
Long Human–Mouse Sequence Alignments Reveal Novel Regulatory Elements: A Reason to Sequence the Mouse Genome
The utility of sequencing entire genomes of bacteria and fungi is amply demonstrated. For instance, as the complete set of genes for each species is catalogued, one can ascertain the full complement of encoded proteins, obtain insights into the function of new proteins by sequence matches to known proteins, and measure the transcriptional levels of all genes in a genome under various environmental conditions or at different stages of the cell cycle (Boguski et al. 1996; Velculescu et al. 1997). The currently sequenced genomes consist primarily of coding regions with little sequence between the genes, and the amount of genetic information in each segment is usually quite high. Larger genomes from more complex organisms have a considerable amount of DNA between the genes and in introns that interrupt the coding regions, and one could question whether it is useful to determine the sequences of all of these noncoding regions. Indeed, the concerted efforts to determine partial sequences of normalized cDNA libraries have generated rich and very useful databases, such as the TIGR database (TDB) and dbEST (Adams et al. 1991; Boguski 1995). Efforts from Schuler and his colleagues to unite the several sequences from each set of cDNA clones representing a unique gene, the UniGene project, will organize this large amount of sequence data. As of late 1996, the UniGene database contained samples of sequences of almost 50,000 genes, which could represent a majority of human genes (Schuler et al. 1996). Of these UniGene clusters, 16,000 have been placed on the human genome map, which will greatly aid in positional cloning of interesting genes.
Although TDB, dbEST, and UniGene are extremely useful, they do have limitations. For instance, comparison of a long genomic DNA sequence with the expressed sequence tag (EST) databases is a very effective method for identifying many of the exons. A recent comparison of programs for predicting gene structures revealed that inclusion of protein sequence similarity searches improved performance (Burset and Guigó 1996), and the most efficient approach to identifying genes in a 284-kb sequence from the end of the short arm of human chromosome 16 was by comparison with the EST databases (Flint et al. 1997). However, it is rare for the EST entries to encompass the complement to an entire mRNA, and hence all exon assignments usually cannot be obtained from the ESTs. In constrast, an alignment of complete sequences of homologous loci between mouse and human will reveal almost all of the exons as regions of particularly high sequence conservation, at least for most loci (see below). This approach is not limited by the abundance of the mRNA in the tissue samples or the completeness of coverage of the cDNA sequence in the EST databases.
A second limitation of the EST databases is the paucity of noncoding sequence in the entries; such sequences are limited to the 5′- and 3′-untranslated regions of the mRNA. Most DNA sequences involved in regulation of gene expression are in noncoding regions; obviously these DNA segments flanking and interrupting the coding regions will have to be sequenced to obtain sequences of regulatory regions. This will occur as the projects determining the complete human genome sequence are completed. But how does one extract information about potential regulatory sequences, and is that information useful in designing experiments to test proposed functions? One standard approach is to determine the DNA sequence from additional species and seek out candidates for conserved sequences, that is, sequences that change only slowly during evolution because such sequence alterations are detrimental to the organism. Clusters of invariant or slowly changing positions in the aligned sequences are “phylogenetic footprints”, which are reliable guides to important regulatory regions (for review, see Gumucio et al. 1996). Historically, these phylogenetic footprints were observed in segments of moderate length (several hundred to a few thousand base pairs) aligned among many species by inspection. Because of the genetic information available and the growing ability to manipulate its genome, mouse has become the most popular mammal for sequencing and comparison with human. We wished to test whether automatically generated pairwise alignments of very long sequences (20–100 kb) between human and rodent species could provide information that generates testable hypotheses.
