
Centromeric instability and chromoanasynthesis observed in nine supernumerary marker chromosomes resolved with long-read genome sequencing
- Kristine Bilgrav Saether1,2,6,
- Angelo Salazar Mantero1,3,6,
- Marlene Ek1,3,
- Maria Pettersson1,3,
- Elisabeth Syk Lundberg1,3,
- Christopher M. Grochowski4,
- Claudia M.B. Carvalho5,
- Jesper Eisfeldt1,2,3 and
- Anna Lindstrand1,2,3
- 1Department of Molecular Medicine and Surgery, Karolinska Institutet, 17176 Stockholm, Sweden;
- 2Science for Life Laboratory, Department of Molecular Medicine and Surgery, Karolinska Institutet, 17176 Stockholm, Sweden;
- 3Department of Clinical Genetics and Genomics, Karolinska University Hospital, 17177 Stockholm, Sweden;
- 4Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas 77030, USA;
- 5Pacific Northwest Research Institute, Seattle, Washington 98122, USA
-
↵6 These authors contributed equally to this work.
Abstract
Small supernumerary marker chromosomes (sSMCs) remain a diagnostic challenge despite sequencing advances. As the field shifts toward cytogenomics, there is a need to establish methodologies to resolve these complex genetic variants at base pair resolution, as well as to identify their chromosomal origin and formation mechanism. Here, we apply long-read genome sequencing (lrGS) in combination with the telomere-to-telomere (T2T-CHM13) assembly to characterize the structure and genomic content of 10 clinically detected sSMCs. We use sequencing data to reconstruct the derivative chromosomes, identify breakpoint junctions (BPJs), and infer formation mechanisms. We resolve the BPJs of nine of the 10 sSMCs at base pair resolution. The analysis reveals six simple intrachromosomal rearrangements (one continuous and five discontinuous) with one to three BPJs, one complex three-way translocation with two BPJs, and two highly complex intrachromosomal rearrangements with five and nine BPJs, respectively. Breakpoint analysis reveals distinct mechanistic signatures: Simple sSMCs show features consistent with microhomology-mediated end joining (MMEJ) or microhomology-mediated break-induced replication (MMBIR), whereas complex sSMCs demonstrate evidence of translocation, chromoanasynthesis, and breakage–fusion–bridge (BFB) cycles. Haplotype analysis supports trisomy rescue in four cases, including all three complex sSMCs. In summary, our study demonstrates that lrGS combined with T2T-CHM13 enables detailed structural and mechanistic characterization of sSMCs, providing experimental support for disruption of trisomy rescue as a key formation mechanism. This work illustrates the feasibility of resolving highly challenging chromosomal abnormalities using long-read sequencing technologies.
Small supernumerary marker chromosomes (sSMCs) are rare but potentially clinically significant when detected. These extra chromosomes may be linear, either as a centric minute or as an inverted duplicated structure, or ring-shaped; the latter are termed small supernumerary ring chromosomes (sSRCs). Their size and genomic content are highly variable, and determining their chromosomal origin is often challenging with G-banding owing to the small size. Although fluorescence in situ hybridization (FISH) has long been the standard tool for characterizing such events, genomic technologies are now emerging as powerful alternatives. However, resolving sSMCs and interpreting their clinical significance remains challenging, in part because they frequently are mosaic and often comprise repetitive or poorly annotated genomic regions (Reddy et al. 2013; Mostovoy et al. 2024).
sSMCs are found in ∼0.044% of the human population, with ∼30% of carriers exhibiting mild to severe phenotypes (Liehr and Weise 2007). They are classified based on shape, genomic content, and degree of mosaicism. About 20%–30% of small supernumerary chromosomes are sSRCs, whereas the remainder are linear sSMCs, often derived from inverted duplications. Approximately 70% of small supernumerary chromosomes originate from acrocentric chromosomes, with Chromosome 15 being the most common source (particularly inv dup(15)/idic(15)). To understand the clinical implications, each case requires careful molecular and clinical evaluation to determine the chromosomal origin and genomic content of the sSMC. Pathogenicity may result from extra copies of triplosensitive genes, as well as epigenetic effects (Anderlid et al. 2001; Mostovoy et al. 2024). Approximately 70% of sSMC carriers are asymptomatic, and a subset is discovered during infertility investigations, in which the increased risk of infertility could be explained by imbalances that arise during gametogenesis. Overall, genomic characterization is relevant, especially for genetic counseling in prenatal diagnostics.
