
Abstract
The exchange and subsequent incorporation of genetic material between distinct lineages, known as introgression, has emerged as a crucial concept in understanding human evolutionary history. With the advent of high-throughput sequencing technologies and the publication of the draft Neanderthal genome in 2010, Green and colleagues were able to demonstrate the presence of Neanderthal DNA in present-day Eurasians, a signature of past interbreeding events with archaic humans. This integration of genetic material from extinct human relatives, such as Neanderthals and Denisovans, into the genomes of modern humans due to historical gene flow events is known as archaic introgression. As new methods and data sets uncover a more complex intermingling between our ancestors and archaic humans than previously thought, the relevance of archaic introgression has only increased, opening exciting new avenues for studying human evolution. Here, we review recent methodological advances in the study of archaic introgression. We begin by providing an overview of the genealogical and genomic signatures left behind by introgression events before reviewing recent methods for studying archaic introgression by outlining their conceptual approaches, data requirements, and types of inferences they support. Finally, we provide recommendations for which methods are most appropriate given a research question and data set, discuss outstanding challenges, and suggest future lines of research to advance the study of archaic introgression.
Prior to high-throughput sequencing, whether archaic humans (e.g., Neanderthals and Denisovans) contributed to the modern human gene pool was hotly debated (Stringer and Andrews 1988; Wolpoff et al. 1994; Stringer 2022). Early analyses of mitochondrial DNA suggested minimal to no Neanderthal contribution to the genetic diversity of modern human populations (Krings et al. 1997, 1999; Caramelli et al. 2003; Currat and Excoffier 2004; Serre et al. 2004). However, in 2010, the publication of the draft Neanderthal genome revealed that non-African populations harbor 1%–4% Neanderthal ancestry (Green et al. 2010). Later that year, a finger bone from Siberia revealed a new archaic human group, the Denisovans, who also interbred with early modern humans (Reich et al. 2010). In contrast to the global distribution of Neanderthal ancestry, Denisovan ancestry occurs at lower levels in most non-African populations, except for Oceanic populations with 4%–6% Denisovan ancestry (Reich et al. 2010, 2011; Meyer et al. 2012). Following thorough investigations of alternative explanations for this signal (Currat and Excoffier 2011; Eriksson and Manica 2012; Yang et al. 2012; Eriksson and Manica 2014; Lohse and Frantz 2014), it is now commonly accepted that archaic and modern human populations interbred at various points in our evolutionary history, and by studying the extent of archaic introgression across global populations (Green et al. 2010; Reich et al. 2010; Meyer et al. 2012; Prüfer et al. 2014; The 1000 Genomes Project Consortium 2015; Mallick et al. 2016; Prüfer et al. 2017; Bergström et al. 2020; Mafessoni et al. 2020; Byrska-Bishop et al. 2022), researchers began to assemble more refined models of human evolutionary history (Wall et al. 2013; Malaspinas et al. 2016; Sankararaman et al. 2016; Vernot et al. 2016; McColl et al. 2018; Bergström et al. 2020; Skov et al. 2020; Witt et al. 2022; Chevy et al. 2023; Iasi et al. 2024; Liu et al. 2024b; Kerdoncuff et al. 2025).
Building on these findings, researchers began to localize introgressed segments and infer demographic parameters such as archaic population sizes and the timing and number of gene flow events (Sankararaman et al. 2012, 2014; Vernot and Akey 2014, 2015; Moorjani et al. 2016; Browning et al. 2018; Steinrücken et al. 2018; Jacobs et al. 2019; Villanea and Schraiber 2019; Massilani et al. 2020; Carlhoff et al. 2021; Choin et al. 2021; Iasi et al. 2021; Larena et al. 2021; Yuan et al. 2021; Chintalapati et al. 2022; Iasi et al. 2024; Li et al. 2024; Kerdoncuff et al. 2025; Sümer et al. 2025). While the precise number of Neanderthal and Denisovan introgression events is an active area of research, there is a growing consensus that multiple waves of archaic introgression have occurred throughout human evolution (for a more detailed discussion, see Gokcumen 2020; Sankararaman 2020; Ahlquist et al. 2021; Gopalan et al. 2021; Reilly et al. 2022; Ongaro and Huerta-Sanchez 2024; Peyrégne et al. 2024; Tagore and Akey 2025). This work also uncovered genomic regions devoid of any archaic ancestry (“introgression deserts”), which commonly reside in regions of high gene density or low recombination (Sankararaman et al. 2014; Vernot and Akey 2014; Sankararaman et al. 2016; Vernot et al. 2016; Schumer et al. 2018; Steinrücken et al. 2018; Chen et al. 2020; Skov et al. 2020; Kerdoncuff et al. 2025). This nonuniform distribution of archaic ancestry likely resulted from natural selection rapidly purging introgressed variants due to the higher mutational load in archaic populations (Harris and Nielsen 2016; Juric et al. 2016; Peter 2016; Schumer et al. 2018; Veller et al. 2023). Despite this trend, studies have shown that archaic ancestry conferred selective advantages in specific environmental or genetic contexts (Racimo et al. 2015; Dannemann and Racimo 2018). This phenomenon of adaptive introgression highlights archaic introgression as an alternative source of new genetic variation, as opposed to de novo mutations, potentially aiding early human populations in adapting to new environments (Racimo et al. 2015; Edelman and Mallet 2021). Classic examples include high-altitude adaptation in Tibetans facilitated by Denisovan-like introgression at EPAS1 (Huerta-Sánchez et al. 2014; Zhang et al. 2021), as well as loci affecting skin pigmentation (Ding et al. 2014b; Sankararaman et al. 2014; Vernot and Akey 2014; Gittelman et al. 2016; Dannemann and Kelso 2017; Racimo et al. 2017), immune function (Mendez et al. 2012, 2013; Sankararaman et al. 2014; Dannemann et al. 2016; Gittelman et al. 2016; Sams et al. 2016; Enard and Petrov 2018; Zhou et al. 2021; Vespasiani et al. 2022; Yermakovich et al. 2024), and dietary adaptations (Khrameeva et al. 2014; Racimo et al. 2017; Setter et al. 2020).
Studying archaic introgression presents unique challenges, particularly because of the limitations of ancient DNA (aDNA) sequencing. Although extensive genomic data exists for modern populations (The 1000 Genomes Project Consortium 2015; Mallick et al. 2016; Bergström et al. 2020; Byrska-Bishop et al. 2022), high-coverage archaic genomes are limited to three Neanderthals (Prüfer et al. 2014, 2017; Mafessoni et al. 2020) and one Denisovan (Meyer et al. 2012). Complementary low-coverage archaic genomes have refined the chronology of archaic introgression while providing a new lens into the social and population structure of archaic populations (Hajdinjak et al. 2018; Bokelmann et al. 2019; Vernot et al. 2021; Skov et al. 2022; Slimak et al. 2024). However, the characteristics of aDNA (e.g., fragmentation, postmortem damage, and contamination) complicate genotype calling and preclude haplotype phasing (Noonan et al. 2006; Skoglund et al. 2014; Orlando et al. 2021). Moreover, the available archaic samples likely represent only a fraction of the genetic diversity present in the true source populations (Green et al. 2010; Reich et al. 2010; Hajdinjak et al. 2018; Bokelmann et al. 2019; Vernot et al. 2021; Skov et al. 2022; Slimak et al. 2024). The methods reviewed here were developed to constrain the signal of archaic introgression while accounting for these technical and biological complexities.
The genealogical and genomic signals of introgression
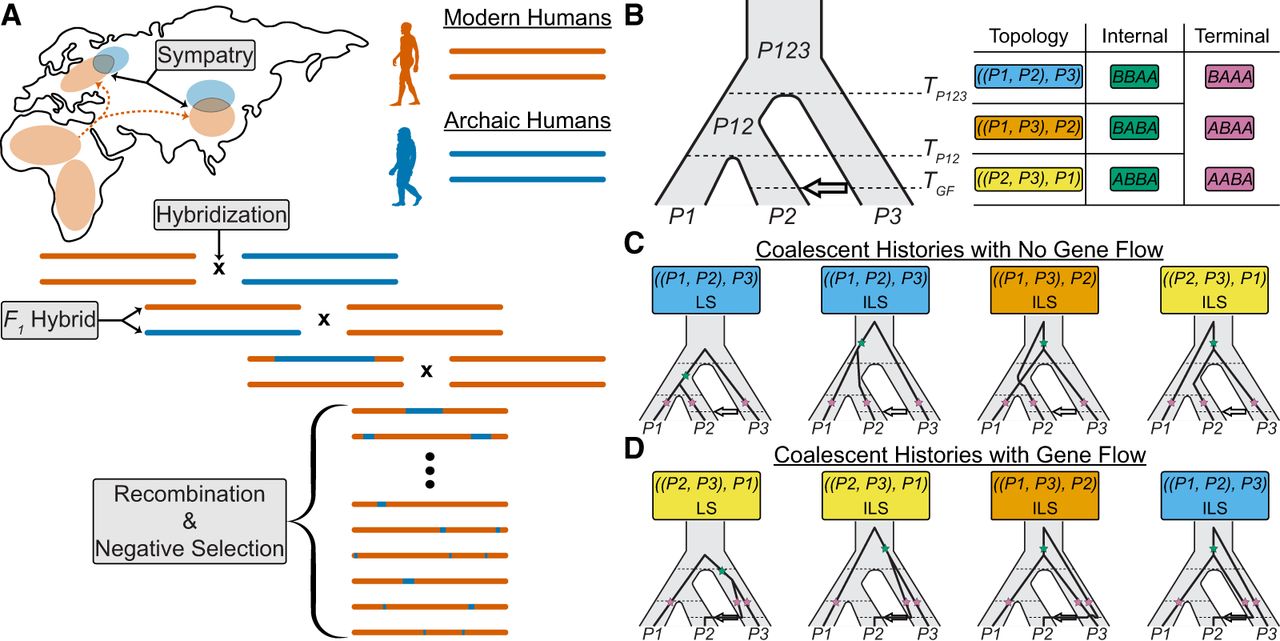
For introgression to occur, two populations must be sympatric (i.e., inhabiting the same geographic region) at some point in their shared history, creating opportunities for interbreeding (Fig. 1A). Hybridization produces first-generation offspring that inherit a uniform mixture of ancestry from both parental populations; however, introgressed segments become established only after repeated backcrossing over subsequent generations (Fig. 1A). Recombination progressively shortens these segments, resulting in an inverse relationship between segment length and time of gene flow (Chakraborty and Weiss 1988; Pool and Nielsen 2009; Gravel 2012; Liang and Nielsen 2014). Therefore, the distribution of introgressed segment lengths is informative regarding the timing of gene flow events (Pugach et al. 2011; Sankararaman et al. 2012; Moorjani et al. 2016; Ni et al. 2016; Corbett-Detig and Nielsen 2017; Jacobs et al. 2019; Shchur et al. 2020; Iasi et al. 2021; Di Santo et al. 2023; Iasi et al. 2024).
The genomic and genealogical signals of introgression. (A) Schematic of the introgression process, where some modern human populations (vermilion) migrated out of Africa, came into contact with archaic humans (blue) in Eurasia followed by subsequent interbreeding events. Over many generations, archaic segments are broken down due to recombination, purged due to negative selection, and incorporated into the genomes of present-day modern humans. (B) Instantaneous Unidirectional Admixture (IUA) model for studying introgression from a donor population (P3) into a hypothesized recipient population (P2) where P1 represents a control population assumed to have no genetic contributions from P3. In this model, the ancestral relationships are traced for a single lineage per population resulting in eight unique coalescent histories with only three possible topologies, that is, branching orders among lineages. Coalescent histories where P1 and P2 coalesce first, P1 and P3 coalesce first, and P2 and P3 coalesce first are depicted in sky blue, orange, and yellow; respectively. Under the IUA model, a site pattern is defined by the configuration of ancestral (A) and derived alleles (B) at a given site for the ordered set {P1, P2, P3, O}, where O represents an outgroup population used to polarized ancestral states in practice. Each topology will result in one unique site pattern resulting from a mutation occurring on the internal branch (green stars), while all topologies can generate three additional site patterns resulting from a mutation occurring on one of the three terminal branches (pink stars). (C) In the absence of gene flow, there are four unique coalescent histories under the IUA model, one with a history of lineage sorting (LS) between P1 and P2, and three equally probable histories of incomplete lineage sorting (ILS). (D) In the presence of gene flow, there are four additional coalescent histories, where the P2 lineage traces its ancestry through P3’s side of the tree, one with a history of LS between P2 and P3, and three equally probable histories of ILS.
Introgressed segments contain both new archaic-derived mutations and reintroduced ancestral alleles, generating characteristic patterns of allele sharing (Green et al. 2010; Reich et al. 2010; Durand et al. 2011; Peede et al. 2022; Dagilis and Matute 2023; Lopez Fang et al. 2024). Non-African populations share more alleles with Neanderthals and Denisovans than African populations do (Green et al. 2010; Durand et al. 2011; Yang et al. 2012; Lohse and Frantz 2014), which suggests that archaic gene flow into non-African populations was generally more extensive. Nonetheless, recent studies have identified instances of gene flow between Neanderthals and African populations (Kuhlwilm et al. 2016; Wall et al. 2019; Chen et al. 2020; Hubisz et al. 2020; Peyrégne et al. 2022; Harris et al. 2023; Li et al. 2024). To date, all known Neanderthal and Denisovan remains have been excavated outside Africa, providing complementary evidence that gene flow from these archaic groups into African populations was likely limited in extent (Green et al. 2010; Meyer et al. 2012; Prüfer et al. 2014, 2017; Hajdinjak et al. 2018; Slon et al. 2018; Mafessoni et al. 2020; Skov et al. 2022; Slimak et al. 2024; Tsutaya et al. 2025).
These allele sharing asymmetries, together with segment length and linkage information, form the conceptual foundation of archaic introgression inference. As detailed below, the distinctions among methods lie in how the genomic signals of introgression are constrained and leveraged.
Site patterns and f-statistics
Coalescent theory has been invaluable for investigating the patterns of genetic variation that arise from introgression. The Instantaneous Unidirectional Admixture (IUA) model (Green et al. 2010; Durand et al. 2011), conceptually akin to earlier multispecies coalescent models with gene flow (Jin et al. 2006; Than et al. 2007; Kubatko 2009; Meng and Kubatko 2009), is a classic framework for modeling introgression (Fig. 1B). The IUA model tracks a lineage from three populations: an African control (P1), a non-African recipient (P2), and an archaic donor (P3), with a single introgression event from P3 into P2 at time TGF, after P1 and P2 split from their most recent common ancestral population P12. In practice, an outgroup population (O) is included that is sufficiently diverged so that the ingroup populations (i.e., P1, P2, P3) always share a most recent common ancestor before coalescing with it. This outgroup serves to polarize ancestral states, as the true ancestral states are rarely known. Each locus in the genome is represented by a genealogical tree categorized into eight distinct coalescent histories (Fig. 1C–D; Hudson 1983; Tajima 1983; Pamilo and Nei 1988).
Without introgression, there are four possible coalescent histories: one lineage sorting (LS), where P1 and P2 lineages coalesce in their most recent common ancestral population P12, and three incomplete lineage sorting (ILS), where P1 and P2 fail to coalesce in P12 (Fig. 1C). Introgression introduces four additional coalescent histories, where P2 traces its ancestry through the P3 side of the population tree, including one additional LS between P2 and P3 and three coalescent histories of ILS (Fig. 1D). In the absence of gene flow, all coalescent histories of ILS are equally probable, where P1 and P2 are equidistant from P3. However, introgression disrupts this expected symmetry, as it introduces more coalescent histories where P2 and P3 coalesce more recently than P1 and P3 (Meng and Kubatko 2009; Durand et al. 2011; Liu et al. 2014a; Hibbins and Hahn 2019).
Although genealogical trees are unobservable, mutations on different branches generate identifiable site patterns used to study introgression (Green et al. 2010; Durand et al. 2011). By polarizing alleles with an outgroup (O), a site pattern is defined by the configuration of ancestral (A) and derived (B) alleles at a given biallelic site for the ordered set {P1, P2, P3, O}. Six site patterns are informative: three indicating derived allele sharing (ABBA, BABA, BBAA) from mutations on internal branches, and three indicating ancestral allele sharing (BAAA, ABAA, AABA) from mutations on terminal branches (Fig. 1B–D). Site pattern-based methods are computationally efficient and require only one genome per population, or allele frequencies, to determine the likelihood of observing the derived allele.
