95%).
Analog hybridization-based methods (Fodor et al. 1991; Lennon and Lehrach 1991; Gress et al. 1992; Southern et al. 1992; Guo et al. 1994;Matson et al. 1995; Nguyen et al. 1995; Schena et al. 1995; Zhao et al. 1995) are traditionally opposed to digital tag-counting methods (Okubo et al. 1992; Matsubara and Okubo 1994; Lee et al. 1995; Okubo et al. 1995; Velculescu et al. 1995) for the analysis of differential gene expression. Both types of methods are sensitive to the quality of the original messenger RNA preparation and/or cDNA libraries. Analog methods promise higher throughput, lower cost, and have the capacity of studying transcripts on a much wider scale of abundance. They are therefore expected to supersede digital methods. On the down side, however, hybridization signals are not easily reproducible, and can be affected by many unknown properties such as the cDNA library complexity, as well as clone and sequence specific features (e.g., insert size, nucleotide composition, presence of repeats, secondary structure, triple helix interaction, etc.). Therefore, the hybridization-based methods require an estimation of the dispersion of the signal associated with each clone (i.e., enough repetitions of each experiment), and multiple standardization and calibration procedures to allow the meaningful comparison of hybridization patterns obtained from various sources (tissues, cell types, etc.) or from different membranes or chips. This is far from routine and has yet to be worked out. In contrast, and thanks to the unique properties of the Poisson distribution, digital methods have the capacity of providing a quantitative assessment of differential expression without the repetition or the standardization of individual tag-counting experiments. The statistical analysis presented here provides an objective method to analyze digital transcript profile data, and adapts it to fit (1) the number of leads one wants to be followed; (2) the fraction of false clues to be tolerated; and (3) the level of modulation in gene expression considered of biological interest.
A program is available on our web site (http://igs-server.cnrs-mrs.fr) to compute the confidence intervals corresponding to arbitrary significance levels and sampling sizeN 1 and N 2.
METHODS
Let us denote p(x) the probability to observex sequence tags of the same gene (i.e., from the 3′ end of the same transcript) when N cDNA clones are picked randomly. For each transcript representing a small (i.e., less than 5%) fraction of the library and N ⩾ 1000, p(x) will closely follow the Poisson distribution:
To compute the confidence intervals listed in Table 1, we made use of the cumulative distributions:
Generalization to Different Sampling Sizes
When different numbers of clones N 1 andN 2 are sequenced from the same library, Equation { label needed for disp-formula[@id='E5'] }becomes
Ricker’s Confidence Interval
The confidence interval computed from Equation { label needed for disp-formula[@id='E1'] } (and its cumulative form, Equation 9, a and b) is different from one introduced previously by Ricker (1937) although, at first, the two may appear to be related.
Given x occurrences of a sequence tag, Ricker’s formula defines a confidence interval [λmin, λmax] x for λ (again the actual number of transcripts of this type per N clones in the library) such as
However, an interesting use of Equation 12, a and b, is the estimation of the range of possible frequencies [λmin, λmax] x = 0 for cDNAs not yet encountered after picking N clones. For example, the 95% confidence interval is given by:
Influence of the Prior Distribution
In the bayesian context, it is prudent to assess the influence of the prior hypothesis used to derive Equation { label needed for disp-formula[@id='E1'] } and Equation { label needed for disp-formula[@id='E2'] }. The flatp(λ) prior allowing equiprobable λ values in the [0 , ∞] range might appear too broad and unrealistic. Nevertheless, it is the most intuitively neutral distribution one can use. The quick convergence of the Poisson distribution rends the contribution of extreme λ values negligible as soon as | λ − x | or | λ − y | increase. To verify this point, more reasonable distributions forp(λ) can be constructed by confining the accessible λ values within a window [λmin, λmax] x centered around the already observed value x. Such a window can for instance be Ricker’s confidence intervals as defined in the previous section (Equation 12, a and b). We then confine the only permitted values of λ to be in this interval, with an equal probability; therefore,
The confidence intervals for the usual 1% and 5% significance levels are given in Table 1 for both the flat and the window priorp(d = λ). There is little difference, with the test derived from using a flat prior being a bit more conservative, as expected. On the down side, Figure 1 shows that the test derived from the window p(λ) prior gives rise to a higher rate of false alarm.
We thank Drs. J. Weissenbach, D. Gautheret, R. Ewing, and C. Abergel for critically reading the manuscript. This work was sponsored by a collaborative research grant from Incyte Pharmaceuticals, Inc.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Notes
[4] Corresponding author.
Notes
[5] E-MAIL [email protected]; FAX 334 91 16 45 49.
REFERENCES
- ↵J.S. AaronsonB. EckmanR.A. BlevinsJ.A. BorkowskiJ. MyersonS. ImranK.O. Elliston(1996) Toward the development of a gene index to the human genome: An assessment of the nature of high-throughput EST sequence data. Genome Res. 6:829–845.
- ↵M.D. Adams(1996) Progress towards a complete set of human genes. in Genomes, molecular biology and drug discovery, eds M.J. BrowneP.L. Thurby(Academic Press, London, UK).
- ↵M.D. AdamsJ.M. KelleyJ.D. GocayneM. DubnickM.H. PolymeropoulosH. XiaoC.R. MerrilA. WuB. OldeR.F. Moreno(1991) Complementary DNA sequencing: Expressed sequence tags and human genome project. Science 252:1651–1656.
- ↵M.D. AdamsM. DubnickA.R. KerlavageR. MorenoJ.M. KelleyT.R. UtterbackJ.W. NagleC. FieldsJ.C. Venter(1992) Sequence identification of 2,375 human brain genes. Nature 355:632–634.