The majority of linear sSMCs possess a centric minute structure, meaning that they are linear with a centrally located centromere (Reddy et al. 2013). For some sSMCs, the mechanisms have been extensively studied, such as the isodicentric Chromosome 15 (idic(15)). In these cases, the extra chromosome consists of two copies of a segment from Chromosome 15q11–q13, arranged in an inverted mirror-image orientation with two centromeres (dicentric) and formed through nonallelic homologous recombination between inverted repeats. The mechanisms underlying other sSMCs are less understood but include postfertilization errors, as well as trisomy and monosomy rescue events following meiotic and mitotic nondisjunction (Grochowski et al. 2018; Kurtas et al. 2019).
Base pair–level resolution of sSMC structures has long been a challenge owing to breakpoints frequently occurring in repetitive regions such as centromeres, telomeres, and acrocentric p-arms, or within low copy repeats (LCRs) (Grochowski et al. 2018; Mostovoy et al. 2024; Schuy et al. 2024). This has limited our understanding of both their formation mechanisms and clinical consequences. Long-read genome sequencing (lrGS) enables high-accuracy analysis of the genome, allowing sSMCs to be resolved at nucleotide-level precision. In addition, the telomere-to-telomere (T2T-CHM13) assembly provides enhanced sequence completeness across satellite and other repetitive regions commonly involved in structural variant (SV) formation (Nurk et al. 2022; Bilgrav Saether et al. 2024).
This study aims to evaluate the capability of emerging genomic technologies, specifically lrGS and optical genome mapping (OGM) in combination with the T2T-CHM13 reference genome, to achieve high-resolution breakpoint mapping of sSMCs. We seek to determine whether these approaches can fully resolve complex chromosomal rearrangements and provide mechanistic insights into their formation, as well as assess their potential clinical relevance.
Results
Genetic characterization of sSMCs
Seven of the sSMCs were ring shaped, two were linear, and one remained unresolved. Nine were intrachromosomal events, consisting of genomic material from single chromosomes, whereas one involved material from three different chromosomes (Table 1).
Overview of the studied sSMCs
By combining short-read genome sequencing (srGS) and lrGS, we resolved all breakpoint junctions (BPJs) of the sSMCs except for three cases with breakpoints in highly repetitive regions (RD_P272, RD_P166, and RD_P274). OGM analysis (performed in two cases lacking srGS data) could verify the ring structure of r(14) (RD_P550) and eight of 10 segments and their order in der(X) (RD_P586) (Supplemental Fig. S1A,B). Six of the resolved sSMCs were classified as simple events, involving one to three BPJs on a single chromosome (RD_P276, RD_P278, RD_P273, RD_P166, RD_P550, RD_P275), and three were complex rearrangements involving either more than three BPJs (RD_P272, RD_P586) or more than one chromosome (RD_P328) (Table 2). In traditional cytogenetics, a simple sSMC is defined as a continuous DNA fragment of a chromosome that has lost its distal ends, whereas a discontinuous sSMC consists of intrachromosomal material with more than one fragment, and complex sSMCs consist of material from more than one chromosome. Using this system, we found that five of the intrachromosomal sSMCs were discontinuous, and one was continuous. However, because the aim of this study is to classify sSMCs based on their characterized BPJs, we will hereafter use a different system that reflects their molecular complexity rather than their cytogenetic appearance.
Characteristics of the resolved breakpoint junctions (BPJs) of marker and ring chromosomes, with coordinates mapped to the T2T-CHM13v2.0 genome build
Twenty-seven out of the 32 sSMC segments involved OMIM genes (range, 1–159). One hundred eighty-five were triplosensitive and 202 intellectual disability (ID) genes (Supplemental Table S2).
Characterization of simple sSMCs
The simple sSMCs consisted of one to three segments (Fig. 1; Supplemental Figs. S2–S7). The simplest sSRC (r(14); RD_P550) consisted of one segment sized 7.2 Mbp; although cytogenetically classified as a linear marker, lrGS revealed a ring structure, as the reads spanned over the BPJ fusing the end and start of the segment. Three sSRCs consisted of two segments (r(7), RD_P278; r(5), RD_P276; and r(8), RD_P273). Of those, r(7) and r(5) had similar structures, with one segment originating from the p-arm and one spanning the centromere; in contrast r(8) consisted of one segment with a centromeric breakpoint, and the other originated from the q-arm. Finally, two sSMCs consisted of three segments: the r(20) (RD_P275) and r(13) (RD_P166). The segment lengths ranged from 8 kbp to 28.4 Mbp, and three of 13 were inverted relative to the centromeric segment.
