
The Role of Genomics in Studying Genetic Susceptibility to Infectious Disease
The notion that selection during epidemics or longer periods of exposure to infectious diseases may have had a major effect in modifying the constitution of the human genome is not new. It was proposed, at least in outline, by A.E. Garrod in 1931. In his remarkable book, The Inborn Factors in Disease, he suggested that infectious diseases may have been a major selective force in human evolution and in shaping our biochemical individuality. In 1948, J.B.S. Haldane, unimpressed with the idea that the extremely high frequency of thalassemia in certain racial groups from the Mediterranean region might reflect an unusually high mutation rate in these populations, proposed that these diseases might have come under intense selection because of heterozygote advantage against malaria. It was, in effect, Haldane’s remarkable insight that opened up the field of the investigation of genetic susceptibility to infection.
Although considerable progress was made in relating the frequency and distribution of different protein polymorphisms to past or present infection, until recently the field was bedeviled by difficulties relating to population homogeneity, founder effects, and gene drift. However, with an increasing ability to analyze human variability at the DNA level, progress has been much more rapid. Comparing the sequences of genes common to rodents and humans, Murphy (1993), found that host defense genes are much more diverse than those for other families of proteins, an observation suggesting that selection in many species has resulted from exposure to different infectious agents. The way in which DNA analysis is transforming our understanding of the reasons for the distribution and high frequency of the human hemoglobin variants, the problem first posed by Haldane, is reviewed by Flint et al. (1993). Clearly, an analysis of the human genome with respect to variable susceptibility to infection is already beginning to provide important new insights into the mechanisms of human diversity.
In considering genetic susceptibility to infectious disease it is important not only to consider the genetic makeup of the host but also that of the infectious agent. Here, we focus mainly on recent studies of the genetic factors that may modify susceptibility to infection in humans and touch only briefly on some recent developments in microbial genetics that may play an important role in this interaction. Mouse models for studying genetic susceptibility to infection have been reviewed recently elsewhere (McLeod et al. 1995).
Genetic Variability of Humans in Response to Infection
Malaria
The inherited disorders of hemoglobin, notably the structural hemoglobin variants S and E, and the α and β thalassemias, are the most common monogenic diseases in man (Fig. 1). The observation that the hemoglobin variants that occur at polymorphic frequencies are unevenly distributed among tropical populations, and the discovery that in every population in which thalassemia is common there is a different pattern of mutation, suggests that these diseases must have arisen independently in different parts of the world and reached their high frequency by locally-acting factors such as selection and drift (Flint et al. 1993). Although earlier studies suggesting that the sickle cell trait might be protective against malaria (Allison 1964) have been confirmed more recently (Hill et al. 1991; Olumese et al. 1997), until the advent of the DNA era it was extremely difficult to demonstrate a relationship between the high frequency of thalassemia and previous or past malarial infection (Flint et al. 1993). Much of the recent information about this question has come from studies of α thalassemia in Melanesia and Polynesia.
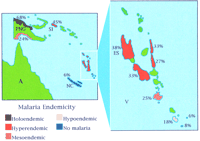
The percentage gene frequencies (numbers on map) of α+thalassemia in Melanesia parallels the intensity of malaria transmission (endemicity). The malaria transmission intensity goes from most intense (holoendemic) to least intense (hypoendemic). (A) Australia; (PNG) Papua New Guinea; (SI) Solomon Islands; (NC) New Caledonia; (V) Vanuatu; (ES) Espiritu Santo—an island in Vanuatu where the study by Williams et al. (1996) was performed. Figure adapted with permission from Miller (1996).