Despite the strong rationale to search for conserved DNA sequences, it is not clear a priori that comparisons of long genomic DNA sequences will reveal regulatory signals. Much regulation of transcription is accomplished by the binding of transcription factors to the DNA (for review, see Mitchell and Tjian 1989). The typical recognition sites are only 6–8 nucleotides long, and limited variants of this short string will bind the protein with high affinity. However, runs of 16 consecutive identical bases can be expected to occur strictly by chance when comparing two 100-kb sequences (Dembo et al. 1994), raising the possibility that single, isolated transcription factor binding sites will be lost in the background noise of spurious matches. Also, biological variation between the two species can confound the approach based on similarity and conservation. For instance, homologous human and mouse transcription factors may have somewhat different specificities, and in some cases humans may use a different set of transcription factors than mice to regulate a homologous gene in a different way. This search for conserved noncoding sequences is further complicated by the differences in patterns of evolution at various loci (Koop 1995). Thus, one needs to test whether or not real regulatory sequences will be detected by this approach. In this perspective we show that the use of new computational tools combined with recent experimental evidence argues strongly that this comparative approach is effective and useful.
Loci with Functional Tests Based on Sequence Alignments
The β-globin gene clusters of humans (HBB) and mice (Hbbd ) are very good models to test the ability of sequence comparisons of noncoding regions to detect regulatory regions. Regulatory elements have been mapped to the proximal DNA sequences extending to as much as 800 bp 5′ to the cap sites of the developmentally regulated genes and to the distal locus control region (LCR) needed for opening an active chromatin domain in erythroid cells and enhancing expression of genes within the locus (for review, seeStamatoyannopoulos and Nienhuis 1994). Recent advances in software development allow comparisons of two long sequences to be carried out automatically. Figures 1 and 2 give pips (percentidentity plots), which provide easily interpreted summaries of such long alignments. In a pip, the percent identity (from 50% to 100%) in each gap-free aligning segment is plotted using the coordinates of the human sequence, and notable features in the human sequence, such as genes, repeats, and DNase hypersensitive sites (HSs) are placed along the horizontal axis. Figure1 shows the region from the LCR through the ε-globin gene and the region encompassing the δ- and β-globin genes. Three general conclusions can be drawn from the human–mouse comparison of this locus. First, as expected (Makalowski et al. 1996), the exons are conserved. Second, not all of the extragenic sequences align (e.g., between δ- and β-globin genes), showing that some regions have diverged significantly. And, finally, there are noteworthy matches both in the proximal 5′-flanking region of each gene, which contains the promoter, and in the distal sequences at the 5′ end of the gene cluster comprising the LCR. Thus, sequence conservation is generally correlated with regions having experimentally determined regulatory function.
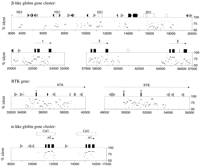
Pips of the human–mouse comparisons from the β-globin gene cluster (HBB) and the Bruton’s tyrosine kinase (BTK) locus, and for a human-rabbit comparison of the α-globin cluster. The human and mouse sequences are first examined by RepeatMasker (A.F.A. Smit and P. Green,http://ftp.genome.washington.edu/cgi-bin/RepeatMasker) to identify known interspersed repeats. Interspersed repeats other than MIR and LINE2 (Smit 1996) are masked (marked as unalignable), and a descendent of the Sim program (Huang et al. 1990) is used to compute all the local alignments that score above a certain approximate significance. The percent identity in each gap-free aligning segment is plotted using the coordinates of the human sequence, and notable features in the human sequence, such as genes (exons are black boxes), repeated DNA sequences (SINEs other than MIR are light gray triangles pointing toward the A-rich 3′ end; L1 repeats are open arrowed boxes; MIR and LINE2 elements are black triangles and pointed boxes, respectively; and other interspersed repeats are dark gray triangles) and DNase hypersensitive sites (white boxes labeled HSn, wheren is an integer) are placed along the top of the plots. Note that the percent identity is only plotted between 50% and 100%, limiting the output to a range of mildly to strongly conserved sequences. The first two rows of panels shows the LCR and ε-globin gene and also the region from δ-globin to β-globin inHBB. The next row shows two portions of the BTKlocus. The last row shows the comparison between α-globin genes of human and rabbit; the published sequences of the mouse α-globin genes are not long enough to be effective in this analysis, and the mouse genes lack the CpG islands.
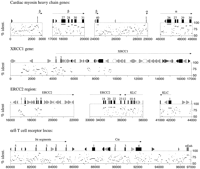
Pips of portions of the human–rodent comparisons for cardiac myosin heavy chain genes MYH6 and MYH7 (top row); the XRCC1 gene (second row); locus with bothERCC2 and KLC (third row); and the α/δ TCR loci (fourth row).