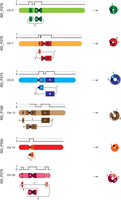
Six simple small supernumerary marker chromosomes (sSMCs). Schematic of the six resolved simple sSMCs and their chromosome copy-number profile. Letters indicate segments, and numbers indicate junctions. The centromere is shown as a black circle. For ring Chromosome 13, the two lines ending in circles represent a breakpoint junction (BPJ) between segments A and B (dotted line on the right) that was not identified.
In total we detected 12 BPJs in six simple sSMCs (range, one to three BPJs per sSMCs). The BPJ architecture revealed seven of 12 with microhomology (2–7 nt) and five of 12 with insertions (range, 4–204 nt; three templated and two random). In eight of the 12 BPJs, one or both breakpoints were located within genes; four of the 12 were in segmental duplications, and 11 of the 12 were in repeat elements, eight of which involved centromeric sequences (Table 2).
Characterization of complex sSMCs
The three complex sSMCs (RD_P272, RD_P586, RD_P328) differed in structure and complexity. The interchromosomal sSMC, der(7)t(X;7;5) (RD_P328), consisted of three segments from three different chromosomes, joined by two BPJs (Fig. 2A–C). The three fragments originated from Chromosome 5 (2.9 Mbp), Chromosome 7 (31.4 Mbp), and Chromosome X (15 Mbp), with the smaller fragments flanking the Chromosome 7 fragment.

A complex sSMC involving Chromosomes X, 7, and 5. (A) Schematic of the chromosomes involved and the resulting structure of the derivative Chromosome 7 identified in RD_P328. (B) Integrative Genomics Viewer images of short-read genome sequencing (srGS) and long-read genome sequencing (lrGS) data for each BPJ. (C) Breakpoint sequences of the two BPJs with the chimeric sequence in the center aligned to the reference sequences shown in pink and blue. Microhomology is indicated in green. Coordinates according to genome build T2T-CHM13v2.0.
The two more complex sSMCs were both intrachromosomal (r(9), RD_P272, der(X); RD_P586) (Fig. 3; Supplemental Figs. S8, S9) consisting of six and 10 segments, respectively. Unlike the other sSMCs, these two incorporate triplicated segments (segments B, G, J in der(X) and C, D, E in r(9)) (Fig. 3).
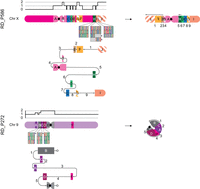
Two complex sSMCs with features of trisomy rescue. der(X) (RD_P586): On the left side, the copy-number profile and affected genomic regions A–J with triplications shown with striped patterns. For regions H, I, and J, the identified haplotypes’ single-nucleotide variant pattern is displayed below with the sSMC haplotype indicated with *. In the middle, a subway plot shows how BPJs 1–9 connect segments A–J. On the right side, the resolved sSMC structure is given. r(9) (RD_P272): as in A. The two lines ending in circles represent a BPJ between segments B and E (dotted line on the right) that was not identified.
Haplotype analysis
Analysis of SNV patterns within the sSMC revealed three distinct haplotypes in all three complex cases, der(7), der(X), and r(9), as well as in r(13), suggesting that the sSMCs originate from different chromatids (Fig. 3; Supplemental Fig. S10).
In total, we detected 16 BPJs in three complex sSMCs (two, five, and nine for der(7), r(9), and der(X), respectively). The BPJ architecture revealed that five of the 16 showed microhomology (1–3 nt) and eight of the 16 insertions (1–224 nt; all random). In eight of the 16 BPJs, one or both breakpoints were located within genes, five of the 16 in segmental duplications, and 15 of the 16 in repeat elements, none involving centromeric sequences. In der(X), matched repeats were observed in five out of nine BPJs (Table 2).
Altogether, the BPJ architecture differed between the simple and complex sSMCs, suggesting distinct mechanisms of formation. In the simple sSMCs, we observed a high frequency of centromeric BPJs.
Discussion
Long-read genomic analysis using the T2T-CHM13 reference resolved nine of 10 sSMCs, identifying six simple intrachromosomal and three complex events, whereas one centromeric ring-shaped sSMC remained unresolved. The simple sSMCs consisted of one to three segments and BPJs, whereas the complex cases involved up to 10 segments, including duplicated fragments, and/or spanned multiple chromosomes. Notably, the resolved sSMCs displayed distinct breakpoint signatures and structural features, suggesting involvement of different mutational mechanisms reflecting diverse paths of sSMC formation.