The most widely used site pattern-based method, Patterson’s D, detects introgression by comparing genome-wide counts of ABBA and BABA, which are expected to be equal in the absence of gene flow (Green et al. 2010; Durand et al. 2011). Instead of using site patterns as proxies for genealogical tree frequencies, the related statistic f4(P1, P2; P3, O) leverages correlations in allele frequencies to test for deviations from tree-like evolution by quantifying the shared genetic drift between clades (P1, P2) and (P3, O) (Reich et al. 2009; Patterson et al. 2012; Peter 2016). In the absence of gene flow, drift is independent between these two clades, and f4(P1, P2; P3, O) is expected to be equal to zero. A significant nonzero value indicates that the drift separating P1 from P3 is partially shared with P2 and O. By including another archaic individual presumed to have no genetic contribution to P2, the proportion of introgressed ancestry from P3 can be quantified using the quotient of two f4 values on permuted tips, that is, f4-ratio (Reich et al. 2009; Patterson et al. 2012; Peter 2016).
Introgression can also reintroduce ancestral variation previously lost in the recipient population (Peede et al. 2022; Dagilis and Matute 2023; Lopez Fang et al. 2024). To leverage this signal, Danc compares the genome-wide counts of BAAA and ABAA to detect introgression through ancestral allele sharing (Peede et al. 2022; Lopez Fang et al. 2024). In the absence of gene flow, the expected terminal branch lengths to P1 and P2 are equal, but with introgression, the terminal branch to P1 is longer than P2, generating excess BAAA site patterns. D+ combines the signal captured from Danc and Patterson’s D by including derived and ancestral allele sharing site patterns in a composite summary statistic, enabling both genome-wide and localized introgression inferences (Peede et al. 2022; Lopez Fang et al. 2024). Likewise, other approaches have been developed to detect locally introgressed regions using site patterns: df (Pfeifer and Kapan 2019) considers all derived-sharing patterns, whereas fd (Martin et al. 2014) accounts for the directionality of gene flow by dynamically switching the donor population label between P2 and P3 based on which harbors the derived allele at a higher frequency—also see fdM (Malinsky et al. 2015) for a symmetrically bounded variant of fd.
In addition to detecting the presence of introgression, site pattern-based methods can estimate the proportion of the recipient’s genome derived from the donor population. For example, fG normalizes the ABBA–BABA excess by the same difference when splitting P3 into two demes P3a and P3b (Durand et al. 2011). Similarly, see fhom (Martin et al. 2014), fanc (Peede et al. 2022), f+ (Peede et al. 2022), and Dp (Hamlin et al. 2020) for additional methods to quantify introgression. For site pattern-based methods and f-statistics, statistical significance is determined through block-jackknifing or bootstrapping with sufficiently large blocks to account for linkage disequilibrium (LD; Green et al. 2010; Durand et al. 2011; Patterson et al. 2012). Importantly, significant values are suggestive of introgression, as alternative demographic scenarios (e.g., ancestral population structure) can mimic these signals (Green et al. 2010; Durand et al. 2011; Peter 2016).
ARG-based methods
The ancestral recombination graph (ARG) represents the complete history of coalescence, recombination, and mutation events for a set of sampled sequences (Griffiths 1991; Griffiths and Marjoram 1996, 1997). As a summary of marginal genealogies across the genome, the ARG captures the evolutionary history of samples, including introgression events (Hubisz and Siepel 2020; Fan et al. 2025). ARG reconstruction is a rapidly evolving area of research, and we direct the reader to several recent reviews that provide overviews of inference approaches, highlighting the trade-offs between accuracy and scalability, along with differences in data requirements and encoding strategies (Brandt et al. 2024; Lewanski et al. 2024; Wong et al. 2024; Nielsen et al. 2025).
Consider a hypothetical ARG (represented as a sequence of local trees) involving a single African, Eurasian, and Neanderthal individual (Fig. 2A). Without introgression, coalescence events more recent than the modern human–Neanderthal split time occur only among modern human lineages. In contrast, gene flow generates local trees where Eurasian and Neanderthal lineages coalesce more recently than their split time, providing a key signal for detecting introgression from the ARG. Quantifying introgression thus involves estimating the proportion of the genome covered by local trees that are consistent with introgression. In practice, this signal is complicated by uncertainty in inferred split times and coalescence events, further compounded by mutation rate heterogeneity (Hubisz and Siepel 2020) and ARG inference approach (e.g., posterior sampling vs. point estimates).
Recent approaches for studying introgression. (A) A hypothetical ancestral recombination graph (ARG) represented as a sequence of three local trees for two African chromosomes in orange (A1/A2), two Eurasian chromosomes in green (E1/E2), and two Neanderthal chromosomes in sky blue (N1/N2). One way to identify introgressed segments from the ARG is to look for local trees where lineages from the recipient and donor populations share a more recent common ancestor than expected, given their known split times. The plot shows the time to the most recent common ancestor (tMRCA) for Eurasian (EUR) and Neanderthal (NEA) chromosomes across the genome (gray line), with the black dotted line indicating their split time. Local trees with a tMRCA younger than this split time (e.g., the middle tree) can be interpreted as evidence of introgression. (B) Reference-based approaches leveraging convolutional neural networks (CNNs) applied to haplotype matrices. The top row depicts an archaic haplotype (blue) alongside haplotype matrices for African and non-African populations, with haplotypes sorted according to their similarity to the archaic haplotype. The combined haplotype matrix (bottom row) is processed by a CNN trained on simulated data, which outputs the probability of introgression in the non-African population for each position (gray line). (C) Reference-free methods do not rely on an archaic reference genome to infer introgressed segments. Instead, they use a control panel with negligible introgressed ancestry (e.g., Africans) and a target panel hypothesized to have received introgression (e.g., non-Africans). The genome is partitioned into windows (gray dashed lines). Within each window, these methods compare the local density of shared mutations (pink) to private mutations; orange and green for the control and target panels, respectively. Segments classified as “Archaic” exhibit a high density of private mutations in the target panel and a low density of shared mutations between the control and target panels. (D) A population tree (left) depicts gene flow from a source to a recipient population, with the black dashed line denoting their split time. A beneficial mutation (blue star) originates in the source population and is introgressed into the recipient population. In the recipient population, lineages carrying the beneficial mutation coalesce rapidly with the source lineage, forming a star-like topology, whereas the black triangle represents a clade of lineages that did not inherit the beneficial allele. The right panel depicts patterns of nucleotide diversity (y-axis) in the recipient population centered on the beneficial mutation (x-axis), with the black dotted line showing the genome-wide average. Under adaptive introgression (solid gray line), a narrow region of reduced diversity flanked by elevated diversity forms a volcano pattern, contrasting with a classical selective sweep (dashed gray line), where the beneficial mutation arises within the recipient population, creates a broader valley of reduced diversity.
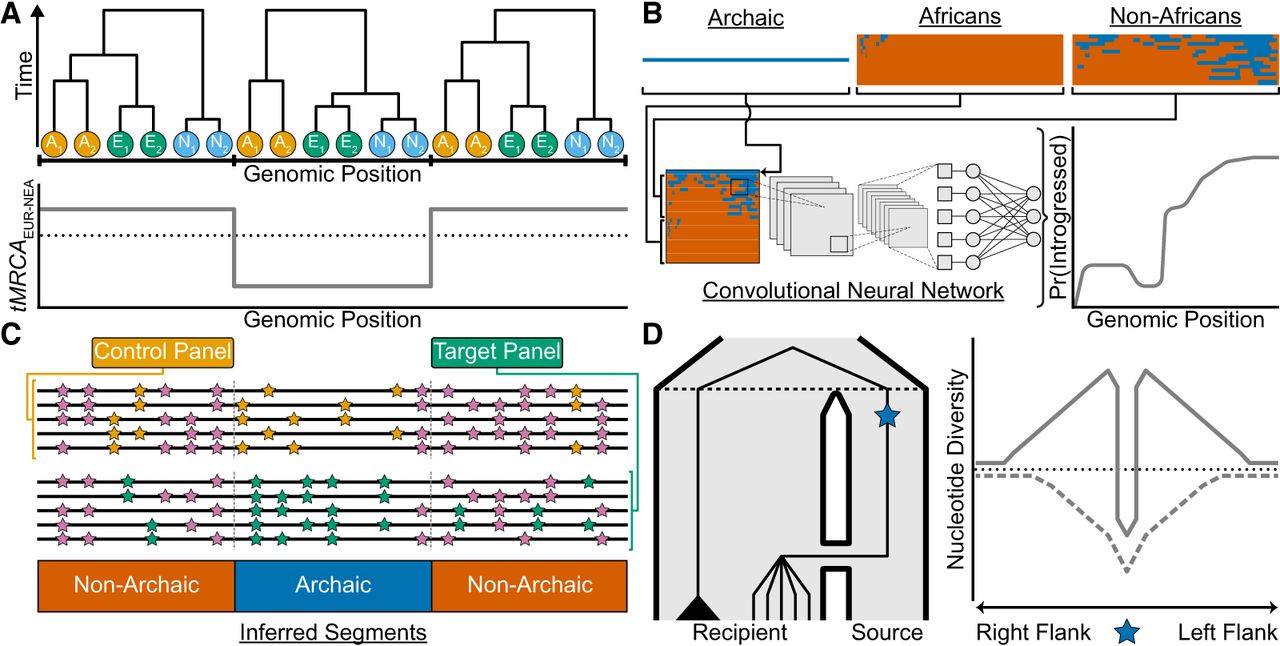
ARGweaver (Table 1; Rasmussen et al. 2014) is a Bayesian method that samples from the posterior distribution of possible ARGs, providing uncertainty estimates for coalescence event times and ARG topology. From this posterior sample, introgressed segments correspond to regions where modern human haplotypes coalesce with archaic lineages consistently more recently than expected (Hubisz and Siepel 2020). ARGweaver-D (Table 1; Hubisz et al. 2020), builds upon this framework by incorporating a user-specified demographic model to track migrating lineages between populations. Identifying introgressed regions involves scanning local trees whose branches follow the migration band above a posterior probability threshold, reflecting the frequency of introgression in the sampled ARGs. Applying ARGweaver-D to modern and archaic genomes, Hubisz et al. (2020) estimated that 3%–7% of the Neanderthal genome originated from an introgression event with early modern humans 200–300 kya. They also inferred that 1% of the Denisovan genome is derived from an unsequenced, highly diverged hominin, with ∼15% of these “super-archaic” regions inherited by modern humans through Denisovan introgression. Unlike other ARG inference methods that require experimentally phased archaic genomes, ARGweaver and ARGweaver-D uniquely support analyses with unphased genotypes by integrating over possible phasings during their sampling procedure (Rasmussen et al. 2014; Hubisz and Siepel 2020; Hubisz et al. 2020). Additionally, these methods account for the archaic individual’s sampling age, enabling the correction of biases in leaf branch lengths and allele age estimates (Hubisz and Siepel 2020; Hubisz et al. 2020). However, their computational demands limit their scalability to ∼50 diploid genomes (Rasmussen et al. 2014; Hubisz and Siepel 2020; Hubisz et al. 2020).
Overview of ancestral recombination graph inference methods for studying introgression
| Method | Scalability | Requires polarized data | Requires phased data | Mutation rate data | Recombination rate data | Demographic parameters |
|---|---|---|---|---|---|---|
| ARGweaver | Up to ∼50 individuals. | No. | No. | Point estimate or map. | Point estimate or map. | Point estimate of Ne or piecewise constant Ne estimates over time. |
| Protocol: Hubisz and Siepel (2020). | ||||||
| ARGweaver-D | Up to ∼50 individuals. | No. | No. | Point estimate or map. | Point estimate or map. | Ne estimates, split times, migration events. |
| Data: Introgression maps from Hubisz et al. (2020). | ||||||
| SARGE | Thousands of individuals. | Yes. | Yes. | No. | No. | N/A |
| N/A | ||||||
| tsinfer/tsdate | Thousands of individuals. | Yes. | Yes. | Point estimate. | Point estimate or map. | Point estimate of Ne or piecewise constant Ne estimates over time. |
| Data: Inferred genealogies from Wohns et al. (2022). | ||||||
[i] For each method, we provide the link to its documentation, the number of individuals to whom it scales, note whether polarizing ancestral states and phasing are required, whether mutation or recombination rate estimates (or maps) are needed, any additional required demographic parameters, and links to publicly available resources. These resources include published protocols and data sets, located in the second row per method. Note that the methods appear in the order in which they are referenced in the text, and all associated URL links are provided in the “Data sets” section.
Alternative methods scale to thousands of genomes by inferring a single “reasonable” ARG, often represented as a sequence of local trees without explicit recombination edges, but at the cost of principled uncertainty estimation and require phased samples. For example, SARGE (Table 1; Schaefer et al. 2021) utilizes a parsimony-based approach that prioritizes scalability over the fine-scale resolution of marginal genealogies, while remaining suitable for studying introgression. Schaefer et al. (2021) used SARGE to identify local trees where non-African and archaic haplotypes formed a clade, excluding trees that span short intervals—likely due to ILS. For the resulting trees, they computed the time to the most recent common ancestor (tMRCA) between non-African and archaic haplotypes, and then scored local trees based on the probability of ILS given the tMRCA and tree span, producing a final set of introgressed regions (Schaefer et al. 2021).
Unlike the previous methods, tsinfer/tsdate (Table 1; Kelleher et al. 2019; Wohns et al. 2022) do not simultaneously infer the ARG for modern and archaic samples. Instead, dated genealogies are first reconstructed from modern human genomes, and subsequently, phased archaic genomes with age estimates are incorporated. Ancestral relationships are then inferred between modern human ancestral nodes and “proxy ancestors” associated with each archaic sample, a construction that prevents any of the samples from being directly descended from another (Kelleher et al. 2019; Wohns et al. 2022). The use of proxy ancestors eliminates the need to compare the local tMRCA against an assumed modern human-archaic population split time, instead enabling the identification of introgressed material by determining whether a modern human lineage descends from an archaic proxy ancestor.
Reference-based methods
One strategy for identifying introgressed segments is to scan a modern human genome and label regions with a strong affinity to a sequenced archaic individual as introgressed. Because these approaches rely on an archaic “reference” genome as the putative source, they are referred to as reference-based methods.
IBDmix (Table 2; Chen et al. 2020) is an identity-by-descent (IBD) approach, requiring only a target population and archaic source genome. For each individual, it computes a running similarity score summarizing the likelihood of IBD between the query and archaic genotypes. When the score becomes negative, IBDmix selects alleles that maximize similarity within that region, labeling them introgressed, and then resumes scanning from that point. Li et al. (2024) used IBDmix to identify Neanderthal segments in African and non-African genomes, and by analyzing these segments, they were able to disentangle the directionality of gene flow, identifying regions in the Neanderthal genome derived from introgression with modern humans (Li et al. 2024). diCal-ADMIX (Table 2; Steinrücken et al. 2018) identifies introgressed segments in target haplotypes using a hidden Markov model (HMM), requiring phased genomes except for the archaic genome, which can be pseudo-haploid. The HMM is based on the sequentially Markovian coalescent conditional sampling distribution (Paul and Song 2010; Paul et al. 2011; Steinrücken et al. 2013) parameterized by a user-defined demographic model of the control (African), target (non-African), and source (archaic) populations. Haplotypes from control and source form “trunk” populations, with target haplotypes iteratively absorbed by these trunks. The hidden state space corresponds to the potential trunk haplotypes and the timing of absorption, whereas the transition and emission probabilities are based on a previously defined model for structured populations with migration and recombination (Steinrücken et al. 2013). After decoding, the probability that a target haplotype is introgressed is computed, resulting in a set of introgressed tracts per target haplotype (Steinrücken et al. 2018).