The β-globin LCR is marked by five major, developmentally stable DNase HSs (Tuan et al. 1985; Forrester et al. 1986), of which the sequences of four are available from human and mouse (HSs 1–4). Naturally, much of the experimental work on the LCR has focused on these major HSs, and the minimal regions needed for position-independent expression in transgenic mice, which we call HS cores, have been mapped (for review, see Grosveld et al. 1993). However, a more detailed look at the pip of the human and mouse β-globin gene clusters shows that sequences outside these well-studied cleavage sites are as conserved as some of the HS cores. Recent studies (Caterina et al. 1991; Jackson et al. 1996a,b) verify that these sequences outside the HS cores do contribute to the ability of the LCR DNA fragment to establish and/or maintain an open chromatin domain after stable integration into the genomic DNA. Furthermore, a detailed examination of the HS2 core, which stands out as being more highly conserved than any other noncoding region of the gene cluster, reveals conserved sequences that had not been implicated previously in its function. Again, recent studies show that one of these, an invariant E-box in the HS2 core, plays an important role in enhancement (Lam and Bresnick 1996; Elnitski et al. 1997).
The value of human–mouse genomic sequence comparisons for locating novel regulatory elements was underscored by a recent analysis of theBTK locus, including the gene for cytoplasmic Bruton’s tyrosine kinase, defects in which lead to X-linked agammaglobulinemia. Again, strongly matching sequences are dispersed throughout the locus, but some regions have diverged too far to be reliably aligned (Oeltjen et al. 1997). Also, exons tend to be among the most highly conserved regions, but extensive, high-scoring matches are also seen in introns and in the 5′-flanking DNA (Fig. 1). The matches in the introns are particularly dramatic around exons 4 and 5 as well as exon 1. In fact, the region from ∼900 bp 5′ of the noncoding exon 1 to ∼3000 bp 3′ to this exon is strikingly well-conserved. Experiments both in vitro and in vivo show that binding sites for PU.1, SpiB, and Sp1 in the 200 bp 5′ to exon 1 contribute to the specific expression ofBTK in the hematopoietic cell lineage (Sideras et al. 1994; Himmelmann et al. 1996; Muller et al. 1996). The pairwise comparison, however, suggested the hypothesis that the much larger region just described has a role in regulation, and this was supported by additional transient transfection experiments (Oeltjen et al. 1997).
Examination of mammalian α-globin genes (HBA) shows that even the absence of expected sequence matches can lead to productive, testable hypotheses. Despite their descent from a common ancestral gene and the requirement for coordinated, tissue-specific regulation, most mammalian α- and β-globin genes are in very different genomic DNA contexts and are regulated in distinctly different ways. In particular, the α-globin gene clusters are highly G+C rich with several CpG islands and are in constitutively active chromatin (Craddock et al. 1995; Fischel-Ghodsian et al. 1987; Hardison et al. 1991), whereas the β-globin gene clusters are more A+T rich, are devoid of CpG islands, and undergo chromatin domain opening only in erythroid tissues (Groudine et al. 1983; Margot et al. 1989;Stamatoyannopoulos and Nienhuis 1994). The flanking and internal sequences of the rabbit and human α-globin gene comprise a prominent CpG-rich island that serves as a strong, enhancer-independent promoter in a variety of transfected mammalian cells (Charnay et al. 1984; James-Pederson et al. 1995). Although the CpG islands are present in orthologous positions in the rabbit and human α-globin gene clusters, sequence alignments show the unexpected result that specific protein binding sites are conserved only in the 100-bp of proximal 5′-flanking regions and not throughout the CpG islands (Hardison et al. 1991; Yost et al. 1993), illustrated in Figure 1. This suggested that the effects of the CpG island outside the proximal promoter were to provide a more permissive environment for promoter activity than do bulk A+T-rich DNAs, but that this effect is not dependent on binding of specific transactivators at discrete locations. The postulated general effect of CpG islands is supported by three lines of evidence: (1) The level of gene expression increases with increasing size of the CpG island included in transfection constructs; (2) deletion of prominent binding sites for Sp1 and YY1 has no effect; and (3) addition of α-globin gene promoter fragments to a transcriptionally inactive CpG island gives a much higher level of expression after integration into the genome than does addition of these fragments to an A+T-rich DNA fragment (Shewchuk and Hardison 1997). This more permissive effect of CpG islands may be exerted at least in part at the level of chromatin structure, as CpG island DNA from the α-globin gene has a much lower affinity for nucleosome reconstitution in vitro than does the A+T-rich DNA fragments from the β-globin gene (Shewchuk 1997).