Traditionally, sSRCs are believed to arise through terminal breaks on each chromosome arm, followed by direct end-to-end fusion via double-strand break repair mechanisms. However, recently it has been shown that sSRCs may form through mechanisms related to the formation of complex genomic rearrangements, such as breakage–fusion–bridge (BFB) cycles (Nazaryan-Petersen et al. 2018) and chromanasynthesis (Grochowski et al. 2018; Kurtas et al. 2018; Schuy et al. 2024).
The breakpoint characteristics of sSMCs studied here correlate with their structural complexity. All simple sSMCs (Fig. 1) had at least one BPJ located within a centromere (Table 2), and except for those centromeric repeats, no matched repeats were observed. Only two BPJs were blunt, both in r(13), and the remaining featured either insertions or microhomology consistent with formation via MMEJ or MMBIR. Whether these simple sSMCs arose in a single event or through a multistep process such as BFB cycles remains uncertain. The observation that all simple sSMCs consisted largely of centromeric material suggests that their formation may be driven by centromere instability. This might be a consequence of sSMCs containing centromeric fragments being more stable, as they can attach to the mitotic spindle. However, centromeric instability itself has been broadly implicated in pathological chromosomal rearrangements and human disease (Barra and Fachinetti 2018). Recent work on individuals with trisomy 21 (Down syndrome, OMIM 190685) has highlighted that centromeres may vary in size and structure and that extreme centromere size asymmetry is a frequent feature in trisomic cells (Mastrorosa et al. 2025). Future studies characterizing centromere size and composition in sSMCs may provide further insights into the exact contribution of centromeres to sSMC formation and stability. Segmental duplications were identified in nine of 28 junctions, but only one (junction 2, r(7)) had breakpoints within matching segmental duplications (Table 1), suggesting they played little role in sSMC formation.
In the complex group, blunt-ends and short microhomologies are present in the der(7) BPJs (Fig. 2; Table 2), suggesting that they likely formed through MMEJ.
The combined data indicate that this interchromosomal sSMC formed through a three-way translocation event. The other two complex sSMCs, r(9) and der(X), are intrachromosomal rearrangements consisting of five and 10 segments, respectively (Fig. 3). These segments include both duplicated and triplicated regions (present once or twice on the resolved sSMC structure). Although their overall structures appear similar, the BPJ features differ (Table 2). The r(9) BPJ characteristics, microhomology and random insertions, are consistent with chromoanasynthesis.
In contrast, der(X) consists of duplicated fragments, inversions, and telomere loss, suggesting that it may have formed through BFB cycles. Five out of nine BPJs in der(X) are located within matched repeat elements, indicating that the rearrangement was LINE-mediated. This is supported by our previous work showing that complex rearrangements clustering on the same chromosome can be mediated by both Alu and LINE elements (Nazaryan-Petersen et al. 2018). However, a highly complex neocentric sSMC that appears to have formed through chromothripsis highlights that chromoanagenesis of complex sSMCs may involve different underlying formation mechanisms (Weber et al. 2022).
Across four sSMCs, one simple and all three complex (r(7), r(9), r(13), and der(X)), three haplotypes were detected, indicating that the sSMC is likely a remnant of a trisomy rescue event. In der(X), however, only three of the 10 segments showed such features, indicating partial trisomy rescue. Previous studies have reported chromothripsis in micronuclei during incomplete trisomy rescue as a formation mechanism for sSMCs (Kurtas et al. 2019; Matsubara et al. 2020), which resembles the patterns observed in r(9), r(13), and der(X). Micronucleus formation and incomplete trisomy rescue involving multiple chromosomes have also been described, leading to complex chromosomal rearrangements (Storchová and Kloosterman 2016), which may explain the r(7) and the presence of three haplotypes. This likely represents a peri-zygotic event, with the mosaic nature of the sSMCs attributable to mitotic instability. This is a similar phenomenon to that described for multiple de novo copy-number variants (CNVs) (Du et al. 2022).
The 10 individuals with sSMCs were initially referred for genetic investigation owing to neurodevelopmental and neurological symptoms, as well as short stature (Table 1). These features are commonly seen in patients undergoing chromosome analysis and consistent with previously reported clinical presentations in individuals with sSMCs (Conlin et al. 2011; Guilherme et al. 2011; Pristyazhnyuk and Menzorov 2018; Kurtas et al. 2019; Li et al. 2022). Even though we have detailed the exact coordinates of involved genomic segments and genes (Supplemental Table S2) whether the duplication or triplication of specific genes contribute to the phenotype remains unclear, as transcriptional activity and downstream effects were not assessed.