Overview of methods for characterizing local patterns of introgression
| Method | Requires polarized data | Control panel | Target panel | Source panel | Additional requirements | Introgression map |
|---|---|---|---|---|---|---|
| IBDmix | No. | N/A | Phased or unphased genotypes for at least 10 individuals. | Phased or unphased genotypes. | Genotyping error rates for the target and source panels. | Per individual from a single source. |
| Data: Neanderthal introgression maps from Li et al. (2024). | ||||||
| diCal-ADMIX | Yes. | Phased genotypes. | Phased genotypes. | Phased or pseudo-haploid genotypes. | Point estimates of the mutation and recombination rates and the demographic history of the control, target, and source panels. | Per haplotype from a single source. |
| Data: Neanderthal introgression maps from Steinrücken et al. (2018). | ||||||
| ArchaicSeeker2.0 | Yes. | Phased genotypes. | Phased genotypes. | Phased or unphased genotypes (per source of introgression). | Ancestral state sequence, outgroup sequence, recombination map, and a population tree (in Newick format) with branch lengths that includes the control, target, and source panels. | Per haplotype from multiple sources. |
| Protocol: Zhang et al. (2022); Data: Introgression maps from Yuan et al. (2021). | ||||||
| admixfrog | No. | Phased or unphased genotypes for at least one individual. | Phased or unphased genotypes (for high-coverage data) or Binary Alignment Map (for low-coverage data). | Phased or unphased genotypes for at least one individual (per source of introgression). | Panel of phased or unphased genotypes representing the source of contamination, initial number of bases to determine if a read is deaminated, initial sequencing error rate, and initial contamination rate (for low-coverage data). | Per individual from multiple sources. |
| Data: Neanderthal introgression maps from Iasi et al. (2024). | ||||||
| CRF | Yes. | Phased genotypes for modern humans and unphased genotypes for archaic humans. | Phased genotypes. | Unphased genotypes. | Recombination map and demographic history of the control, target, and source panels to simulate training data for parameter estimation. | Per haplotype from multiple sources. |
| Data: Introgression maps from Sankararaman et al. (2016). | ||||||
| IntroUNET | No. | Phased or unphased genotypes (only when the source of introgression is unknown). | Phased or unphased genotypes. | Phased or unphased genotypes. | Demographic history of the target and source panels or the demographic history of the control, target, and source panels (only when the source of introgression is unknown). | Per haplotype (for phased data) or per individual (for unphased data) from a single source. |
| N/A | ||||||
| SPrime | No. | Phased or unphased genotypes for at least 15 individuals. | Phased or unphased genotypes for at least 15 individuals. | Phased or unphased genotypes (only for computing matching rates). | Recombination map. | Per population from an unknown source. |
| Protocol: Zhou and Browning (2021); Data: Introgression maps from Browning et al. (2018). | ||||||
| sstar | Yes. | Phased or unphased genotypes for at least 10 individuals. | Phased or unphased genotypes for at least 10 individuals. | Phased or unphased genotypes (optional). | Point estimate of the recombination rate or recombination map and demographic history of the control, target, and source panels. | Per individual from an unknown source or multiple sources (per optional source panel). |
| N/A | ||||||
| ArchIE | Yes. | Phased genotypes for at least 50 individuals. | Phased genotypes for at least 50 individuals. | N/A | Demographic history of the control, target, and source panels. | Per haplotype from an unknown source. |
| Data: Introgression maps from Durvasula and Sankararaman (2020). | ||||||
| hmmix | Yes. | Phased or unphased genotypes. | Phased or unphased genotypes. | Phased or unphased genotypes (optional). | Point estimate of the mutation rate or mutation map, point estimate of the recombination rate, and estimates of: split times between the control and target panels and the control and source panels, time of the gene flow event, and admixture proportion. | Per haplotype (for phased data) or per individual (for unphased data) from an unknown source. |
| Data: Introgression maps from Skov et al. (2018). | ||||||
| DAIseg | Yes. | Phased or unphased genotypes. | Phased or unphased genotypes. | Phased or unphased genotypes per source (optional). | Point estimate of the mutation rate or mutation map, point estimate of the recombination rate, and estimates of: split times between the control and target panels and the control and source panels, time of the gene flow events, and associated admixture proportion per gene flow event. | Per haplotype (for phased data) or per individual (for unphased data) from multiple unknown sources. |
| Data: Introgression maps from Planche et al. (2025). | ||||||
[i] For each method, we provide a link to its documentation, note whether polarizing ancestral states is required, and describe the data requirements for a control panel (assumed to lack any genetic contributions from the source panel), a target panel (the putative recipient), and a source panel (the putative donor). We also provide any additional data requirements, the resolution of the inferred introgression maps (e.g., population-, individual-, or haplotype-level, and the number of possible sources), and links to publicly available resources (located in the second row per method) including published protocols and data sets. Note that the methods appear in the order in which they are referenced in the text, and all associated URL links are provided in the “Data sets” section.
ArchaicSeeker2.0 (Table 2; Yuan et al. 2021) also uses an HMM, but allows inference of multiple introgression events from multiple archaic sources. Given input genomes for Neanderthal, Denisovan, and African populations, ArchaicSeeker2.0 first identifies candidate introgressed sequences in query genomes. These candidate sequences are then matched to a set of tree topologies corresponding to introgression, ILS, or false positives. The parameters for introgression events are then inferred based on sequences that match a tree topology consistent with introgression. Applying ArchaicSeeker2.0 to modern and archaic human genomes, Yuan et al. (2021) inferred two waves of Neanderthal- and Denisovan-like introgression events across Eurasia and Oceania and revealed how additional gene flow events among modern human populations have shaped the global distribution of introgressed ancestry.
The growing availability of sequenced ancient modern human genomes presents an opportunity to explore the temporal and spatial dynamics of archaic introgression, although technical limitations (e.g., sequencing artifacts, contamination, and low sequencing coverage) complicate such inferences. To overcome this, admixfrog (Table 2; Peter 2020) combines a genotype likelihood model, explicitly accounting for contamination and sequencing errors, with an HMM to infer introgressed ancestry in a target individual as a mixture of multiple high-quality source panels. By integrating these two models, admixfrog can simultaneously infer multiple ancestry states (from different archaic sources), genotype likelihoods, sequencing error rates, and contamination rates, even for genomes with coverage as low as 0.03× (Peter 2020). Notably, admixfrog has been used to infer instances of gene flow between Neanderthals and Denisovans (Peter 2020).
Departing from HMMs, Sankararaman et al. (2014) developed a Conditional Random Field (CRF) for identifying Neanderthal segments in non-African populations, later extending it to infer both Neanderthal and Denisovan segments (Table 2; Sankararaman et al. 2016). Unlike HMMs, CRFs model the conditional probability of hidden states given a set of features computed from the observed data, allowing multiple emission functions that relate observed features to hidden states (i.e., introgressed or not). As discriminative models, CRFs require training data, for example, from simulations, to learn their parameters (Sankararaman et al. 2014, 2016). Sankararaman et al. (2016) used a CRF to infer archaic segments per haplotype in a target panel by comparing them to an archaic source individual and a control panel lacking introgressed ancestry. Their implementation uses two emission functions: one compares patterns of derived allele sharing between the target haplotype, archaic source, and control panel, and the other compares haplotype distances between the target haplotype and the archaic source versus the minimum distance to the control panel (Sankararaman et al. 2016). To identify Neanderthal segments, Africans and Denisovans serve as the control panel, whereas Africans and Neanderthals are used to detect Denisovan segments (Sankararaman et al. 2016). Sankararaman et al.’s (2016) CRF rarely misclassified modern human ancestry as archaic, but misclassification between Neanderthal and Denisovan ancestry was more frequent and comparable to the correct classification probabilities, underscoring the difficulty of attributing archaic sources to introgressed segments.
Recently, deep learning methods have gained popularity in evolutionary biology owing to advances in simulation software for generating synthetic training data (Baumdicker et al. 2022; Haller and Messer 2023), user-friendly ecosystems for implementing machine learning architectures (Chen et al. 2019; Zeng et al. 2021; Sanchez et al. 2023; Wang et al. 2023), and specialized computing hardware. Convolutional Neural Networks (CNNs), originally developed for computer vision tasks, have been widely adopted for population genetic inferences because of the similarities between image data and SNP matrices (Fig. 2B; Flagel et al. 2019). One application of CNNs in introgression inference is IntroUNET (Table 2; Ray et al. 2024), which takes matrices with rows corresponding to sampled haplotypes and columns corresponding to the derived allele count as input. Because 2D convolutions are dependent on row ordering, haplotypes are sorted to minimize the genetic distance between adjacent rows (Ray et al. 2024). Using SNP matrices constructed from putative source and recipient populations and large-scale simulations based on their shared demographic history for training, IntroUNET estimates the likelihood of introgression per SNP per haplotype or individual if genotypes are unphased. When the source population is unknown or unsequenced, IntroUNET can be trained using a control population assumed to lack introgressed ancestry, which can be used to identify segments derived from super-archaic introgression (Ray et al. 2024).
Reference-free methods
The methods discussed so far primarily rely on available archaic “reference” genomes to infer introgressed segments. However, because of the limited number of high-coverage archaic genomes, a proportion of the genetic variation present in the donor populations is likely to be unrepresented, especially true for unsampled archaic source populations (Durvasula and Sankararaman 2020). Fortunately, archaic introgression leaves detectable genomic signatures that enable inference without requiring an archaic genome; these approaches are known as reference-free methods (Fig. 2C). For instance, the S* statistic (Plagnol and Wall 2006) provides a measure of tightly linked SNPs in the target population that are absent in a closely related control population, which is assumed to lack introgressed ancestry. Typically, African populations serve as the control, although alternatives (e.g., European populations for inferring Denisovan segments) may be more appropriate depending on the context (Zhang et al. 2021).
SPrime (Table 2; Browning et al. 2018) is a popular extension of S* that accounts for how low levels of introgressed ancestry in the control population and heterogeneity in mutation and recombination rates affect S* scores. SPrime iteratively finds alleles in a query genotype that maximize an S*-like score, classifies these putative archaic alleles as forming an introgressed segment, and removes them for the next iteration until the segment’s score falls below a user-specified threshold. Notably, this method was used to provide the first line of evidence for two distinct pulses of Denisovan introgression into modern humans (Browning et al. 2018).
Also relying on S*, sstar (Table 2; Huang et al. 2022), partitions the genome into windows, computes S* scores per window, and then filters these scores for significance. If no genomes for the putative source population exist, sstar requires a demographic model of the control, target, and source populations to simulate and determine the S* threshold for classifying introgressed segments. If genomic data are available, sstar uses the sequence similarity between the query and source sequences as the final discriminator. Huang et al.’s (2022) simulation study indicates that sstar is robust to a number of complex introgression scenarios (e.g., introgression from multiple sources and unsampled sources) and maintains power to identify introgressed segments given limited sample sizes.
ArchIE (Table 2; Durvasula and Sankararaman 2019) implements a logistic regression model trained on simulated data, to classify genomic windows in a focal haplotype as archaic or not. To do so, ArchIE uses a suite of summary statistics that capture different aspects of introgression signals: the individual site frequency spectrum (SFS), the first four moments of the pairwise difference distribution for the target population, the number of private SNPs in a focal haplotype, the minimum pairwise differences between a focal haplotype and control population, and S* scores (Plagnol and Wall 2006; Chen et al. 2007; Hammer et al. 2011). By incorporating population- and individual-level summary statistics, ArchIE can more accurately identify introgressed windows than S* or SPrime alone (Durvasula and Sankararaman 2019). Although limited to haplotype data and requiring prior knowledge of the joint demographic history for the control, target, and source populations, ArchIE has been used to identify introgressed segments in West African genomes, which are believed to have been inherited from an unsampled archaic population (Durvasula and Sankararaman 2020). hmmix (Table 2; Skov et al. 2018; Macià et al. 2025) takes an approach conceptually similar to S* to classify introgressed segments based on the local density of variants absent in the control population. Using an HMM where the observations correspond to the number of private derived alleles in the query haplotype (or unphased diploid genotype) per window, hmmix first finds the optimal set of model parameters before inferring a decoded sequence of ancestry predictions (i.e., archaic or not) across the query genome. The only prior knowledge required are initial estimates of the model’s parameters, as the optimal set is learned, making inference applicable to Neanderthal, Denisovan, or even super-archaic introgression (Teixeira et al. 2021). DAIseg (Table 2; Planche et al. 2025) further enhances inferences of introgressed segments and their most likely donor by extending the hmmix framework to an arbitrary number of control populations and archaic hidden states. Notably, this more flexible reference-free approach is particularly useful for simultaneously identifying Neanderthal and Denisovan introgressed tracts without the need for additional post hoc filtering steps (Planche et al. 2025).
Methods to detect adaptive introgression
Although most archaic ancestry found in modern humans is assumed to be neutral, adaptively introgressed material provides fitness advantages to the recipient population (Racimo et al. 2015; Edelman and Mallet 2021). Such segments will rise in frequency, reducing the opportunity for recombination to break them apart, and thus tend to be longer than predicted by neutrality (Shchur et al. 2020). Genealogically, adaptively introgressed regions exhibit a high density of recent coalescent events among lineages carrying the beneficial introgressed allele, producing star-like topologies (Fig. 2D; Coop and Griffiths 2004; Kim and Nielsen 2004; Hejase et al. 2020). Accordingly, these introgressed alleles often appear in strong LD and at high frequencies in the recipient population but remain rare or absent in populations that did not experience the same selective advantage. However, similar patterns can arise from alternative scenarios, such as post-introgression bottlenecks (Kerdoncuff et al. 2025) or heterosis masking recessive deleterious mutations, which subsequently inflate the frequency of introgressed ancestry when donor populations carry higher mutational loads (Ingvarsson and Whitlock 2000; Kim et al. 2018; Zhang et al. 2020).
Researchers have historically identified adaptively introgressed regions using outlier-based approaches (Racimo et al. 2015; Edelman and Mallet 2021), conducting genome-wide scans for positive selection and introgression, and then intersecting outlier regions to identify candidates. However, the genomic signature of adaptive introgression differs from that of classical selective sweeps, leaving a narrower valley of reduced genetic diversity flanked by regions harboring alleles at intermediate frequencies in the recipient population (Fig. 2D; Santiago and Caballero 2005; Setter et al. 2020). Furthermore, methods designed to detect positive selection are typically optimized for specific scenarios, making their selection nontrivial and influencing downstream power (for a recent review of methods, see: Hejase et al. 2020). Therefore, methods that explicitly leverage the unique genomic signatures of adaptive introgression are desirable to avoid potential false inferences from intersecting multiple genomic scans.
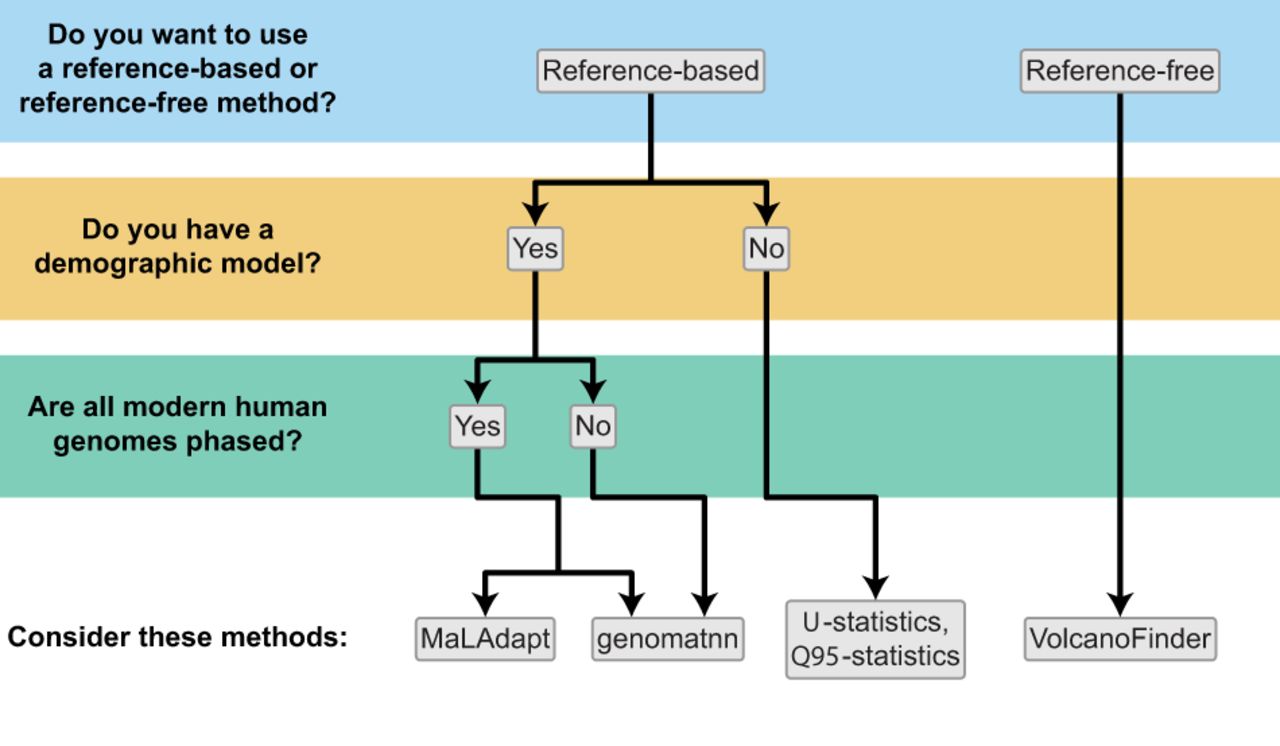
To address these challenges, Racimo et al. (2016) introduced the U- and Q95-statistics, window-based summaries designed to detect adaptive introgression. The key idea behind these statistics is that regions shaped by archaic adaptive introgression are expected to harbor many sites where the archaic allele has risen to high frequency in the target population, while remaining rare or absent in a control population presumed to lack archaic genetic contributions. UA,B,C(w, x, y) counts sites where the archaic allele (in C) is rare (<w) in the control population A, common (>x) in the target population B, and at frequency y in the archaic individual C. The main drawback of U-statistics is that researchers must define a threshold for what constitutes a “high” archaic allele frequency in the target population. As an alternative, Q95A,B,C(w, y) computes the 95th percentile of the archaic allele frequency distribution in the target population B, conditioned on the alleles being rare (<w) in the control population A and present at frequency y in the archaic individual C. To distinguish Neanderthal from Denisovan adaptive introgression, Racimo et al. (2016) also developed the UA,B,C,D(w, x, y, z) and Q95A,B,C,D(w, y, z) statistics, requiring specific allele frequencies in both archaic individuals C (=y) and D (=z). Although these summary statistics still rely on outlier-based genome-wide scans, multiple studies have demonstrated that they are highly effective at identifying regions of adaptive introgression while minimizing confounding effects from demographic processes and heterosis (Racimo et al. 2016; Zhang et al. 2020; Gower et al. 2021; Zhang et al. 2023; Romieu et al. 2025).