There are two main forms of α thalassemia, α+ and αO, which are usually caused by the deletion of one or both of the linked pairs of α-globin genes on chromosome 16, respectively (Higgs et al. 1989). The normal human genotype is represented by αα/αα. Heterozygotes for α+thalassemia (−α/αα) are clinically normal, whereas homozygotes (−α/−α) have mild anemia with reduced levels of hemoglobin in their red cells. Surveys of malaria prevalence have shown that before eradication campaigns were initiated, the disease was endemic below 2500 meters in Papua New Guinea and in parts of Melanesia. It has been found that in these regions, α+thalassemia has a gene frequency proportional to the prevalence of malaria (Flint et al. 1986). As the disease becomes less frequent in the interior of Papua New Guinea, so does α+ thalassemia, and there is a steady decline in the gene frequency across Vanuatu down to New Caledonia, where malaria is absent. It is always possible that this distribution simply reflects the fact that α+ thalassemia was introduced to these island populations by founders from the mainland and that the gene frequency was gradually diluted as they migrated south. However, this explanation was excluded by the finding that the molecular forms of α+ thalassemia and the restriction fragment length polymorphisms associated with them in the island populations are completely different from those of the Asian mainland (Flint et al. 1986). Thus there is a clear altitude and north–south correlation between α+ thalassemia and present or past malaria in this region.
Recently, a case control study carried out in Madang, on the north coast of Papua New Guinea, has demonstrated a highly significant protective effect of the homozygous state for α+ thalassemia against the serious complications of Plasmodium falciparummalaria (S.J. Allen, A. O’Donnell, N.D.E. Alexander, M.P. Alpers, T.E.A. Peto, J.B. Clegg, and D.J. Weatherall, in prep.). Evidence about the possible mechanisms involved has also come recently from studies in Vanuatu (Williams et al. 1996). In a large cohort of babies of different α-globin genotypes, followed carefully over the first years of life, it was found that those homozygous for α+thalassemia have a higher frequency of both Plasmodium vivaxand P. falciparum malaria in the first 2 years of life, after which this effect is not seen. These observations were strengthened by the finding of significantly higher frequencies of enlarged spleens in the homozygous α+ thalassemic infants. These quite unexpected observations offer a conceptual framework for understanding how α+ thalassemia might protect against malaria. It appears that homozygotes are more susceptible to malaria but only at a time when the disease rarely kills. Thus, it is possible that this may provide them with an immunizing dose of malaria that offers later protection. Interestingly, in areas in which both types of malaria occur, the earliest infections in life are usually attributable toP. vivax. Furthemore, there is some evidence that there may be cross-immunization between this form of malaria and that attributable to P. falciparum (Maitland et al. 1997). The fact that babies with α+ thalassemia are more susceptible to P. vivaxinfections early in life may explain their later resistance to the disease.
If the homozygous state for α+ thalassemia has no effect on fitness, this may be an example of a transient rather than a balanced polymorphism, whereby if malaria persists, the α+ thalassemia gene will go to fixation. However, insufficient information is yet known about the phenotypic expression of α+ thalassemia to be certain that it has no deleterious effects.
It is now clear that many other red cell polymorphisms have been shaped by malaria. The protective effect of a deficiency of the red cell enzyme, glucose-6-phosphate dehydrogenase (G6PD), has been confirmed recently by Ruwende et al. (1995), who have found that both female heterozygotes and male hemizygotes have a reduced risk, ∼50%, of having severe malaria. Protection is also mediated by variation in the structure or synthesis of a variety of red cell-surface antigens (Miller 1994); the molecular basis for the absence of the Duffy antigen, found many years ago to be associated with resistance toP. vivax malaria, has now been defined as a substitution in the binding site for the GATA 1 erythroid transcription factor at position −46 to the promoter (Tournamille et al. 1995). By gene mapping analysis for the Melanesian form of ovalocytosis, which is sometimes difficult to identify phenotypically, it has been found that this polymorphism provides complete protection against cerebral malaria (S. Allen, pers. comm.). It is also possible that differences in the Na/K composition of red cells between races may have resulted from exposure to malaria in the past, although so far the molecular basis for this phenomenon has not been investigated (Miller 1994).