We and others have previously shown that lrGS is an effective genomic technique for identifying and resolving large chromosomal rearrangements within highly repetitive regions (Porubsky et al. 2022; Bilgrav Saether et al. 2024; Eisfeldt et al. 2024; Mostovoy et al. 2024; Schuy et al. 2024; Ten Berk de Boer et al. 2024). In this study, lrGS was critical for resolving the breakpoints of sSMCs with BPJs located in highly repetitive and complex regions. In contrast, srGS, performed in eight of the 10 sSMCs, detected and even resolved two events, r(7) and the complex interchromosomal der(7), but could not phase the remaining sSMC structures with BPJs in large repeats (Table 2). Therefore, in this study, srGS data were primarily useful for CNV analysis. OGM confirmed eight of the 10 involved segments and the overall structure of the most complex sSMC (der(X)) and was a valuable tool for validation in this case.
The T2T-CHM13 assembly added >250 Mbp of new sequence to the reference genome compared with GRCh38 (Nurk et al. 2022; Bilgrav Saether et al. 2024). We and others have shown that these sequences can be clinically relevant, especially for SV analysis (Porubsky et al. 2023; Bilgrav Saether et al. 2024; Ten Berk de Boer et al. 2024; Paulin et al. 2025). Of note, in four of the 28 sSMC BPJs reported here, one breakpoint was located in genomic sequence missing from GRCh38 but present in T2T-CHM13 (Table 2). The specific sSMCs (r(5), r(13), r(14), der(X)) could therefore not have been resolved with lrGS aligned to GRCh38; hence, T2T-CHM13 was essential. However, for r(13) and r(14), with breakpoints in the p-arm of acrocentric chromosomes, T2T-CHM13 still did not enable full characterization. Specifically, although lrGS allowed us to identify the breakpoints in these regions, we were not able to fully characterize the BPJs. Similarly, for r(9), two segments remain unplaced in the final sSMC structure, most likely owing to polymorphic repetitive sequences at the breakpoints. Although one BPJ cannot be resolved with lrGS aligned to T2T-CHM13, cytogenetic investigations have shown that it has a ring structure. This highlights the continued importance of cytogenetics as a complementary method to genomics for resolving highly complex rearrangements, particularly when breakpoints are located in polymorphic or repetitive regions. Some of these limitations may be addressed using pangenomes, which can represent polymorphic sequences more comprehensively (Liao et al. 2023).
In summary, we demonstrate the power of lrGS, in combination with the T2T-CHM13 assembly, to resolve complex and repetitive breakpoint regions and capture variation within centromeres and acrocentric p-arms, allowing a detailed molecular characterization of sSMCs. This approach delineates distinct formation mechanisms for simple and complex markers and reveals evidence of trisomy rescue in both groups. It also points toward improved diagnostics by enabling more complete capture and interpretation of cytogenetic aberrations with higher resolution and accuracy compared with traditional karyotyping and array-based methods.
Methods
Ethics approval and consent
Ethical approval was given by the regional ethical review board in Stockholm, Sweden (ethical permit nos. 2012/222-31/3 and 2024-05341-01). These ethics permits allow the use of clinical samples for analysis of scientific importance as part of clinical development. The IRB approval does not require us to get written consent for clinical testing. The research conformed to the principles of the Helsinki Declaration.
Short-read genome sequencing
srGS was performed using the HiSeq Xten platform. The samples were sequenced using 2 × 150 bp paired-end reads, on one flow cell for each sample, which resulted in 30× median coverage. The data were aligned to T2T-CHM13 (chm13v2.0) (Nurk et al. 2022) using BWA-MEM (v0.7.17) (Li and Durbin 2009). SVs were detected using CNVnator (v0.4.1) (Abyzov et al. 2011) and TIDDIT (v2.8.1) (Eisfeldt et al. 2017). The variant calls were annotated and filtered based on allele frequency using SVDB (2.8.2), and relevant SV calls were visualized and quality-controlled using the Integrative Genomics Viewer (IGV) (Robinson et al. 2011).