As an alternative to summary statistics, VolcanoFinder (Table 3; Setter et al. 2020) uses a coalescent-based model of adaptive introgression, requiring only genetic data from the recipient population. Leveraging the distinctive volcano-shaped pattern of genetic diversity left by adaptive introgression (Fig. 2D), VolcanoFinder implements a composite likelihood ratio test to assess whether the local SFS in the recipient population is consistent with adaptive introgression. Notably, this method is especially effective when the donor and recipient populations are highly divergent, making it particularly useful for detecting introgression from unsampled archaic populations lacking genomic data (Setter et al. 2020).
Overview of methods for studying adaptive introgression
| Method | Requires polarized data | Control panel | Target panel | Source panel | Additional data requirements |
|---|---|---|---|---|---|
| VolcanoFinder | No. | N/A | Phased or unphased genotypes. | N/A | Outgroup sequence to polarize ancestral states (optional). |
| Tutorial: Example workflow from Setter et al. (2020). | |||||
| genomatnn | No. | Phased or unphased genotypes. | Phased or unphased genotypes. | Phased or unphased genotypes. | Demographic history of the control, target, and source panels (at minimum). |
| Data: Pretrained models from Gower et al. (2021); Data: Adaptive introgression maps from Gower et al. (2021). | |||||
| MaLAdapt | Yes. | Phased genotypes. | Phased genotypes. | Phased or unphased genotypes. | Recombination map, genomic annotations (for computing exon density), and demographic history of the control, target, and source panels (at minimum). |
| Data: Adaptive introgression maps from Zhang et al. (2023). | |||||
[i] For each method, we provide a link to its documentation, note whether polarizing ancestral states is required, and describe the data requirements for a control panel (assumed to lack any genetic contributions from the source panel), a target panel (the putative recipient), and a source panel (the putative donor). We also provide any additional data requirements, along with links to publicly available resources (located in the second row per method) including published protocols and data sets. Note that the methods appear in the order in which they are referenced in the text, and all associated URL links are provided in the “Data sets” section.
Building on the growing application of deep learning models for population genetic inferences, genomatnn (Table 3; Gower et al. 2021) uses a CNN trained on simulated data to detect adaptive introgression by classifying local genotype matrices as adaptively introgressed or not. genomatnn takes a 2D SNP matrix as input (haplotypes x sites), representing the presence or absence of the derived allele at a given site per haplotype (or per unphased diploid genotype). Haplotypes in the matrix are sorted based on similarity to a genome from the donor population and consequently require an archaic genome as input. Although its power to detect adaptive introgression is sensitive to the demographic parameters used for training, namely the timing and strength of selection, genomatnn has successfully recovered previously proposed candidates of both Neanderthal and Denisovan adaptive introgression, in addition to identifying novel candidates (Gower et al. 2021).
Another machine learning method to detect adaptive introgression, MaLAdapt (Table 3; Zhang et al. 2023) uses an Extra-Trees classifier (Geurts et al. 2006) trained on simulated data to classify genomic windows based on summary statistics calculated across donor, recipient, and control populations. The advantage of this ensemble learning technique is that, although the simulated data do not precisely match the empirical data, the relationships between them—as encoded by summary statistics—are similar, and this approach has proved effective for studying introgression in nonhuman systems (Schrider et al. 2018). However, MaLAdapt assumes that the demographic model used for the simulations is representative of the demographic model underlying the empirical data. Despite MaLAdapt recovering previously proposed candidates of adaptive introgression, Zhang et al. (2023) did not find evidence of adaptive introgression in HLA genes (Abi-Rached et al. 2011; Ding et al. 2014a), aligning with recent evidence that these were likely false positives resulting from heterosis (Zhang et al. 2020).
Practical considerations
The proliferation of methods for studying archaic introgression makes it challenging to decide which method is best suited for a given analysis. As no systematic benchmarking study has yet compared the power and precision of these methods, it is premature to broadly recommend one method over another. Instead, based on the methods discussed in this review, we provide a set of recommendations for studying archaic introgression to highlight potential use cases, depending on the genomic scale of interest (i.e., characterizing global patterns of introgression, identifying introgressed regions along the genome, or pinpointing adaptively introgressed regions) and data available (Figs. 3–5). Additionally, to help researchers sift through the wide array of resources available to them, we provide easy-to-access references for all methods reviewed, including the key data requirements, targets and resolution of inference, links to documentation, and links to publicly available resources, that is, published protocols, tutorials, and data sets (Tables 1–3). In the following subsections, we discuss practical considerations that researchers must navigate when studying archaic introgression, highlight outstanding challenges, and provide recommendations for future research directions.
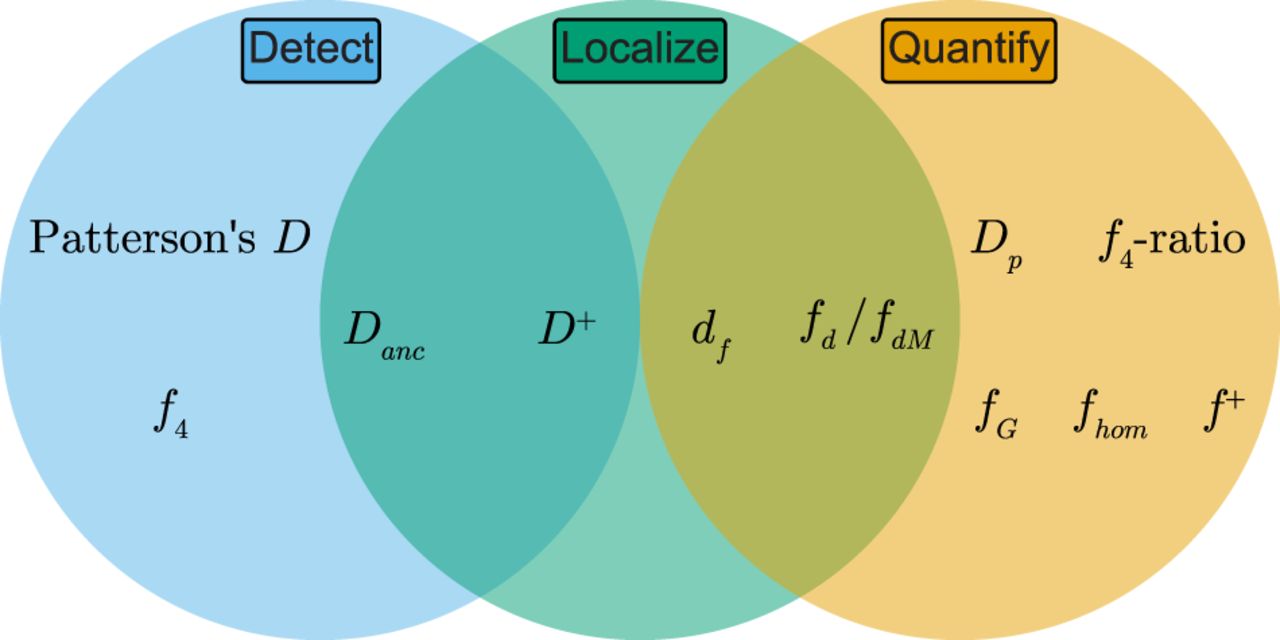
Recommendations for detecting and quantifying patterns of introgression using site pattern-based methods and f-statistics. Venn diagram summarizing site pattern-based methods and f-statistics used to detect and quantify introgression. Methods to detect introgression on a genome-wide scale are shown in the sky blue circle, those for quantifying introgression on a genome-wide scale are shown in the orange circle, and methods designed to localize signals of introgression in nonoverlapping genomic windows are shown in the green circle.
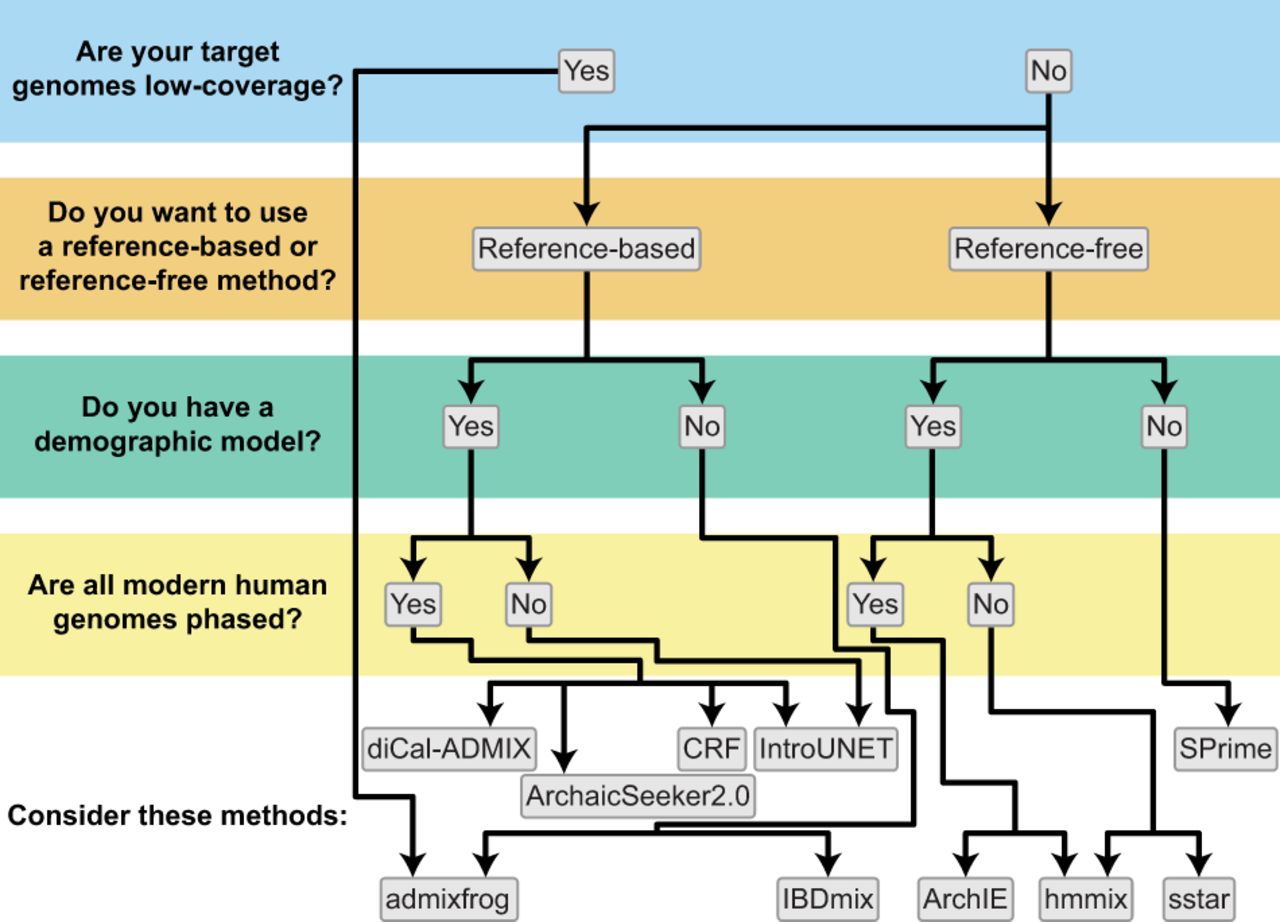
Recommendations for identifying introgressed segments along the genome. Decision tree to determine which inference methods are appropriate for characterizing local patterns of introgression based on data availability.
Recommendations for detecting adaptive introgression. Decision tree to determine which inference methods are appropriate for detecting adaptive introgression based on data availability.
Genome availability and quality
All methods for studying archaic introgression are inherently constrained by the number and quality of publicly available archaic genomes. To date, three high-coverage Neanderthal genomes (Prüfer et al. 2014, 2017; Mafessoni et al. 2020) and one high-coverage Denisovan genome (Meyer et al. 2012) are publicly available; however, they likely do not fully capture the amount of genetic variation present in the Neanderthal and Denisovan populations that interbred with the ancestors of present-day humans. In the case of studying Denisovan introgression, this limitation is exacerbated by the finding that the only high-coverage sequenced Denisovan individual is distantly related to the introgressing Denisovan populations that interbred with modern humans (Skoglund and Jakobsson 2011; Jacobs et al. 2019; Ongaro and Huerta-Sanchez 2024; Peyrégne et al. 2024).
Although the progression of publicly available high-coverage archaic genomes has been slow, the recent growth in the number of published ancient modern human genomes (Mallick et al. 2024) provides an opportunity to study how archaic ancestry has changed across time and space. The majority of these data have been sequenced using capture-enrichment methods or are low-coverage, restricting researchers to primarily analyzing pseudo-haploid genotypes. Although this poses little concern for site pattern-based methods and f-statistics, this data type tends to be limited to methods aimed at characterizing global patterns of introgression. One increasingly popular alternative involves imputing and/or phasing ancient genomes using present-day genomes as a reference panel, which has been shown to be accurate on a genome-wide level (Hui et al. 2020; Ausmees et al. 2022; Ausmees and Nettelblad 2023; Sousa da Mota et al. 2023; Çubukcu and Kılınç 2024; Escobar-Rodríguez and Veeramah 2024; Garrido Marques et al. 2024; Medina-Tretmanis et al. 2024; Kumar et al. 2025) and particularly accurate in introgressed regions of the genome (Capodiferro et al. 2025). However, the accuracy of imputing ancient genomes is influenced by multiple factors, including the age of the sample (Sousa da Mota et al. 2023; Escobar-Rodríguez and Veeramah 2024), aDNA sequencing approach (Hui et al. 2020; Sousa da Mota et al. 2023; Çubukcu and Kılınç 2024; Garrido Marques et al. 2024), overall sequencing coverage (Hui et al. 2020; Ausmees et al. 2022; Ausmees and Nettelblad 2023; Sousa da Mota et al. 2023; Çubukcu and Kılınç 2024; Escobar-Rodríguez and Veeramah 2024), the genetic ancestry composition and size of the reference panel (Hui et al. 2020; Ausmees et al. 2022; Sousa da Mota et al. 2023; Çubukcu and Kılınç 2024; Escobar-Rodríguez and Veeramah 2024; Kumar et al. 2025), the minor allele frequency spectrum of the reference panel (Hui et al. 2020; Ausmees et al. 2022; Ausmees and Nettelblad 2023; Sousa da Mota et al. 2023; Çubukcu and Kılınç 2024; Escobar-Rodríguez and Veeramah 2024), the choice of imputation software (Ausmees and Nettelblad 2023; Escobar-Rodríguez and Veeramah 2024; Garrido Marques et al. 2024; Kumar et al. 2025), and postimputation quality control steps (Hui et al. 2020; Sousa da Mota et al. 2023; Çubukcu and Kılınç 2024; Escobar-Rodríguez and Veeramah 2024). Although imputing ancient genomes has proven effective for studying archaic introgression, inferred introgressed segments are often shorter than their true lengths (Capodiferro et al. 2025). Developing strategies to correct this bias should be a focus of future work, as doing so will improve the accuracy for inferring the timing of introgression and, in turn, our understanding of the distribution of archaic ancestry across time and space.