Work over recent years has shown that genetic variability attributable to selection by malaria is not confined to the red blood cell. Certain polymorphisms of the HLA–DR system are associated with substantial protection against both cerebral malaria and severe malarial anemia (Hill et al. 1992). Furthermore, as in the case of the thalassemias, the particular pattern of polymorphisms varies among different populations. Whereas earlier studies of HLA associations with disease viewed the histocompatibility antigens as informative markers for closely linked genes that might encode for genuine immune response determinants, more recent studies at the molecular level have suggested that the class 1 and 2 genes and their products may, in themselves, function as immune response agents through determinant selection. It is now believed, particularly in the case of the infectious disease associations, that the products of these loci act through differential selection of particular peptide epitopes from a pathogen for presentation to T lymphocytes. Studies of the HLA–B53 association with malaria have provided an excellent example of how this might be mediated through the identification of HLA–B53 cytotoxic lymphocytes that recognize a particular parasite epitope (Hill et al. 1992;Hill 1996).
Another interesting malaria association that has been uncovered recently involves the gene encoding tumor necrosis factor (TNF). Gambian children who are homozygous for a single-base change at position −308 of the TNF promoter have a markedly increased risk of death from cerebral malaria (McGuire et al. 1994). Reporter gene analysis indicates that this polymorphism can increase levels of TNF expression (Wilson et al. 1997), an observation that is compatible with a substantial body of clinical and experimental evidence that excessive TNF production is a major factor in the pathogenesis of cerebral malaria (for review, see Kwiatkowski 1995). Associations of the −308 variant with other protozoal and bacterial infections, mentioned below, provide further evidence of an important genetic effect at this locus.
The case of TNF, although compelling, raises a general problem about the application of the genomic approach to the analysis of susceptibility to disease. On one hand, there is a strong clinical association with a candidate gene that is known to be involved in the disease process, and for which there is good evidence that its level of expression is a critical determinant of clinical outcome (Tracey 1995). On the other hand, the evidence that the polymorphism is functional relies on reporter gene experiments that may or may not be relevant to the in vivo situation. Attempts to demonstrate that the polymorphism affects the levels of TNF production in blood taken from uninfected individuals have failed (Westendorp et al. 1997), although it could be argued that relatively strong phenotypic effects might easily be missed if the experimental conditions for TNF stimulation were not exactly right or if, as seems likely, the genetic control of TNF production is multifactorial. Furthermore, it is important to prove beyond doubt that a polymorphism, whether functional or not, is the actual cause of the disease association rather than simply a genetic marker. The TNF gene resides in the class III region of the MHC, densely surrounded by genes of fundamental importance to host defense, so the dissection of disease associations across this region and replication of these associations in different populations will be of great importance.
If the high gene frequencies for red cell and other polymorphisms reflect protection against malaria, it is difficult at first sight to understand why they have not become more evenly distributed in the tropical populations of the world. Why, for example, is hemoglobin S not seen in Southeast Asia or hemoglobin E in Africa? Why is Melanesian ovalocytosis not more widespread, and why do different HLA–DR polymorphisms appear to be protective in particular populations? It seems likely that these observations reflect the fairly recent appearance of malaria as the principal agent that has maintained these polymorphisms, a notion that is strengthened by an analysis of the relationship between globin gene polymorphisms and their associated restriction fragment length polymorphism haplotypes (Flint et al. 1993). They also suggest that malaria must have been a particularly powerful selective force to have recruited so many deleterious traits in such a short time.
Other Parasitic Infections
Although less progress has been made than in the case of malaria, there is increasing evidence that variability in host responsiveness to other parasitic infections may have a strong genetic basis (Hill 1996). A major recent advance has been the use of segregation and linkage analysis in 11 Brazilian families to localize a gene, termedSM1, that governs intensity of infection by Schistosoma mansoni (Marquet et al. 1996). It lies at chromosome 5q31–q33, a region encoding interleukin-4 (IL-4), IL-5, and several other immunological mediators that are thought to contribute to host defense in this disease. The result is of general importance because it illustrates the feasibility of making a genome-wide search to discover critical susceptibility loci for common infections. Reasonably strong HLA–DR associations have been found in cases of leishmaniasis (Cabrera et al. 1995), onchocerciasis (Meyer et al. 1994), and filariasis (Yazdanbaksh et al. 1995). Similarly, associations between the TNF −308 promoter variant have been found in leishmaniasis (Cabrera et al. 1995). This is of particular interest because, as in the case of malaria, mucocutaneous leishmaniasis has been found to be accompanied by high circulating levels of TNF.