Loci With Alignments That Lead to Testable Hypotheses
Earlier comparisons of long human and rodent genomic regions did not report the formulation and testing of hypotheses about gene regulation and thus were less informative about the value of long human–rodent sequence alignments. These loci included (1) the α- and β-myosin heavy chains used in cardiac muscle, (2)XRCC1, an X-ray repair–complementing gene whose product is involved in ligation, (3) ERCC2, an excision repair–complementing gene that encodes a single-stranded DNA-dependent ATPase and DNA helicase (this gene will also complement defective nucleotide excision repair in xeroderma pigmentosum cells of complementation group D), and (4) a region encoding portions of the α- and δ-subunits of the T-cell receptor (TCR) required for the cellular immune response. Portions of the pips for these regions are shown in Figure 2. One general and key point is that the patterns of conservation vary considerably between the loci, probably reflective of the differences in rate of divergence in different regions of the genome (Li et al. 1990; Hardison et al. 1991; Koop 1995). Thus, criteria as to what constitutes “conserved” may have to be adjusted, depending on the genomic context of the sequences examined.
The comparison of human and hamster genes encoding the cardiac myosin heavy chains shows matches throughout the entire loci, with the expected highest-scoring matches in exons and consistently lower scores in the introns, a pattern that contrasts with the BTKcomparison. High-scoring matches are also seen 5′ to exon 1 of both the β- and the α-myosin heavy chain genes, extending for >2000 bp 5′ to exon 1 in the case of the α-myosin heavy chain gene. One could hypothesize that the latter corresponds to a regulatory region. Experimental tests to date are controversial on this point, with experiments in transfected cells indicating that all regulatory elements (e.g., the thyroid hormone response) are located within 300 bp 5′ to exon 1, whereas experiments in transgenic mice implicate several thousand base pairs 5′ to exon 1 in regulation (for review, see Robbins 1996). Further experiments could test whether these disparate conclusions result from differences in the types of regulatory elements revealed by the different assays. More analysis could also test the correspondence with conserved sequences. One important complication illustrated by this locus is that these genes are regulated somewhat differently in humans and rodents. In particular, the β-myosin heavy chain is the major isoform in the adult ventricle of humans but not hamsters. Obviously, the particularcis-acting elements involved in that aspect of regulation would be expected to differ in the two species. Another intriguing match is found both in intron 30 of the β-myosin heavy chain gene and intron 29 of the α-myosin heavy chain gene, which are homologous introns. A portion of this intron is as about as highly conserved as the surrounding exons, which is suggestive of some important function. This is a distinctive feature of only this intron in this locus.
Comparison of the XRCC1 genes (Lamerdin et al. 1995) shows very little match in the introns, but long, high-scoring alignments extend for 3000 bp 5′ to exon 1, as well as some shorter matches at the 3′ end of the gene. At the present time, no experimental tests of role of this substantial 5′-flanking region have been reported. We tested the hypothesis that some other gene could be located in this region by searching for matches in dbEST. A very strong match to one cDNA sequence is found with the mouse 5′-flanking region (positions 7732–8500, which is homologous to human positions 1001–1800). As shown in Figure 3, the presumptive exon sequence is identical to that of the cDNA, and we conclude that one of the reasons for the strong conservation is the presence of a coding region of a currently unknown gene. Presumably the homologous cDNA in human has not yet been sequenced.