Disambiguating the most likely donor
A central challenge in archaic introgression research is identifying the most likely donor of the inferred introgressed ancestry. Most non-African populations harbor Neanderthal ancestry and some populations from Asia and Oceania also harbor Denisovan ancestry in varying proportions, necessitating the development of strategies to distinguish between these archaic sources. On a genome-wide scale, this differentiation is relatively straightforward using site pattern-based methods or f-statistics. For example, Qin and Stoneking (2015) leveraged the fact that the Han Chinese and Oceanic populations harbor similar amounts of Neanderthal ancestry to infer Denisovan ancestry proportions in Oceanic populations using an f4-ratio approach. In the numerator, an excess of shared drift between an Oceanic population X and Neanderthal, f4 (Yoruba, Neanderthal; Han, X), is attributable to Denisovan introgression and normalized by the total shared drift between the Neanderthal and the Denisovan in the denominator, f4 (Yoruba, Neanderthal; Han, Denisovan), one can constrain the Denisovan ancestry proportion for X.
Disambiguating between potential donors for local introgressed regions is more challenging, although Sankararaman et al.’s (2016) CRF, ARGweaver-D (Hubisz et al. 2020), admixfrog (Peter 2020), ArchaicSeeker2.0 (Yuan et al. 2021), and DAIseg (Planche et al. 2025) are notable exceptions that explicitly account for this complexity. Reference-based methods inherently identify the donor based on the archaic genome used but face two key limitations: they depend on the availability and quality of archaic genomes and typically require additional postprocessing steps unless they jointly infer segments from multiple archaic sources. For example, to generate their final callset of introgressed Neanderthal segments, Chen et al. (2020) used IBDmix to infer introgressed Denisovan segments and subsequently masked any introgressed segment that fell within the intersection of these two archaic segment sets.
On the other hand, although reference-free approaches do not require an archaic genome for inference, their use is necessary for an additional post hoc step to annotate the most probable archaic source for each segment. Although this task is conceptually straightforward, the field lacks a standardized, principled population genetics approach to do so. Common strategies include counting the number of derived alleles shared between a candidate segment and archaic genomes or computing a so-called match rate, the proportion of alleles within the segment that match those in the archaic genome. Researchers must then define thresholds to classify segments as Neanderthal, Denisovan, or ambiguous. Although these thresholds can be defined rigorously (e.g., using simulations), the lack of standardization across studies has resulted in a patchwork of strategies for annotating putative introgressed segments.
Future studies should develop more principled approaches for annotating inferred introgressed tracts. A straightforward post hoc solution involves computing sequence divergence, the average number of pairwise differences normalized by the total number of callable positions, which has a direct coalescent interpretation related to the tMRCA between an introgressed segment and archaic genome (Peyrégne et al. 2022). This approach would eliminate arbitrary thresholds by comparing tMRCAs to inferred modern-archaic human split times and using the segment length to distinguish introgression from ILS. Alternatively, taking inspiration from Vernot et al. (2016), a hybrid two-step approach that integrates the strengths of both reference-free and reference-based methods presents a promising path forward. In this framework, a reference-free method first generates a proposal set of unlabeled introgressed segments, which is then passed to a reference-based probabilistic model to identify false positives and assign the most likely archaic donor.
Reconciling assumptions
Archaic introgression is often modeled as an instantaneous pulse of gene flow. Yet, growing evidence suggests multiple waves of Neanderthal (Sankararaman et al. 2014; Vernot and Akey 2014; Kim and Lohmueller 2015; Vernot and Akey 2015; Prüfer et al. 2017; Villanea and Schraiber 2019; Iasi et al. 2024; Li et al. 2024) and Denisovan (Reich et al. 2010, 2011; Meyer et al. 2012; Sankararaman et al. 2016; Vernot et al. 2016; Browning et al. 2018; Jacobs et al. 2019; Choin et al. 2021; Larena et al. 2021) introgression, gene flow between Neanderthals and Denisovans (Prüfer et al. 2014; Slon et al. 2018; Peter 2020; Villanea et al. 2025), and enigmatic gene flow from unsampled super-archaic populations (Hubisz et al. 2020; Mafessoni et al. 2020; Rogers et al. 2020). This evidence points to a more complex history of gene flow that must be addressed to avoid biased interpretations of introgression patterns. This unaccounted for complexity may be of particular concern for machine learning methods that rely on training data from large-scale simulations under prespecified demographic histories, as demographic mis-specification appears to be a major confounder (Durvasula and Sankararaman 2019; Gower et al. 2021; Ray et al. 2024; Cobb and Smith 2025; Hackl and Huang 2025). Encouragingly, there are machine learning techniques (e.g., domain adaptation; Mo and Siepel 2023; Cobb and Smith 2025) that could provide a potential solution for addressing the mismatch between the synthetic training data generated under a demographic model and the real data where the true demographic history is unknown. In contrast, natural selection and demographic processes (e.g., migration between control and recipient populations, bottlenecks, expansions, and population structure) can generate spurious signals that mimic introgression or dampen genuine signals (Durand et al. 2011; Peter 2016; Martin and Amos 2021; Smith and Hahn 2024; Kerdoncuff et al. 2025; Tournebize and Chikhi 2025). For example, Tournebize and Chikhi (2025) recently demonstrated that both allele frequency- and LD-based methods can produce spurious signals of archaic introgression under models of complex population structure that lack archaic gene flow events. These provocative findings emphasize the importance of carefully scrutinizing results for possible confounding factors and exploring alternative models to ensure that any evidence of archaic introgression is the most parsimonious explanation.
Similarly, African populations are commonly used as a control panel, presumed to lack archaic ancestry from Neanderthals and Denisovans, to identify which variants in non-African populations are likely of an archaic origin. However, mounting evidence of gene flow events between African populations and archaic humans challenges this assumption (Hammer et al. 2011; Kuhlwilm et al. 2016; Wall et al. 2019; Chen et al. 2020; Durvasula and Sankararaman 2020; Hubisz et al. 2020; Peyrégne et al. 2022; Harris et al. 2023; Ragsdale et al. 2023; Li et al. 2024). This calls for the further development of methods that do not rely on control panels to make inferences about archaic introgression. IBDmix exemplifies this approach, and has been used to detect Neanderthal ancestry across African populations (Chen et al. 2020; Harris et al. 2023; Li et al. 2024). IBDmix infers introgressed segments at the individual level for a single archaic source, with its power increasing alongside the sample size of the target panel. ARGweaver-D, in contrast, circumvents this reliance on a control panel by instead requiring a predefined demographic model, enabling inferences of introgressed segments at the haplotype level for an arbitrary number of potential archaic sources but its computational demands limit scalability (Hubisz et al. 2020). Future methods should draw inspiration from both IBDmix and ARGweaver-D to develop new inference frameworks for identifying introgressed segments at the haplotype level, which can accommodate multiple archaic sources, remain robust to sample size variation, and are computationally scalable.
Conclusion
Over the past 15 years, there has been a steady increase in the number of studies describing the extent and evolutionary impact of archaic introgression across populations worldwide. The continued effort to extend existing approaches and develop new methods will not only improve the resolution of our understanding of the complex demographic history of our species but also deepen our understanding and appreciation of the role archaic introgression has played in shaping both genetic and phenotypic variation throughout human evolution.
Although most studies on archaic introgression have focused on European or Asian populations, studying the landscape of archaic ancestry in recently admixed populations remains an ongoing challenge (Villanea and Witt 2022; Witt et al. 2023; Villanea et al. 2025). For instance, individuals from the Americas may have genetic ancestry that can be traced to populations commonly labeled as Indigenous American, European, or African due to the history of European colonization and the transatlantic slave trade (Medina-Muñoz et al. 2023). Consequently, there is a need for methods that can distinguish introgressed segments originating from different genetic backgrounds. One potential avenue to address these complexities is to develop methods that jointly infer both local ancestry and introgressed tracts. Because introgressed tracts would be sequestered within the particular genetic background from which they originated, local ancestry calls should provide greater resolution for identifying introgressed tracts, as recombination events coincide with the boundaries of introgressed tracts and induce transitions between genetic backgrounds.
Clear areas for progress also exist in loosening the data requirements for archaic introgression inference by developing methods that explicitly accommodate the challenges inherent to low-coverage aDNA sequencing. Because archaic variants on the same introgressed segment are expected to be in strong LD (Chakraborty and Weiss 1988; Wall 2000), and many of these variants are represented in modern human reference panels, imputation offers a promising avenue for studying archaic introgression in such contexts (Capodiferro et al. 2025). Nonetheless, imputation alone cannot fully capture the underlying complexities of aDNA sequencing, as indeed, both empirical and simulation-based benchmarking have shown that imputation accuracy is sensitive to the overall sequencing quality and coverage of the aDNA data (Hui et al. 2020; Ausmees et al. 2022; Ausmees and Nettelblad 2023; Sousa da Mota et al. 2023; Çubukcu and Kılınç 2024; Escobar-Rodríguez and Veeramah 2024; Garrido Marques et al. 2024). A future direction for overcoming these technical limitations is to integrate methods that explicitly account for the aforementioned sequencing complexities (e.g., admixfrog’s genotype likelihood model; Peter 2020) into an aDNA imputation framework, thereby modeling both genotype uncertainty and the underlying characteristics of aDNA that generate this uncertainty. Alternatively, other strategies could bypass imputation entirely. For example, Colate (Speidel et al. 2021) and Twigstats (Speidel et al. 2025) infer demographic and population history parameters from low-coverage ancient genomes by leveraging mutation age estimates obtained from Relate’s (Speidel et al. 2019) genome-wide genealogies reconstructed from modern human data. Future work could draw on these approaches to harness the potential of inferred ARGs for studying archaic introgression in low-coverage ancient individuals not included in the reconstructed genealogies.
These exciting avenues for future work will only encourage the development and further exploration of possible methods for investigating the nature of archaic introgression. We hope that by keeping both practitioners and developers abreast of this ever-expanding toolkit, the field will continue to deepen our understanding of our shared and tangled evolutionary history.
Data sets
All links to methods reported in Tables 1–3 were last accessed on January 7, 2026:
ARGweaver: https://github.com/mdrasmus/argweaver,
ARGweaver-D: https://github.com/CshlSiepelLab/argweaver-d-analysis,
tsinfer: https://tskit.dev/tsinfer/docs/stable,
tsdate: https://tskit.dev/tsdate/docs/stable,
diCal-ADMIX: https://diCal-ADMIX.sourceforge.net,
ArchaicSeeker2.0: https://github.com/Shuhua-Group/ArchaicSeeker2.0,
admixfrog: https://github.com/BenjaminPeter/admixfrog,
Sankararaman et al.’s (2016) CRF: https://drive.google.com/file/d/1Kz9jDP1tm8291tUZr0Zk2ahvglRnTJ0d,
IntroUNET: https://github.com/SchriderLab/introNets,
ArchIE: https://github.com/sriramlab/ArchIE,
hmmix: https://github.com/LauritsSkov/Introgression-detection,
DAIseg: https://github.com/LeoPlanche/DAIseg,
VolcanoFinder: https://degiorgiogroup.fau.edu/vf.html,
genomatnn: https://github.com/grahamgower/genomatnn,
MaLAdapt: https://github.com/xzhang-popgen/maladapt.
All links to publicly available data sets referenced in Tables 1–3 were last accessed on January 7, 2026:
Introgression maps from Hubisz et al. (2020): http://compgen.cshl.edu/ARGweaver/introgressionHub/files,
Inferred genealogies from Wohns et al. (2022): https://zenodo.org/records/5512994,
Neanderthal introgression maps from Li et al. (2024): https://zenodo.org/records/5512994,
Neanderthal introgression maps from Steinrücken et al. (2018): https://drive.google.com/drive/folders/0B_RxlKHkUXRDflZDd0xWTEVNWVF2OFppUmNka3ZWN19pbU5qVjBVV2lGVVBuQTJkWE5KTDg?resourcekey=0-LAWuR89grC372Hd572CGag&usp=drive_link,
Introgression maps from Yuan et al. (2021): https://github.com/Shuhua-Group/ArchaicSeeker2.0/tree/master/IntrogressedSeg,
Neanderthal introgression maps from Iasi et al. (2024): https://doi.org/10.5061/dryad.zw3r228gg,
Introgression maps from Sankararaman et al. (2016): https://drive.google.com/drive/folders/1DyhMw0E1mXQUDNeGQbQrcQbHe0uTyK2h,
Introgression maps from Browning et al. (2018): https://doi.org/10.17632/y7hyt83vxr.1,
Introgression maps from Durvasula and Sankararaman (2020): https://github.com/sriramlab/afr_archaic_maps,
Introgression maps from Skov et al. (2018): https://doi.org/10.1371/journal.pgen.1007641.s012,
Introgression maps from Planche et al. (2025): https://github.com/LeoPlanche/DAIseg/blob/main/src/IntrogressionMap,
Example workflow from Setter et al. (2020): https://doi.org/10.5061/dryad.7h44j0zr7,
Pretrained models from Gower et al. (2021): https://github.com/grahamgower/genomatnn/tree/main/pre-trained,
Adaptive introgression maps from Gower et al. (2021): https://github.com/grahamgower/genomatnn/tree/main/predictions,
Adaptive introgression maps from Zhang et al. (2023): https://github.com/xzhang-popgen/maladapt/tree/main/empirical_scores.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank the members of the Ramachandran and Huerta-Sánchez lab for their insightful comments and discussions. D.P. and E.H.-S. were supported by National Institutes of Health (NIH) grant no. R35GM128946 (to E.H.-S.). D.P. is also a trainee supported by the Brown University Predoctoral Training Program in Biological Data Science (NIH T32 GM128596) and the Blavatnik Family Foundation Graduate Fellowship. M.M.B. was supported by the Howard Hughes Medical Institute Gilliam Fellowship. J.M.T. was supported by the Young Investigator’s grant from the Human Frontier Science Program (to E.H.-S.). M.M. was supported by National Science Foundation grant no. PRFB-2305910 (to M.M.). E.H.-S. was also supported by the Alfred P. Sloan Award.
Notes
[4] Article published online before print. Article and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278993.124.
[5] Freely available online through the Genome Research Open Access option.