Bacterial Disease
It has been suspected for some time that there may be individual susceptibility to bacterial disease, and progress in investigating this possibility has been increased dramatically by the availability of probes for a wide variety of candidate genes. The importance of genetic variability in TNF production in response to infection, as noted in the case of malaria, has been further emphasised by the recent observation that TNF promoter polymorphisms are related to susceptibility to some important bacterial infections, notably meningogoccal meningitis (Nadel et al. 1996), lepromatous leprosy (Roy et al. 1997), and trachoma (Conway et al. 1997). Although it is too early to say precisely how these associations have arisen, it is clear that the further exploration of TNF variants and their receptors in relationship to bacterial disease may play an important role in elucidating the mechanisms of individual susceptibility to these conditions.
The mannose-binding protein (MBP), a serum lectin that plays an important role in immunity, is emerging as another interesting candidate. MBP is involved in the activation of complement and acts directly as an opsonin, using the C1q receptor on macrophages. Several mutations have been described at the MBP locus that lead to low levels of serum MBP (Lipscombe et al. 1992; Madsen et al. 1994). Because MBP deficiency has been implicated in an increased susceptibility to bacterial and fungal infections in children, it is clear that the association between these variants and infection is well worth exploring by the analysis of large population samples.
It has been suspected for some years that the ability to secrete the soluble forms of the ABO blood group antigens into saliva and other body fluids, which is the secretor status of an individual, may be associated with varying susceptibility to bacterial infection. Recently, the molecular basis for the nonsecretor phenotype has now been determined (Kelly et al. 1995). The gene involved, Sec2,encodes an α-(1,2)-fucosyl transferase. It turns out that nonsecretors are homozygous for a nonsense allele at this locus. It has been found recently that this variant occurs at a high frequency in sub-Saharan Africans, suggesting that it may be the predominant nonsecretor mutation in many populations (Hill 1996). If so, screening for this mutation will facilitate the study of population associations of secretor status and bacterial infection.
Evidence for the associations between bacterial infection and polymorphisms of other candidate genes, including natural resistance-associated macrophage protein 1 and the FcγRII receptor, is summarized by Hill (1996).
Recently, a Maltese village has been reported in which four children had marked susceptibility to atypical mycobacterial infection. Immunological studies suggested that they had defective production of TNF in response to endotoxin and a failure to up-regulate the cytokine in response to γ interferon (IFN-γ). Sequence analysis of the gene for the IFN-γ receptor 1 revealed a point mutation at nucleotide 395 that introduces a stop codon and results in a truncated protein that lacks the transmembrane and cytoplasmic domains of the receptor (Newport et al. 1996). Although this may be an extreme example of genetic susceptibility to mycobacterial infection, it provides another important indication of the remarkable potential for variability in host susceptibility related to genetic variability of the TNF response.
Viral Disease
The molecular basis for individual susceptibility to viral disease is becoming clearer, at least in a few diseases. Between 5% and 20% of individuals infected by the hepatitis B virus develop a chronic carrier state that may be associated with liver disease and hepatocellular carcinoma. It has been found recently that different HLA–DR polymorphisms are associated with either chronic carriage or rates of viral clearance (Almari and Batchelor 1994; Thursz et al. 1995).