Long-read genome sequencing
lrGS was conducted using the Pacific Biosciences (PacBio) Revio system, with average read lengths ranging from 10 to 19 kbp and average coverage between 22× and 67× (Supplemental Table S1). In brief, reads were aligned to T2T-CHM13 (chm13v2.0) using minimap2 (2.24-r1122) (Li 2018); SNVs were called using DeepVariant (1.5.0) (Poplin et al. 2018); SVs were called using Sniffles (1.0.12-1) (Sedlazeck et al. 2018); and CNVs were called using HiFiCNV (v1.0.1-4de7d40) (https://github.com/PacificBiosciences/HiFiCNV).
A variant frequency database consisting of nine samples was used to annotate each variant with population frequency. WhatsHap (1.7) (Martin et al. 2023) was used to phase variants and haplotag the BAM file. All commands are given in the Supplemental Code. De novo assembly was performed using Hifiasm (0.23.0–h5ca1c30_0) (Cheng et al. 2021), and SVs were called using SVIM-asm (v1.0.3) (Supplemental Code; Heller and Vingron 2021).
Optical Genome Mapping
Optical Genome Mapping (OGM) was performed as described previously (Bilgrav Saether et al. 2024). In brief, ultra-high-molecular-weight genomic DNA from cases RD_P550 and RD_P586 were loaded onto a flow cell on the Saphyr optical mapping system (Bionano Genomics). Data were aligned to an in silico T2T-CHM13 reference genome using the Bionano Solve v3.7 RefAligner module.
Identification and characterization of sSMCs
SV calls (srGS, lrGS, OGM) were intersected with CNV patterns (srGS) and manually searched for informative signals involving the chromosomal regions of interest. Their obtained list was visualized in IGV (Robinson et al. 2011).
To identify regions present in T2T-CHM13 but absent from GRCh38, breakpoint positions were intersected using BEDTools (Quinlan and Hall 2010) with a list of regions not present in GRCh38 (Bilgrav Saether et al. 2024).
To understand the function and biological consequences of the sSMCs, lists of disease-associated genes were downloaded on November 14, 2024, from OMIM (Amberger et al. 2015) (Morbid OMIM version 31.0, 4629 genes), ClinGen (1281 triplosensitivity genes) (Rehm et al. 2015), and Genomics England PanelApp (2792 ID genes) (Martin et al. 2019). Gene positions in GRCh38 were converted to T2T-CHM13 coordinates by extracting gene information from a gene annotation file downloaded from the UCSC Table Browser (Perez et al. 2025). Finally, the coordinates were intersected with the genomic locations of the segments in the sSMCs.
Haplotype analysis was performed by manual inspection of informative SNVs using IGV. Trisomy rescue was considered when heterozygous SNVs were observed that did not align with either of the two haplotypes defined by WhatsHap, suggesting the presence of a third haplotype.
Data access
The long-read sequencing data generated in this study have been submitted to the European Genome-phenome Archive (EGA; https://ega-archive.org) under accession number EGAS50000001466.
Competing interest statement
A.L. has received speakers’ honoraria from Pacific Biosciences and Illumina. All other authors declare no competing interests.
Acknowledgments
We acknowledge the support from the National Genomics Infrastructure (NGI) at Science for Life Laboratory in providing assistance in massive parallel sequencing. The computations were performed on resources provided by SNIC through the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under project SNIC sens2017106, sens2020021, and sens2023005. An author of this publication is a member of the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability ERN-ITHACA (EU framework partnership agreement ID: 3HP-HP-FPA ERN-01-2016/739516). A.L. was supported by grants from the Swedish Research Council (2019-02078), Region Stockholm (FoUI-1000468 and FoUI-978581), the Rare Diseases Research Foundation (Sällsynta Fonden), the Swedish Brain Foundation (FO2024-0128-HK-44), and the Swedish Cancer Society (24 3504 Pj).
Author contributions: A.L. and J.E. conceptualized the study, administered the project, and provided resources, supervision, and funding. K.B.S. performed bioinformatic analysis. K.B.S. and A.S.M. performed initial data curation and formal analysis, and M.E., A.L., J.E. C.M.B.C., C.M.G., and E.S.L. helped with clinical and genomic interpretations of results. M.E. did wet-laboratory experiments. K.B.S., A.L., and J.E. wrote the original draft. K.B.S. made the figures supported by A.S.M. A.S.M, M.E., M.P., E.S.L., C.M.G., and C.M.B.C. reviewed and edited the original draft.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.281175.125.
-
Freely available online through the Genome Research Open Access option.
- Received July 9, 2025.
- Accepted March 2, 2026.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