References
- ↵The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. 10.1038/nature15393
- ↵Abi-Rached L, Jobin MJ, Kulkarni S, McWhinnie A, Dalva K, Gragert L, Babrzadeh F, Gharizadeh B, Luo M, Plummer FA, 2011. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science 334: 89–94. 10.1126/science.1209202
- ↵Ahlquist KD, Bañuelos MM, Funk A, Lai J, Rong S, Villanea FA, Witt KE. 2021. Our tangled family tree: new genomic methods offer insight into the legacy of archaic admixture. Genome Biol Evol 13: evab115. 10.1093/gbe/evab115
- ↵Ausmees K, Sanchez-Quinto F, Jakobsson M, Nettelblad C. 2022. An empirical evaluation of genotype imputation of ancient DNA. G3 (Bethesda) 12: jkac089. 10.1093/g3journal/jkac089
- ↵Ausmees K, Nettelblad C. 2023. Achieving improved accuracy for imputation of ancient DNA. Bioinformatics 39: btac738. 10.1093/bioinformatics/btac738
- ↵Baumdicker F, Bisschop G, Goldstein D, Gower G, Ragsdale AP, Tsambos G, Zhu S, Eldon B, Ellerman EC, Galloway JG, 2022. Efficient ancestry and mutation simulation with msprime 1.0. Genetics 220: iyab229. 10.1093/genetics/iyab229
- ↵Bergström A, McCarthy SA, Hui R, Almarri MA, Ayub Q, Danecek P, Chen Y, Felkel S, Hallast P, Kamm J, 2020. Insights into human genetic variation and population history from 929 diverse genomes. Science 367: eaay5012. 10.1126/science.aay5012
- ↵Bokelmann L, Hajdinjak M, Peyrégne S, Brace S, Essel E, de Filippo C, Glocke I, Grote S, Mafessoni F, Nagel S, 2019. A genetic analysis of the Gibraltar Neanderthals. Proc Natl Acad Sci 116: 15610–15615. 10.1073/pnas.1903984116
- ↵Brandt DYC, Huber CD, Chiang CWK, Ortega-Del Vecchyo D. 2024. The promise of inferring the past using the ancestral recombination graph. Genome Biol Evol 16: evae005. 10.1093/gbe/evae005
- ↵Browning SR, Browning BL, Zhou Y, Tucci S, Akey JM. 2018. Analysis of human sequence data reveals two pulses of archaic Denisovan admixture. Cell 173: 53–61.e9. 10.1016/j.cell.2018.02.031
- ↵Byrska-Bishop M, Evani US, Zhao X, Basile AO, Abel HJ, Regier AA, Corvelo A, Clarke WE, Musunuri R, Nagulapalli K, 2022. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185: 3426–3440.e19. 10.1016/j.cell.2022.08.004
- ↵Capodiferro MR, Planche L, Breslin EM, Ongaro L, Ávila-Arcos MC, Jay F, Cassidy LM, Huerta-Sanchez E. 2025. Archaic ancestry inference in imputed ancient human genomes. bioRxiv 10.1101/2025.07.23.664192
- ↵Caramelli D, Lalueza-Fox C, Vernesi C, Lari M, Casoli A, Mallegni F, Chiarelli B, Dupanloup I, Bertranpetit J, Barbujani G, 2003. Evidence for a genetic discontinuity between Neandertals and 24,000-year-old anatomically modern Europeans. Proc Natl Acad Sci 100: 6593–6597. 10.1073/pnas.1130343100
- ↵Carlhoff S, Duli A, Nägele K, Nur M, Skov L, Sumantri I, Oktaviana AA, Hakim B, Burhan B, Syahdar FA, 2021. Genome of a middle holocene hunter-gatherer from wallacea. Nature 596: 543–547. 10.1038/s41586-021-03823-6
- ↵Chakraborty R, Weiss KM. 1988. Admixture as a tool for finding linked genes and detecting that difference from allelic association between loci. Proc Natl Acad Sci 85: 9119–9123. 10.1073/pnas.85.23.9119
- ↵Chen H, Green RE, Pääbo S, Slatkin M. 2007. The joint allele-frequency spectrum in closely related species. Genetics 177: 387–398. 10.1534/genetics.107.070730
- ↵Chen KM, Cofer EM, Zhou J, Troyanskaya OG. 2019. Selene: a PyTorch-based deep learning library for sequence data. Nat Methods 16: 315–318. 10.1038/s41592-019-0360-8
- ↵Chen L, Wolf AB, Fu W, Li L, Akey JM. 2020. Identifying and interpreting apparent Neanderthal ancestry in African individuals. Cell 180: 677–687.e16. 10.1016/j.cell.2020.01.012
- ↵Chevy ET, Huerta-Sánchez E, Ramachandran S. 2023. Integrating sex-bias into studies of archaic introgression on chromosome X. PLoS Genet 19: e1010399. 10.1371/journal.pgen.1010399
- ↵Chintalapati M, Patterson N, Moorjani P. 2022. The spatiotemporal patterns of major human admixture events during the European Holocene. eLife 11: e77625. 10.7554/eLife.77625
- ↵Choin J, Mendoza-Revilla J, Arauna LR, Cuadros-Espinoza S, Cassar O, Larena M, Ko AM-S, Harmant C, Laurent R, Verdu P, 2021. Genomic insights into population history and biological adaptation in Oceania. Nature 592: 583–589. 10.1038/s41586-021-03236-5
- ↵Cobb KA, Smith ML. 2025. The reasonable effectiveness of domain adaptation for inference of introgression. bioRxiv 10.1101/2025.01.17.633659
- ↵Coop G, Griffiths RC. 2004. Ancestral inference on gene trees under selection. Theor Popul Biol 66: 219–232. 10.1016/j.tpb.2004.06.006
- ↵Corbett-Detig R, Nielsen R. 2017. A hidden Markov model approach for simultaneously estimating local ancestry and admixture time using next generation sequence data in samples of arbitrary ploidy. PLoS Genet 13: e1006529. 10.1371/journal.pgen.1006529
- ↵Çubukcu H, Kılınç GM. 2024. Evaluation of genotype imputation using glimpse tools on low coverage ancient DNA. Mamm Genome 35: 461–473. 10.1007/s00335-024-10053-4
- ↵Currat M, Excoffier L. 2004. Modern humans did not admix with Neanderthals during their range expansion into Europe. PLoS Biol 2: e421. 10.1371/journal.pbio.0020421
- ↵Currat M, Excoffier L. 2011. Strong reproductive isolation between humans and Neanderthals inferred from observed patterns of introgression. Proc Natl Acad Sci 108: 15129–15134. 10.1073/pnas.1107450108
- ↵Dagilis AJ, Matute DR. 2023. The fitness of an introgressing haplotype changes over the course of divergence and depends on its size and genomic location. PLoS Biol 21: e3002185. 10.1371/journal.pbio.3002185
- ↵Dannemann M, Andrés AM, Kelso J. 2016. Introgression of Neandertal- and Denisovan-like haplotypes contributes to adaptive variation in human toll-like receptors. Am J Hum Genet 98: 22–33. 10.1016/j.ajhg.2015.11.015
- ↵Dannemann M, Kelso J. 2017. The contribution of Neanderthals to phenotypic variation in modern humans. Am J Hum Genet 101: 578–589. 10.1016/j.ajhg.2017.09.010
- ↵Dannemann M, Racimo F. 2018. Something old, something borrowed: admixture and adaptation in human evolution. Curr Opin Genet Dev 53: 1–8. 10.1016/j.gde.2018.05.009
- ↵Di Santo LN, Quilodrán CS, Currat M. 2023. Temporal variation in introgressed segments’ length statistics computed from a limited number of ancient genomes sheds light on past admixture pulses. Mol Biol Evol 40: msad252. 10.1093/molbev/msad252
- ↵Ding Q, Hu Y, Xu S, Wang J, Jin L. 2014a. Neanderthal introgression at chromosome 3p21.31 was under positive natural selection in East Asians. Mol Biol Evol 31: 683–695. 10.1093/molbev/mst260
- ↵Ding Q, Hu Y, Xu S, Wang C-C, Li H, Zhang R, Yan S, Wang J, Jin L. 2014b. Neanderthal origin of the haplotypes carrying the functional variant Val92Met in the MC1R in modern humans. Mol Biol Evol 31: 1994–2003. 10.1093/molbev/msu180
- ↵Durand EY, Patterson N, Reich D, Slatkin M. 2011. Testing for ancient admixture between closely related populations. Mol Biol Evol 28: 2239–2252. 10.1093/molbev/msr048
- ↵Durvasula A, Sankararaman S. 2019. A statistical model for reference-free inference of archaic local ancestry. PLoS Genet 15: e1008175. 10.1371/journal.pgen.1008175
- ↵Durvasula A, Sankararaman S. 2020. Recovering signals of ghost archaic introgression in African populations. Sci Adv 6: eaax5097. 10.1126/sciadv.aax5097
- ↵Edelman NB, Mallet J. 2021. Prevalence and adaptive impact of introgression. Annu Rev Genet 55: 265–283. 10.1146/genet.2021.55.issue-1
- ↵Enard D, Petrov DA. 2018. Evidence that RNA viruses drove adaptive introgression between Neanderthals and modern humans. Cell 175: 360–371.e13. 10.1016/j.cell.2018.08.034
- ↵Eriksson A, Manica A. 2012. Effect of ancient population structure on the degree of polymorphism shared between modern human populations and ancient hominins. Proc Natl Acad Sci 109: 13956–13960. 10.1073/pnas.1200567109
- ↵Eriksson A, Manica A. 2014. The doubly conditioned frequency spectrum does not distinguish between ancient population structure and hybridization. Mol Biol Evol 31: 1618–1621. 10.1093/molbev/msu103
- ↵Escobar-Rodríguez M, Veeramah KR. 2024. Evaluation of ancient DNA imputation: a simulation study. Hum Popul Genet Genom 4: 1–23. 10.47248/hpgg
- ↵Fan C, Cahoon JL, Dinh BL, Ortega-Del Vecchyo D, Huber CD, Edge MD, Mancuso N, Chiang CWK. 2025. A likelihood-based framework for demographic inference from genealogical trees. Nat Genet 57: 865–874. 10.1038/s41588-025-02129-x
- ↵Flagel L, Brandvain Y, Schrider DR. 2019. The unreasonable effectiveness of convolutional neural networks in population genetic inference. Mol Biol Evol 36: 220–238. 10.1093/molbev/msy224
- ↵Garrido Marques A, Rubinacci S, Malaspinas A-S, Delaneau O, Sousa da Mota B. 2024. Assessing the impact of post-mortem damage and contamination on imputation performance in ancient DNA. Sci Rep 14: 6227. 10.1038/s41598-024-56584-3
- ↵Geurts P, Ernst D, Wehenkel L. 2006. Extremely randomized trees. Mach Learn 63: 3–42. 10.1007/s10994-006-6226-1
- ↵Gittelman RM, Schraiber JG, Vernot B, Mikacenic C, Wurfel MM, Akey JM. 2016. Archaic hominin admixture facilitated adaptation to out-of-Africa environments. Curr Biol 26: 3375–3382. 10.1016/j.cub.2016.10.041
- ↵Gokcumen O. 2020. Archaic hominin introgression into modern human genomes. Am J Phys Anthropol 171: 60–73. 10.1002/ajpa.v171.S70
- ↵Gopalan S, Atkinson EG, Buck LT, Weaver TD, Henn BM. 2021. Inferring archaic introgression from hominin genetic data. Evol Anthropol 30: 199–220. 10.1002/evan.v30.3
- ↵Gower G, Picazo PI, Fumagalli M, Racimo F. 2021. Detecting adaptive introgression in human evolution using convolutional neural networks. eLife 10: e64669. 10.7554/eLife.64669
- ↵Gravel S. 2012. Population genetics models of local ancestry. Genetics 191: 607–619. 10.1534/genetics.112.139808
- ↵Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH-Y, 2010. A draft sequence of the neandertal genome. Science 328: 710–722. 10.1126/science.1188021
- ↵Griffiths R. 1991. The two-locus ancestral graph. Lect Notes Monogr Ser 18: 100–117. 10.1214/lnms/1215459289
- ↵Griffiths RC, Marjoram P. 1996. Ancestral inference from samples of DNA sequences with recombination. J Comput Biol 3: 479–502. 10.1089/cmb.1996.3.479
- ↵Griffiths RC, Marjoram P. 1997. An ancestral recombination graph. Inst Math Appl 87: 257.
- ↵Hackl J, Huang X. 2025. Revisiting adaptive introgression at the HLA genes in lithuanian genomes with machine learning. Infect Genet Evol 127: 105708. 10.1016/j.meegid.2024.105708
- ↵Hajdinjak M, Fu Q, Hübner A, Petr M, Mafessoni F, Grote S, Skoglund P, Narasimham V, Rougier H, Crevecoeur I, 2018. Reconstructing the genetic history of late Neanderthals. Nature 555: 652–656. 10.1038/nature26151
- ↵Haller BC, Messer PW. 2023. SLiM 4: multispecies eco-evolutionary modeling. Am Nat 201: E127–E139. 10.1086/723601
- ↵Hamlin JAP, Hibbins MS, Moyle LC. 2020. Assessing biological factors affecting postspeciation introgression. Evol Lett 4: 137–154. 10.1002/evl3.159
- ↵Hammer MF, Woerner AE, Mendez FL, Watkins JC, Wall JD. 2011. Genetic evidence for archaic admixture in Africa. Proc Natl Acad Sci 108: 15123–15128. 10.1073/pnas.1109300108
- ↵Harris K, Nielsen R. 2016. The genetic cost of Neanderthal introgression. Genetics 203: 881–891. 10.1534/genetics.116.186890
- ↵Harris DN, Platt A, Hansen MEB, Fan S, McQuillan MA, Nyambo T, Mpoloka SW, Mokone GG, Belay G, Fokunang C, 2023. Diverse African genomes reveal selection on ancient modern human introgressions in Neanderthals. Curr Biol 33: 4905–4916.e5. 10.1016/j.cub.2023.09.066
- ↵Hejase HA, Dukler N, Siepel A. 2020. From summary statistics to gene trees: methods for inferring positive selection. Trends Genet 36: 243–258. 10.1016/j.tig.2019.12.008
- ↵Hibbins MS, Hahn MW. 2019. The timing and direction of introgression under the multispecies network coalescent. Genetics 211: 1059–1073. 10.1534/genetics.118.301831
- ↵Huang X, Kruisz P, Kuhlwilm M. 2022. Sstar: a python package for detecting archaic introgression from population genetic data with S. Mol Biol Evol 39: msac212. 10.1093/molbev/msac212
- ↵Hubisz M, Siepel A. 2020. Inference of ancestral recombination graphs using ARGweaver. Methods Mol Biol 2090: 231–266. 10.1007/978-1-0716-0199-0
- ↵Hubisz MJ, Williams AL, Siepel A. 2020. Mapping gene flow between ancient hominins through demography-aware inference of the ancestral recombination graph. PLoS Genet 16: e1008895. 10.1371/journal.pgen.1008895
- ↵Hudson RR. 1983. Testing the constant-rate neutral allele model with protein sequence data. Evolution 37: 203. 10.1111/evo.1983.37.issue-1
- ↵Huerta-Sánchez E, Jin X, Asan B, Vinckenbosch N, Liang Y, Yi X, He M, Somel M, 2014. Altitude adaptation in tibetans caused by introgression of Denisovan-like DNA. Nature 512: 194–197. 10.1038/nature13408
- ↵Hui R, D’Atanasio E, Cassidy LM, Scheib CL, Kivisild T. 2020. Evaluating genotype imputation pipeline for ultra-low coverage ancient genomes. Sci Rep 10: 18542. 10.1038/s41598-020-75387-w
- ↵Iasi LNM, Ringbauer H, Peter BM. 2021. An extended admixture pulse model reveals the limitations to human-Neandertal introgression dating. Mol Biol Evol 38: 5156–5174. 10.1093/molbev/msab210
- ↵Iasi LNM, Chintalapati M, Skov L, Mesa AB, Hajdinjak M, Peter BM, Moorjani P. 2024. Neanderthal ancestry through time: insights from genomes of ancient and present-day humans. Science 386: eadq3010. 10.1126/science.adq3010
- ↵Ingvarsson PK, Whitlock MC. 2000. Heterosis increases the effective migration rate. Proc Biol Sci 267: 1321–1326. 10.1098/rspb.2000.1145
- ↵Jacobs GS, Hudjashov G, Saag L, Kusuma P, Darusallam CC, Lawson DJ, Mondal M, Pagani L, Ricaut F-X, Stoneking M, 2019. Multiple deeply divergent Denisovan ancestries in papuans. Cell 177: 1010–1021.e32. 10.1016/j.cell.2019.02.035