Another puzzling observation about the natural history of viral illness, that is, why some patients who are regularly exposed to the human immunodeficiency virus (HIV) do not become infected, recently has been explained, at least in part, at the molecular level. Liu et al. (1996) observed that the CD4+ T cells of two such individuals were highly resistant in vitro to the entry of primary macrophage–tropic virus but were readily infected with transformed T-cell-line-adapted viruses. It was found that these individuals were homozygous for a defect in the CKR-5 gene, which codes for the coreceptor for primary HIV-1 isolates. It turned out that they had inherited a defective CRK-5 allele containing an internal 32-bp deletion. The resulting protein is truncated and cannot be detected at the cell surface. This mutation has been observed in other patients who were resistant to HIV infection, although not in all.
Further possible examples of polymorphisms that may modify the response to virus infection are summarized by Hill (1996).
Selection of Other Common Monogenic Diseases by Infection
Though no other monogenic diseases approach the global frequency of the thalassemias; there are, nevertheless, a few that are particularly common in some populations. It has been suggested, for example, that the very high frequency of the common cystic fibrosis allele in Northern Europeans might reflect selection against a major infectious disease, possibly one of the diarrheal illnesses that swept across Europe in the past. Similarly, genetic and epidemiological studies have provided some evidence that the high frequency of Tay–Sachs disease in some Jewish populations reflects heterozygote resistance to tuberculosis in some of the ghettos of Eastern Europe (for review, see Weatherall 1996). Although these are interesting speculations, and there have been similar hypotheses proposed to explain the distribution of the ABO blood groups, they must be viewed with caution. In a recent theoretical paper Thompson and Neel (1997)show that there is no need to postulate positive selection with respect to the more common disease-associated alleles.
Genetic Variability in Pathogens
Although it has long been realized that infectious agents can rapidly change their genetic makeup and, hence, avoid host defense mechanisms or therapeutic agents, recent theoretical (Taddei et al. 1997) and experimental (Sniegowski et al. 1997) studies, reviewed byMoxon and Thaler (1997), provide some interesting insights into the dynamics of this process. These new concepts may have considerable importance to the further understanding of drug resistance and virulence, and may have implications beyond the field of infectious disease.
As pointed out by Moxon and Thaler (1997), pathogenic bacteria face a particularly arduous task because infections occur within a very short time during which pathogens encounter varying host polymorphisms, a wide range of immune mechanisms, and microenvironments, which may already be modified by the administration of potent antibiotics. It is becoming clear that bacteria have a remarkable repertoire of different genetic systems with which to tackle these problems.
One such mechanism is the generation of so-called mutator alleles, which can lead to an increase in mutation rate. Such alleles may predispose many different genes to potentially beneficial mutations and may “hitch hike” because the mutator allele and the selected alleles of other genes may be linked, especially in asexual clonal populations. Furthermore, certain pathogens, including viruses, bacteria, and even parasites, have subsets of genes that are excessively prone to mutation through, for example, slipped-strand mispairings, conversions, or point mutations. These hypermutable genes encode surface molecules, such as adhesins and invasins, which are involved in interactions with host molecules. Hence, populations of microorganisms can use combinatorial systems rapidly to generate phenotypic variation, which can influence antigenicity, motility, chemotaxis, attachment to host cells, and many other qualities that can alter their virulence. The molecular mechanisms for these remarkable adaptive changes, which are only just starting to be worked out, are summarized by Moxon and Thaler (1997).
Comment
The exploration of host susceptibility to infectious agents at the molecular level has only just started. Although recent work on globin, the histocompatibility loci and the genetic basis for variable TNF response to infections, and host resistance to viral infections is starting to provide some indications of the complexity of the mechanisms involved, for the most part knowledge of the field is restricted to the demonstration of simple associations between the incidence or severity of disease and particular protein and DNA polymorphisms. So far there have been very few attempts to try to define susceptibility genes by whole genome searches, and the list of definite susceptibility loci is relatively small. Although it will undoubtedly unravel biological systems of enormous complexity, further work in this field, involving both host and bacterial genomes, has important implications, not just for a better understanding of human diversity, but for directing work toward more effective control and management of infectious diseases.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL janet.watt%mailgate.JR2{at}ox.ac.uk; FAX 44 (0) 1865 222501.
- Cold Spring Harbor Laboratory Press