- ↵M.D. AdamsA.R. KerlavageR.D. FleischmannR.A. FuldnerC.J. BultN.H. LeeE.F. KirknessK.G. WeinstockJ.D. GocayneO. White(1995) Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. (The Genome Directory, Suppl.) Nature 377:3–174.
- ↵A. Agresti(1996) An introduction to categorical data analysis. (John Wiley, New York, NY).
- ↵W. Bains(1996) Virtually sequenced: The next genomic generation. Nature Biotechnol. 14:711–713.
- ↵M.S. BoguskiG.D. Schuler(1995) ESTablishing a human transcript map [News]. Nature Genet. 10:369–371.
- ↵Editorial (1996) Capitalizing on the genome. Nature Genet. 13:1–5.
- ↵S.P.A. FodorJ.L. ReadM.C. PirrungL. StryerA.T. LuD. Solas(1991) Light-directed, spatially addressable parallel chemical synthesis. Science 251:467–470.
- ↵T.M. GressJ.D. HoheiselG.G. LennonG. ZehetnerH. Lehrach(1992) Hybridization fingerprinting of high-density cDNA-library arrays with cDNA pools derived from whole tissues. Mamm. Genome 3:609–619.
- ↵Z. GuoR.A. GuilfoyleA.J. ThielR. WangL.M. Smith(1994) Direct fluorescence analysis of genetic polymorphisms by hybridization with oligonucleotides arrays on glass supports. Nucleic Acids. Res. 22:5456–5465.
- ↵A.S. KhanA.S. WilcoxM.H. PolymeropoulosJ.A. HopkinsT.J. StevensM. RobinsonA.K. OrpanaJ.M. Sikela(1992) Single pass sequencing and physical and genetic mapping of human brain cDNAs [see Comments]. Nature Genet. 2:180–185.
- ↵B. Kuska(1996) Cancer genome anatomy project set for take-off. J. Natl. Cancer Inst. 88:1801–1803.
- ↵L. HillierG. LennonM. BeckerM.F. BonaldoB. ChiapelliS. ChissoeN. DietrichT. DuBuqueA. FavelloW. Gish(1996) Generation and analysis of 280,000 human expressed sequence tags. Genome Res. 6:807–828.
- ↵N.H. LeeK.G. WeinstockE.F. KirknessJ.A. Earle-HuguesR.A. FuldnerS. MarmarosA. GlodekJ.D. GocayneM.D. AdamsA.R. Kerlavage(1995) Comparative expressed-tag analysis of differential gene expression profiles in PC-12 cells before and after nerve growth factor treatment. Proc. Natl. Acad. Sci. 92:8303–8307.
- ↵G.G. LennonH. Lehrach(1991) Hybridization analyses of arrayed cDNA libraries. Trends Genet. 7:314–317.
- ↵R.S. MatsonJ. RampalS.L. Pentoney JrP.D. AndersonP. Coassin(1995) Biopolymer synthesis on polypropylene supports: Oligonucleotide arrays. Anal. Biochem. 224:110–116.
- ↵K. MatsubaraK. Okubo(1994) Identification of new genes by systematic analysis of cDNAs and database construction. Curr. Opin. Biotechnol. 4:672–677.
- ↵C. NguyenD. RochaS. GranjeaudM. BalditK. BernardP. NaquetB.R. Jordan(1995) Differential gene expression in the murine thymus assayed by quantitative hybridization of arrayed cDNA clones. Genomics 29:207–216.
- ↵R. Nowak(1995) Entering the postgenome era. Science 270:368–369.
- ↵C. O’Brien(1997) Cancer genome anatomy project launched. Mol. Med. Today 3:94.
- ↵K. OkuboN. HoriR. MatobaT. NiiyamaA. FukushimaY. KojimaK. Matsubara(1992) Large scale cDNA sequencing for analysis of quantitative and qualitative aspects of gene expression. Nature Genet. 2:173–179.
- ↵K. OkuboK. ItohA. FukushimaJ. YoshiiK. Matsubara(1995) Monitoring cell physiology by expression profiles and discovering cell type-specific genes by compiled expression profiles. Genomics 30:178–186.
- ↵W.E. Ricker(1937) The concept of confidence or fiducial limits applied to the Poisson frequency distribution. J. Am. Statist. Assoc. 32:349–357.
- ↵S. Siegel(1956) Nonparametric methods for the behavioral sciences. (McGraw-Hill, New York, NY).
- ↵M. SchenaD. ShalonR.W. DavisP.O. Brown(1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270:467–470.
- ↵E.M. SouthernU. MaskosJ.K. Elder(1992) Analyzing and comparing nucleic acid sequences by hybridization to arrays of oligonucleotides: Evaluation using experimental models. Genomics 13:1008–1017.
- ↵K.D. Tocher(1950) Extension of the Neyman-Pearson theory of tests to discontinuous variates. Biometrika 37:130–144.
- ↵V.E. VelculescuL. ZhangB. VogelsteinK.W. Kinzler(1995) Serial analysis of gene expression. Science 270:484–487.
- ↵A.S WilcoxA.S. KhanJ.A. HopkinsJ.M. Sikela(1991) Use of 3′ untranslated sequences of human cDNAs for rapid chromosome assignment and conversion to STSs: Implications for an expression map of the genome. Nucleic Acids Res. 19:1837–1843.
- ↵N. ZhaoH. HashidaN. TakahashiY. MisumiY. Sakaki(1995) High-density cDNA filter analysis: A novel approach for large-scale, quantitative analysis of gene expression. Gene 156:207–213.