- ↵Jin G, Nakhleh L, Snir S, Tuller T. 2006. Maximum likelihood of phylogenetic networks. Bioinformatics 22: 2604–2611. 10.1093/bioinformatics/btl452
- ↵Juric I, Aeschbacher S, Coop G. 2016. The strength of selection against Neanderthal introgression. PLoS Genet 12: e1006340. 10.1371/journal.pgen.1006340
- ↵Kelleher J, Wong Y, Wohns AW, Fadil C, Albers PK, McVean G. 2019. Inferring whole-genome histories in large population datasets. Nat Genet 51: 1330–1338. 10.1038/s41588-019-0483-y
- ↵Kerdoncuff E, Skov L, Patterson N, Banerjee J, Khobragade P, Chakrabarti SS, Chakrawarty A, Chatterjee P, Dhar M, Gupta M, 2025. 50,000 years of evolutionary history of India: impact on health and disease variation. Cell 188: 3389–3404.e6. 10.1016/j.cell.2025.04.027
- ↵Khrameeva EE, Bozek K, He L, Yan Z, Jiang X, Wei Y, Tang K, Gelfand MS, Prufer K, Kelso J, 2014. Neanderthal ancestry drives evolution of lipid catabolism in contemporary Europeans. Nat Commun 5: 3584. 10.1038/ncomms4584
- ↵Kim BY, Lohmueller KE. 2015. Selection and reduced population size cannot explain higher amounts of neandertal ancestry in East Asian than in European human populations. Am J Hum Genet 96: 454–461. 10.1016/j.ajhg.2014.12.029
- ↵Kim BY, Huber CD, Lohmueller KE. 2018. Deleterious variation shapes the genomic landscape of introgression. PLoS Genet 14: e1007741. 10.1371/journal.pgen.1007741
- ↵Kim Y, Nielsen R. 2004. Linkage disequilibrium as a signature of selective sweeps. Genetics 167: 1513–1524. 10.1534/genetics.103.025387
- ↵Krings M, Stone A, Schmitz RW, Krainitzki H, Stoneking M, Pääbo S. 1997. Neandertal DNA sequences and the origin of modern humans. Cell 90: 19–30. 10.1016/S0092-8674(00)80310-4
- ↵Krings M, Geisert H, Schmitz RW, Krainitzki H, Pääbo S. 1999. DNA sequence of the mitochondrial hypervariable region II from the neandertal type specimen. Proc Natl Acad Sci 96: 5581–5585. 10.1073/pnas.96.10.5581
- ↵Kubatko LS. 2009. Identifying hybridization events in the presence of coalescence via model selection. Syst Biol 58: 478–488. 10.1093/sysbio/syp055
- ↵Kuhlwilm M, Gronau I, Hubisz MJ, de Filippo C, Prado-Martinez J, Kircher M, Fu Q, Burbano HA, Lalueza-Fox C, de la Rasilla Marco, 2016. Ancient gene flow from early modern humans into eastern Neanderthals. Nature 530: 429–433. 10.1038/nature16544
- ↵Kumar KH, Rubinacci S, Zöllner S. 2025. MetaGLIMPSE: meta imputation of low coverage sequencing data for modern and ancient genomes. bioRxiv 10.1101/2025.06.24.660721
- ↵Larena M, McKenna J, Sanchez-Quinto F, Bernhardsson C, Ebeo C, Reyes R, Casel O, Huang J-Y, Hagada KP, Guilay D, 2021. Philippine ayta possess the highest level of Denisovan ancestry in the world. Curr Biol 31: 4219–4230.e10. 10.1016/j.cub.2021.07.022
- ↵Lewanski AL, Grundler MC, Bradburd GS. 2024. The era of the ARG: an introduction to ancestral recombination graphs and their significance in empirical evolutionary genomics. PLoS Genet 20: e1011110. 10.1371/journal.pgen.1011110
- ↵Li L, Comi TJ, Bierman RF, Akey JM. 2024. Recurrent gene flow between Neanderthals and modern humans over the past 200,000 years. Science 385: eadi1768. 10.1126/science.adi1768
- ↵Liang M, Nielsen R. 2014. The lengths of admixture tracts. Genetics 197: 953–967. 10.1534/genetics.114.162362
- ↵Liu KJ, Dai J, Truong K, Song Y, Kohn MH, Nakhleh L. 2014a. An HMM-based comparative genomic framework for detecting introgression in eukaryotes. PLoS Comput Biol 10: e1003649. 10.1371/journal.pcbi.1003649
- ↵Liu X, Koyama S, Tomizuka K, Takata S, Ishikawa Y, Ito S, Kosugi S, Suzuki K, Hikino K, Koido M, 2024b. Decoding triancestral origins, archaic introgression, and natural selection in the Japanese population by whole-genome sequencing. Sci Adv 10: eadi8419. 10.1126/sciadv.adi8419
- ↵Lohse K, Frantz LAF. 2014. Neandertal admixture in Eurasia confirmed by maximum-likelihood analysis of three genomes. Genetics 196: 1241–1251. 10.1534/genetics.114.162396
- ↵Lopez Fang L, Peede D, Ortega-Del Vecchyo D, McTavish EJ, Huerta-Sánchez E. 2024. Leveraging shared ancestral variation to detect local introgression. PLoS Genet 20: e1010155. 10.1371/journal.pgen.1010155
- ↵Macià MC, Skov L, Damgaard Bæk ZE, Hobolth A. 2025. Enhancement of hidden markov model analyses for improved inference of archaic introgression in modern humans. bioRxiv 10.1101/2025.04.22.649993
- ↵Mafessoni F, Grote S, de Filippo C, Slon V, Kolobova KA, Viola B, Markin SV, Chintalapati M, Peyrégne S, Skov L, 2020. A high-coverage neandertal genome from chagyrskaya cave. Proc Natl Acad Sci 117: 15132–15136. 10.1073/pnas.2004944117
- ↵Malaspinas A-S, Westaway MC, Muller C, Sousa VC, Lao O, Alves I, Bergström A, Athanasiadis G, Cheng JY, Crawford JE, 2016. A genomic history of aboriginal Australia. Nature 538: 207–214. 10.1038/nature18299
- ↵Malinsky M, Challis RJ, Tyers AM, Schiffels S, Terai Y, Ngatunga BP, Miska EA, Durbin R, Genner MJ, Turner GF. 2015. Genomic islands of speciation separate cichlid ecomorphs in an east African crater lake. Science 350: 1493–1498. 10.1126/science.aac9927
- ↵Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, Racimo F, Zhao M, Chennagiri N, Nordenfelt S, Tandon A, 2016. The simons genome diversity project: 300 genomes from 142 diverse populations. Nature 538: 201–206. 10.1038/nature18964
- ↵Mallick S, Micco A, Mah M, Ringbauer H, Lazaridis I, Olalde IN, Patterson N, Reich D. 2024. The Allen ancient DNA resource (AADR) a curated compendium of ancient human genomes. Sci Data 11: 182. 10.1038/s41597-024-03031-7
- ↵Martin SH, Davey JW, Jiggins CD. 2014. Evaluating the use of ABBA–BABA statistics to locate introgressed loci. Mol Biol Evol 32: 244–257. 10.1093/molbev/msu269
- ↵Martin SH, Amos W. 2021. Signatures of introgression across the allele frequency spectrum. Mol Biol Evol 38: 716–726. 10.1093/molbev/msaa239
- ↵Massilani D, Skov L, Hajdinjak M, Gunchinsuren B, Tseveendorj D, Yi S, Lee J, Nagel S, Nickel B, Devièse T, 2020. Denisovan ancestry and population history of early East Asians. Science 370: 579–583. 10.1126/science.abc1166
- ↵McColl H, Racimo F, Vinner L, Demeter F, Gakuhari T, Moreno-Mayar JV, van Driem G, Gram Wilken U, Seguin-Orlando A, 2018. The prehistoric peopling of southeast Asia. Science 361: 88–92. 10.1126/science.aat3628
- ↵Medina-Muñoz SG, Ortega-Del Vecchyo D, Cruz-Hervert LP, Ferreyra-Reyes L, García-García L, Moreno-Estrada A, Ragsdale AP. 2023. Demographic modeling of admixed Latin American populations from whole genomes. Am J Hum Genet 110: 1804–1816. 10.1016/j.ajhg.2023.08.015
- ↵Medina-Tretmanis J, Jay F, Ávila-Arcos MC, Huerta-Sanchez E. 2024. Simulation-based benchmarking of ancient haplotype inference for detecting population structure. Hum Popul Genet Genom 4: 1–25. 10.47248/hpgg
- ↵Mendez FL, Watkins JC, Hammer MF. 2012. A haplotype at STAT2 introgressed from Neanderthals and serves as a candidate of positive selection in Papua New Guinea. Am J Hum Genet 91: 265–274. 10.1016/j.ajhg.2012.06.015
- ↵Mendez FL, Watkins JC, Hammer MF. 2013. Neandertal origin of genetic variation at the cluster of OAS immunity genes. Mol Biol Evol 30: 798–801. 10.1093/molbev/mst004
- ↵Meng C, Kubatko LS. 2009. Detecting hybrid speciation in the presence of incomplete lineage sorting using gene tree incongruence: a model. Theor Popul Biol 75: 35–45. 10.1016/j.tpb.2008.10.004
- ↵Meyer M, Kircher M, Gansauge M-T, Li H, Racimo F, Mallick S, Schraiber JG, Jay F, Prüfer K, de Filippo C, 2012. A high-coverage genome sequence from an archaic Denisov3an individual. Science 338: 222–226. 10.1126/science.1224344
- ↵Mo Z, Siepel A. 2023. Domain-adaptive neural networks improve supervised machine learning based on simulated population genetic data. PLoS Genet 19: e1011032. 10.1371/journal.pgen.1011032
- ↵Moorjani P, Sankararaman S, Fu Q, Przeworski M, Patterson N, Reich D. 2016. A genetic method for dating ancient genomes provides a direct estimate of human generation interval in the last 45,000 years. Proc Natl Acad Sci 113: 5652–5657. 10.1073/pnas.1514696113
- ↵Ni X, Yang X, Guo W, Yuan K, Zhou Y, Ma Z, Xu S. 2016. Length distribution of ancestral tracks under a general admixture model and its applications in population history inference. Sci Rep 6: 20048. 10.1038/srep20048
- ↵Nielsen R, Vaughn AH, Deng Y. 2025. Inference and applications of ancestral recombination graphs. Nat Rev Genet 26: 47–58. 10.1038/s41576-024-00772-4
- ↵Noonan JP, Coop G, Kudaravalli S, Smith D, Krause J, Alessi J, Chen F, Platt D, Pääbo S, Pritchard JK, 2006. Sequencing and analysis of Neanderthal genomic DNA. Science 314: 1113–1118. 10.1126/science.1131412
- ↵Ongaro L, Huerta-Sanchez E. 2024. A history of multiple Denisovan introgression events in modern humans. Nat Genet 56: 2612–2622. 10.1038/s41588-024-01960-y
- ↵Orlando L, Allaby R, Skoglund P, Der Sarkissian C, Stockhammer PW, Ávila-Arcos MC, Fu Q, Krause J, Willerslev E, Stone AC, 2021. Ancient DNA analysis. Nat Rev Methods Primers 1: 1–26. 10.1038/s43586-020-00011-0
- ↵Pamilo P, Nei M. 1988. Relationships between gene trees and species trees. Mol Biol Evol 5: 568–583.
- ↵Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, Genschoreck T, Webster T, Reich D. 2012. Ancient admixture in human history. Genetics 192: 1065–1093. 10.1534/genetics.112.145037
- ↵Paul JS, Song YS. 2010. A principled approach to deriving approximate conditional sampling distributions in population genetics models with recombination. Genetics 186: 321–338. 10.1534/genetics.110.117986
- ↵Paul JS, Steinrücken M, Song YS. 2011. An accurate sequentially Markov conditional sampling distribution for the coalescent with recombination. Genetics 187: 1115–1128. 10.1534/genetics.110.125534
- ↵Peede D, Ortega-Del Vecchyo D, Huerta-Sanchez E. 2022. The utility of ancestral and derived allele sharing for genome-wide inferences of introgression. bioRxiv 10.1101/2022.12.02.518851
- ↵Peter BM. 2016. Admixture, population structure, and F-statistics. Genetics 202: 1485–1501. 10.1534/genetics.115.183913
- ↵Peter BM. 2020. 100,000 years of gene flow between Neandertals and Denisovans in the Altai Mountains. bioRxiv 10.1101/2020.03.13.990523
- ↵Peyrégne S, Kelso J, Peter BM, Pääbo S. 2022. The evolutionary history of human spindle genes includes back-and-forth gene flow with Neandertals. eLife 11: e75464. 10.7554/eLife.75464
- ↵Peyrégne S, Slon V, Kelso J. 2024. More than a decade of genetic research on the Denisovans. Nat Rev Genet 25: 83–103. 10.1038/s41576-023-00643-4
- ↵Pfeifer B, Kapan DD. 2019. Estimates of introgression as a function of pairwise distances. BMC Bioinformatics 20: 207. 10.1186/s12859-019-2747-z
- ↵Plagnol V, Wall JD. 2006. Possible ancestral structure in human populations. PLoS Genet 2: e105. 10.1371/journal.pgen.0020105
- ↵Planche L, Ilina A, Ávila-Arcos MC, Jay F, Huerta-Sanchez E, Shchur V. 2025. An archaic reference-free method to jointly infer Neanderthal and Denisovan introgressed segments in modern human genomes. bioRxiv 10.1101/2025.03.17.643330
- ↵Pool JE, Nielsen R. 2009. Inference of historical changes in migration rate from the lengths of migrant tracts. Genetics 181: 711–719. 10.1534/genetics.108.098095
- ↵Prüfer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, de Filippo C, 2014. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505: 43–49. 10.1038/nature12886
- ↵Prüfer K, de Filippo C, Grote S, Mafessoni F, Korlević P, Hajdinjak M, Vernot B, Skov L, Hsieh P, Peyrégne S, 2017. A high-coverage neandertal genome from vindija cave in Croatia. Science 358: 655–658. 10.1126/science.aao1887
- ↵Pugach I, Matveyev R, Wollstein A, Kayser M, Stoneking M. 2011. Dating the age of admixture via wavelet transform analysis of genome-wide data. Genome Biol 12: R19. 10.1186/gb-2011-12-2-r19
- ↵Qin P, Stoneking M. 2015. Denisovan ancestry in east eurasian and native American populations. Mol Biol Evol 32: 2665–2674. 10.1093/molbev/msv141
- ↵Racimo F, Sankararaman S, Nielsen R, Huerta-Sánchez E. 2015. Evidence for archaic adaptive introgression in humans. Nat Rev Genet 16: 359–371. 10.1038/nrg3936
- ↵Racimo F, Marnetto D, Huerta-Sánchez E. 2016. Signatures of archaic adaptive introgression in present-day human populations. Mol Biol Evol 34: 296–317. 10.1093/molbev/msw216
- ↵Racimo F, Gokhman D, Fumagalli M, Ko A, Hansen T, Moltke I, Albrechtsen A, Carmel L, Huerta-Sánchez E, Nielsen R. 2017. Archaic adaptive introgression in TBX15/WARS2. Mol Biol Evol 34: 509–524. 10.1093/molbev/msw283
- ↵Ragsdale AP, Weaver TD, Atkinson EG, Hoal EG, Möller M, Henn BM, Gravel S. 2023. A weakly structured stem for human origins in Africa. Nature 617: 755–763. 10.1038/s41586-023-06055-y
- ↵Rasmussen MD, Hubisz MJ, Gronau I, Siepel A. 2014. Genome-wide inference of ancestral recombination graphs. PLoS Genet 10: e1004342. 10.1371/journal.pgen.1004342
- ↵Ray DD, Flagel L, Schrider DR. 2024. IntroUNET: identifying introgressed alleles via semantic segmentation. PLoS Genet 20: e1010657. 10.1371/journal.pgen.1010657
- ↵Reich D, Thangaraj K, Patterson N, Price AL, Singh L. 2009. Reconstructing Indian population history. Nature 461: 489–494. 10.1038/nature08365
- ↵Reich D, Green RE, Kircher M, Krause J, Patterson N, Durand EY, Viola B, Briggs AW, Stenzel U, Johnson PLF, 2010. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468: 1053–1060. 10.1038/nature09710
- ↵Reich D, Patterson N, Kircher M, Delfin F, Nandineni MR, Pugach I, Ko AM-S, Ko Y-C, Jinam TA, Phipps ME, 2011. Denisova admixture and the first modern human dispersals into southeast Asia and Oceania. Am J Hum Genet 89: 516–528. 10.1016/j.ajhg.2011.09.005