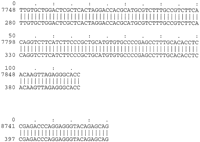
Alignments of the mouse XRCC1 region and a mouse EST (the reverse complement of GenBank accession no. W14361). Aligning regions 7748–7864 and 8741–8765 of the mouse sequence (accession no. L34078) correspond to positions 280–396 and 397–421 of the EST and to positions 1020–1136 and 2465–2489 of human (accession no. L34079). The alignment terminates at the end of the EST, and consensus splice junctions (intron = CT...AC), conserved between human and mouse, indicate a gene transcribed from right to left.
In contrast, the comparison of the ERCC2 locus (Lamerdin et al. 1996) shows matches in the 5′ flanking region of exon 1 for only ∼200 bp, indicative of a very small regulatory region. One plausible explanation is that ERCC2 may be expressed at about the same level in all cells, given the ubiquitous need for excision repair of the DNA. Thus, it may be under relatively simple control, manifested in this analysis as a limited number ofcis-regulatory sequences. The adjacent, oppositely transcribedKLC2 gene shows a series of short matching segments for ∼1000 bp 5′ to the cap site. In such cases where the matching sequences are primarily restricted to exons, and especially when the pattern of expression differs in some respects between human and rodent, examination of the homologous locus in a species more closely related to humans, such as a prosimian primate, could be informative. For instance, regulatory elements that are conserved in primates but divergent in some other mammalian order should be readily detectable.
The α- and δ-TCR locus shows a dramatic pattern of high-scoring sequence matches throughout the almost 100 kb sequenced in human and mouse (Koop and Hood 1994; Koop et al. 1994). Surprisingly, many introns are more similar than the surrounding exons, not only those encoding the J segments but also those encoding the constant regions of the TCRs. The results indicate that much of the entire locus is conserved, but it is not yet clear what the constraints are that prevent substantial sequence alteration in this locus. Some possibilities are the need for programmed DNA rearrangements, a dispersion of an extraordinary number of regulatory regions throughout the locus, or protection of this region of the chromosome from nucleotide substitutions. Elucidating the basis for this extensive sequence similarity in the TCR locus will require considerably more work. It would be helpful in this case to examine the sequences of a more distantly related species, such as the chicken. Given the slower rate of evolution in this region, allowing for a longer time or phylogenetic distance in the comparison may eliminate the less important sequences from the alignments and bring out the truly functional ones.
Conclusion: Sequence the Mouse Genome
Recent experimental results for BTK and both the β-like and α-like globin genes show that comparison of human and mouse genomic sequences is effective at discovering novel regulatory elements. This case was not so obvious even 12 months ago. Although single, isolated protein-binding sites are not detected by current methods, the ordered clusters of binding sites characteristic of many regulatory elements are readily seen. It is also clear that genomic sequence data can be effectively exploited using existing software. For instance, the pips for complete human and mouse genomic DNA sequences (each being ∼3 billion bp) could be computed in a month on an inexpensive 1997 workstation. This survey shows that in most cases, human–rodent sequence comparisons should be sufficient to highlight conserved regions possibly contributing to gene regulation. Thus, sequencing the mouse genome along with that of human will allow discovery of regulatory elements in both species. Without the mouse sequence for comparison, little of the information contained in noncoding segments of the human genome will be accessible by computational approaches. Furthermore, two other considerations argue for obtaining additional sequences for particular loci. The pattern of expression differs between human and mouse for some genes, probably resulting from changes in regulatory elements. In these cases, alignments with a sequence from a more closely related species will likely be informative. In other loci, the mutation rate for that genomic region may be so low that human–mouse comparisons are not optimally informative. Alignments with sequences from a more distantly related species should be sought for such loci.
Complete pips for the genomic regions discussed above are available on the World Wide Web at http://globin.cse.psu.edu/. That site also contains links to GenBank records for the sequences and to Medline abstracts of relevant literature citations. Software to prepare pips is still evolving and will be the subject of a later report.
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵5 Corresponding author.
-
E-MAIL webb{at}cse.psu.edu; FAX (814) 865-3176.
-
- Received May 6, 1997.
- Accepted July 30, 1997.
- Cold Spring Harbor Laboratory Press