- ↵Reilly PF, Tjahjadi A, Miller SL, Akey JM, Tucci S. 2022. The contribution of Neanderthal introgression to modern human traits. Curr Biol 32: R970–R983. 10.1016/j.cub.2022.08.027
- ↵Rogers AR, Harris NS, Achenbach AA. 2020. Neanderthal-Denisovan ancestors interbred with a distantly related hominin. Sci Adv 6: eaay5483. 10.1126/sciadv.aay5483
- ↵Romieu J, Camarata G, Crochet P-A, de Navascués M, Leblois R, Rousset F. 2025. Performance evaluation of adaptive introgression classification methods. Peer Community J 5: e95. 10.24072/pcjournal.617
- ↵Sams AJ, Dumaine A, Nédélec Y, Yotova V, Alfieri C, Tanner JE, Messer PW, Barreiro LB. 2016. Adaptively introgressed Neandertal haplotype at the OAS locus functionally impacts innate immune responses in humans. Genome Biol 17: 246. 10.1186/s13059-016-1098-6
- ↵Sanchez T, Bray EM, Jobic P, Guez J, Letournel A-C, Charpiat G, Cury J, Jay F. 2023. dnadna: a deep learning framework for population genetics inference. Bioinformatics 39: btac765. 10.1093/bioinformatics/btac765
- ↵Sankararaman S. 2020. Methods for detecting introgressed archaic sequences. Curr Opin Genet Dev 62: 85–90. 10.1016/j.gde.2020.05.026
- ↵Sankararaman S, Patterson N, Li H, Pääbo S, Reich D. 2012. The date of interbreeding between Neandertals and modern humans. PLoS Genet 8: e1002947. 10.1371/journal.pgen.1002947
- ↵Sankararaman S, Mallick S, Dannemann M, Prüfer K, Kelso J, Pääbo S, Patterson N, Reich D. 2014. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507: 354–357. 10.1038/nature12961
- ↵Sankararaman S, Mallick S, Patterson N, Reich D. 2016. The combined landscape of Denisovan and Neanderthal ancestry in present-day humans. Curr Biol 26: 1241–1247. 10.1016/j.cub.2016.03.037
- ↵Santiago E, Caballero A. 2005. Variation after a selective sweep in a subdivided population. Genetics 169: 475–483. 10.1534/genetics.104.032813
- ↵Schaefer NK, Shapiro B, Green RE. 2021. An ancestral recombination graph of human, Neanderthal, and Denisovan genomes. Sci Adv 7: eabc0776. 10.1126/sciadv.abc0776
- ↵Schrider DR, Ayroles J, Matute DR, Kern AD. 2018. Supervised machine learning reveals introgressed loci in the genomes of Drosophila simulans and D. sechellia. PLoS Genet 14: e1007341. 10.1371/journal.pgen.1007341
- ↵Schumer M, Xu C, Powell DL, Durvasula A, Skov L, Holland C, Blazier JC, Sankararaman S, Andolfatto P, Rosenthal GG, 2018. Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science 360: 656–660. 10.1126/science.aar3684
- ↵Serre D, Langaney A, Chech M, Teschler-Nicola M, Paunovic M, Mennecier P, Hofreiter M, Possnert G, Pääbo S. 2004. No evidence of Neandertal mtDNA contribution to early modern humans. PLoS Biol 2: E57. 10.1371/journal.pbio.0020057
- ↵Setter D, Mousset S, Cheng X, Nielsen R, DeGiorgio M, Hermisson J. 2020. VolcanoFinder: genomic scans for adaptive introgression. PLoS Genet 16: e1008867. 10.1371/journal.pgen.1008867
- ↵Shchur V, Svedberg J, Medina P, Corbett-Detig R, Nielsen R. 2020. On the distribution of tract lengths during adaptive introgression. G3 (Bethesda) 10: 3663–3673. 10.1534/g3.120.401616
- ↵Skoglund P, Jakobsson M. 2011. Archaic human ancestry in east Asia. Proc Natl Acad Sci 108: 18301–18306. 10.1073/pnas.1108181108
- ↵Skoglund P, Northoff BH, Shunkov MV, Derevianko AP, Pääbo S, Krause J, Jakobsson M. 2014. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc Natl Acad Sci 111: 2229–2234. 10.1073/pnas.1318934111
- ↵Skov L, Hui R, Shchur V, Hobolth A, Scally A, Schierup MH, Durbin R. 2018. Detecting archaic introgression using an unadmixed outgroup. PLoS Genet 14: e1007641. 10.1371/journal.pgen.1007641
- ↵Skov L, Coll Macià M, Sveinbjörnsson G, Mafessoni F, Lucotte EA, Einarsdóttir MS, Jonsson H, Halldorsson B, Gudbjartsson DF, Helgason A, 2020. The nature of Neanderthal introgression revealed by 27,566 icelandic genomes. Nature 582: 78–83. 10.1038/s41586-020-2225-9
- ↵Skov L, Peyrégne S, Popli D, Iasi LNM, Devièse T, Slon V, Zavala EI, Hajdinjak M, Sümer AP, Grote S, 2022. Genetic insights into the social organization of Neanderthals. Nature 610: 519–525. 10.1038/s41586-022-05283-y
- ↵Slimak L, Vimala T, Seguin-Orlando A, Metz L, Zanolli C, Joannes-Boyau R, Frouin M, Arnold LJ, Demuro M, Devièse T, 2024. Long genetic and social isolation in Neanderthals before their extinction. Cell Genomics 4: 100593. 10.1016/j.xgen.2024.100593
- ↵Slon V, Mafessoni F, Vernot B, de Filippo C, Grote S, Viola B, Hajdinjak M, Peyrégne S, Nagel S, Brown S, 2018. The genome of the offspring of a Neanderthal mother and a Denisovan father. Nature 561: 113–116. 10.1038/s41586-018-0455-x
- ↵Smith ML, Hahn MW. 2024. Selection leads to false inferences of introgression using popular methods. Genetics 227: iyae089. 10.1093/genetics/iyae089
- ↵Sousa da Mota B, Rubinacci S, Cruz Dávalos DI, G Amorim CE, Sikora M, Johannsen NN, Szmyt MH, Włodarczak P, Szczepanek A, Przybyła MM, 2023. Imputation of ancient human genomes. Nat Commun 14: 3660. 10.1038/s41467-023-39202-0
- ↵Speidel L, Forest M, Shi S, Myers SR. 2019. A method for genome-wide genealogy estimation for thousands of samples. Nat Genet 51: 1321–1329. 10.1038/s41588-019-0484-x
- ↵Speidel L, Cassidy L, Davies RW, Hellenthal G, Skoglund P, Myers SR. 2021. Inferring population histories for ancient genomes using genome-wide genealogies. Mol Biol Evol 38: 3497–3511. 10.1093/molbev/msab174
- ↵Speidel L, Silva M, Booth T, Raffield B, Anastasiadou K, Barrington C, Götherström A, Heather P, Skoglund P. 2025. High-resolution genomic history of early medieval Europe. Nature 637: 118–126. 10.1038/s41586-024-08275-2
- ↵Steinrücken M, Paul JS, Song YS. 2013. A sequentially Markov conditional sampling distribution for structured populations with migration and recombination. Theor Popul Biol 87: 51–61. 10.1016/j.tpb.2012.08.004
- ↵Steinrücken M, Spence JP, Kamm JA, Wieczorek E, Song YS. 2018. Model-based detection and analysis of introgressed Neanderthal ancestry in modern humans. Mol Ecol 27: 3873–3888. 10.1111/mec.2018.27.issue-19
- ↵Stringer CB, Andrews P. 1988. Genetic and fossil evidence for the origin of modern humans. Science 239: 1263–1268. 10.1126/science.3125610
- ↵Stringer C. 2022. The development of ideas about a recent African origin for homo sapiens. Rivista di antropologia [J Anthropol Sci] 100: 5–18. 10.4436/JASS.10009
- ↵Sümer AP, Rougier H, Villalba-Mouco V, Huang Y, Iasi LNM, Essel E, Bossoms Mesa A, Furtwaengler A, Peyrégne S, de Filippo C, 2025. Earliest modern human genomes constrain timing of Neanderthal admixture. Nature 638: 711–717. 10.1038/s41586-024-08420-x
- ↵Tagore D, Akey JM. 2025. Archaic hominin admixture and its consequences for modern humans. Curr Opin Genet Dev 90: 102280. 10.1016/j.gde.2024.102280
- ↵Tajima F. 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics 105: 437–460. 10.1093/genetics/105.2.437
- ↵Teixeira JAC, Jacobs GS, Stringer C, Tuke J, Hudjashov G, Purnomo GA, Sudoyo H, Cox MP, Tobler R, Turney CSM, 2021. Widespread Denisovan ancestry in Island Southeast Asia but no evidence of substantial super-archaic hominin admixture. Nat Ecol Evol 5: 616–624. 10.1038/s41559-021-01408-0
- ↵Than C, Ruths D, Innan H, Nakhleh L. 2007. Confounding factors in HGT detection: statistical error, coalescent effects, and multiple solutions. J Comput Biol 14: 517–535. 10.1089/cmb.2007.A010
- ↵Tournebize R, Chikhi L. 2025. Ignoring population structure in hominin evolutionary models can lead to the inference of spurious admixture events. Nat Ecol Evol 9: 225–236. 10.1038/s41559-024-02591-6
- ↵Tsutaya T, Sawafuji R, Taurozzi AJ, Fagernäs Z, Patramanis I, Troché G, Mackie M, Gakuhari T, Oota H, Tsai C-H, 2025. A male Denisovan mandible from pleistocene Taiwan. Science 388: 176–180. 10.1126/science.ads3888
- ↵Veller C, Edelman NB, Muralidhar P, Nowak MA. 2023. Recombination and selection against introgressed DNA. Evolution 77: 1131–1144. 10.1093/evolut/qpad021
- ↵Vernot B, Akey JM. 2014. Resurrecting surviving neandertal lineages from modern human genomes. Science 343: 1017–1021. 10.1126/science.1245938
- ↵Vernot B, Akey JM. 2015. Complex history of admixture between modern humans and Neandertals. Am J Hum Genet 96: 448–453. 10.1016/j.ajhg.2015.01.006
- ↵Vernot B, Tucci S, Kelso J, Schraiber JG, Wolf AB, Gittelman RM, Dannemann M, Grote S, McCoy RC, Norton H, 2016. Excavating Neandertal and Denisovan DNA from the genomes of Melanesian individuals. Science 352: 235–239. 10.1126/science.aad9416
- ↵Vernot B, Zavala EI, Gómez-Olivencia A, Jacobs Z, Slon V, Mafessoni F, Romagné F, Pearson A, Petr M, Sala N, 2021. Unearthing Neanderthal population history using nuclear and mitochondrial DNA from cave sediments. Science 372: eabf1667. 10.1126/science.abf1667
- ↵Vespasiani DM, Jacobs GS, Cook LE, Brucato N, Leavesley M, Kinipi C, Ricaut F-X, Cox MP, Gallego Romero I. 2022. Denisovan introgression has shaped the immune system of present-day Papuans. PLoS Genet 18: e1010470. 10.1371/journal.pgen.1010470
- ↵Villanea FA, Schraiber JG. 2019. Multiple episodes of interbreeding between Neanderthal and modern humans. Nat Ecol Evol 3: 39–44. 10.1038/s41559-018-0735-8
- ↵Villanea FA, Witt KE. 2022. Underrepresented populations at the archaic introgression frontier. Front Genet 13: 821170. 10.3389/fgene.2022.821170
- ↵Villanea FA, Peede D, Kaufman EJ, Añorve Garibay V, Chevy ET, Villa-Islas V, Witt KE, Zeloni R, Marnetto D, Moorjani P, 2025. The MUC19 gene: an evolutionary history of recurrent introgression and natural selection. Science 389: eadl0882. 10.1126/science.adl0882
- ↵Wall JD. 2000. Detecting ancient admixture in humans using sequence polymorphism data. Genetics 154: 1271–1279. 10.1093/genetics/154.3.1271
- ↵Wall JD, Yang MA, Jay F, Kim SK, Durand EY, Stevison LS, Gignoux C, Woerner A, Hammer MF, Slatkin M. 2013. Higher levels of Neanderthal ancestry in east asians than in Europeans. Genetics 194: 199–209. 10.1534/genetics.112.148213
- ↵Wall JD, Ratan A, Stawiski E, GenomeAsia 100K Consortium. 2019. Identification of African-specific admixture between modern and archaic humans. Am J Hum Genet 105: 1254–1261. 10.1016/j.ajhg.2019.11.005
- ↵Wang Y, Wang W, Liu D, Hou W, Zhou T, Ji Z. 2023. GeneSegNet: a deep learning framework for cell segmentation by integrating gene expression and imaging. Genome Biol 24: 235. 10.1186/s13059-023-03054-0
- ↵Witt KE, Villanea F, Loughran E, Zhang X, Huerta-Sanchez E. 2022. Apportioning archaic variants among modern populations. Philos Trans R Soc Lond B Biol Sci 377: 20200411. 10.1098/rstb.2020.0411
- ↵Witt KE, Funk A, Añorve Garibay V, Fang LL, Huerta-Sánchez E. 2023. The impact of modern admixture on archaic human ancestry in human populations. Genome Biol Evol 15: evad066. 10.1093/gbe/evad066
- ↵Wohns AW, Wong Y, Jeffery B, Akbari A, Mallick S, Pinhasi R, Patterson N, Reich D, Kelleher J, McVean G. 2022. A unified genealogy of modern and ancient genomes. Science 375: eabi8264. 10.1126/science.abi8264
- ↵Wolpoff MH, Thorne AG, Smith FH, Frayer DW, Pope GG. 1994. Multiregional evolution: a world-wide source for modern human populations. In Origins of anatomically modern humans, pp. 175–199. Springer US, Boston.
- ↵Wong Y, Ignatieva A, Koskela J, Gorjanc G, Wohns AW, Kelleher J. 2024. A general and efficient representation of ancestral recombination graphs. Genetics 228: iyae100. 10.1093/genetics/iyae100
- ↵Yang MA, Malaspinas A-S, Durand EY, Slatkin M. 2012. Ancient structure in Africa unlikely to explain Neanderthal and non-African genetic similarity. Mol Biol Evol 29: 2987–2995. 10.1093/molbev/mss117
- ↵Yermakovich D, André M, Brucato N, Kariwiga J, Leavesley M, Pankratov V, Mondal M, Ricaut F-X, Dannemann M. 2024. Denisovan admixture facilitated environmental adaptation in Papua New Guinean populations. Proc Natl Acad Sci 121: e2405889121. 10.1073/pnas.2405889121
- ↵Yuan K, Ni X, Liu C, Pan Y, Deng L, Zhang R, Gao Y, Ge X, Liu J, Ma X, 2021. Refining models of archaic admixture in eurasia with ArchaicSeeker 2.0. Nat Commun 12: 6232. 10.1038/s41467-021-26503-5
- ↵Zeng M, Li M, Fei Z, Wu F-X, Li Y, Pan Y, Wang J. 2021. A deep learning framework for identifying essential proteins by integrating multiple types of biological information. IEEE/ACM Trans Comput Biol Bioinform 18: 296–305. 10.1109/TCBB.2019.2897679
- ↵Zhang R, Yuan K, Xu S. 2022. Detecting archaic introgression and modeling multiple-wave admixture with ArchaicSeeker 2.0. STAR Protoc 3: 101314. 10.1016/j.xpro.2022.101314
- ↵Zhang X, Kim B, Lohmueller KE, Huerta-Sánchez E. 2020. The impact of recessive deleterious variation on signals of adaptive introgression in human populations. Genetics 215: 799–812. 10.1534/genetics.120.303081
- ↵Zhang X, Witt KE, Bañuelos MM, Ko A, Yuan K, Xu S, Nielsen R, Huerta-Sanchez E. 2021. The history and evolution of the Denisovan-EPAS1 haplotype in Tibetans. Proc Natl Acad Sci 118: e2020803118. 10.1073/pnas.2020803118
- ↵Zhang X, Kim B, Singh A, Sankararaman S, Durvasula A, Lohmueller KE. 2023. MaLAdapt reveals novel targets of adaptive introgression from Neanderthals and Denisovans in worldwide human populations. Mol Biol Evol 40: msad001. 10.1093/molbev/msad001
- ↵Zhou Y, Browning SR. 2021. Protocol for detecting introgressed archaic variants with SPrime. STAR Protoc 2: 100550. 10.1016/j.xpro.2021.100550
- ↵Zhou S, Butler-Laporte G, Nakanishi T, Morrison DR, Afilalo J, Afilalo M, Laurent L, Pietzner M, Kerrison N, Zhao K, 2021. A Neanderthal OAS1 isoform protects individuals of European ancestry against COVID-19 susceptibility and severity. Nat Med 27: 659–667. 10.1038/s41591-021-01281-1