
Abstract
Gene coexpression networks (GCNs) describe relationships among genes that maintain cellular identity and homeostasis. However, typical RNA-seq experiments often lack sufficient sample sizes for reliable GCN inference. recount3, a data set with 316,443 processed human RNA-seq samples, provides an opportunity to improve network reconstruction. However, GCN inference from public data is challenged by confounders and inconsistent labeling. To address this, we develop a pipeline to annotate samples based on cell-type composition. By comparing aggregation strategies, we find that regressing confounders within studies and prioritizing larger studies optimizes network reconstruction. We apply these findings to infer three consensus networks (universal, cancer, noncancer) and 27 context-specific networks. Central genes in consensus networks are enriched for evolutionarily constrained genes and ubiquitous biological pathways, whereas context-specific central nodes include tissue-specific transcription factors. The increased statistical power from data aggregation facilitates the derivation of variant annotations from context-specific networks, which are significantly enriched for complex-trait heritability independent of overlap with baseline functional genomic annotations. Although data aggregation led to strictly increasing held-out log-likelihood, we observe diminishing marginal improvements, suggesting that integrating complementary modalities, such as Hi-C and ChIP-seq, can further refine network reconstruction. Our approach outlines best practices for GCN inference and highlights both the strengths and limitations of data aggregation.
Critical cellular processes including the maintenance of cellular identity, homeostasis, and the cellular response to external stimuli are orchestrated through complex transcriptional coregulation of multiple genes (Hartwell et al. 1999; Hasty et al. 2002; Oltvai and Barabási 2002; Alon 2003). Gene coexpression networks (GCNs) are a commonly used framework to describe gene-gene relationships and are comprised of nodes that represent genes and edges linking coexpressed genes (Stuart et al. 2003). A comprehensive catalog of gene coexpression relationships has the potential to characterize genes with unknown functions (Schlitt et al. 2003), identify regulatory genes (Narang et al. 2015), determine changes in regulatory mechanisms that are key to cellular identity (Wang et al. 2021), and prioritize genes that drive phenotypic variability (van Dam et al. 2017).
Despite the utility of GCNs in understanding biological systems, network inference is still a challenging problem and suffers from both false positive and negative edges (Diaz and Stumpf 2022). In particular, the typical sample size of most RNA-seq studies is orders of magnitude smaller than the number of gene pairs over which regulatory relationships are inferred, making network inference an underdetermined problem. Additionally, factors such as the stochastic nature of gene expression, experimental noise, missing data, and unobserved technical confounders make it difficult to avoid false positives or negatives.
Because the number of possible gene-gene interactions scales with the square of the number of genes examined, a potential solution to increase statistical power by reducing network complexity has been to utilize methods such as WGCNA that infer modules or groups of coexpressed genes that are regulated by one or more transcription factors rather than individual gene interactions (Segal et al. 2003). Although this approach has been successful at decreasing the number of hypotheses tested and thereby increasing statistical power (Wolf et al. 2014), it does not identify detailed network structure or distinguish between direct and indirect gene interactions. In contrast, network inference by graphical lasso (Friedman et al. 2008; Hastie et al. 2013) results in the identification of pairwise edges reflecting direct effects, such that the absence of an edge implies the conditional independence of the genes when all other genes are observed. Further, the formulation of graphical lasso enables flexible penalization based on the number of edges found in the network, which aids in the identification of a network structure that improves the discovery of true gene-gene interactions while reducing false positives (Huang et al. 2020). Here, we focus on improving the statistical power of network inference by significantly increasing the number of samples used in network inference, leveraging large-scale publicly available and uniformly processed RNA-seq data from recount3 (Wilks et al. 2021) which includes human RNA-seq samples from GTEx (The GTEx Consortium 2020), TCGA (Tomczak et al. 2015), and the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) (Kodama et al. 2012; Katz et al. 2022).
Public RNA-seq data resources such as recount3 offer an unprecedented opportunity to improve GCN inference but also present major challenges due to heterogeneity in sample preparation, sequencing protocols, missing or inconsistent metadata, and variation in biological and technical factors across studies. To address these challenges, we set out to develop a data preprocessing pipeline that can identify and exclude outliers, harmonize expression measurements across studies, and group samples into biologically meaningful contexts appropriate for network reconstruction.
Although statistical methods for confounder correction have been widely applied within individual studies (Parsana et al. 2019), it remains unclear how best to account for confounding and integrate data across multiple studies in a way that preserves true biological signal and enhances the quality of network inference. This motivated us to systematically evaluate strategies for confounder adjustment and data aggregation across studies. Finally, to assess the utility of these networks for downstream biological discovery, we examined the relevance of inferred network features, such as node centrality, to known regulatory roles and their enrichment for complex trait heritability using stratified LD score regression (S-LDSC) (Finucane et al. 2015). In summary, our study seeks to provide a carefully annotated RNA-seq data set, outline best practices for GCN inference by leveraging publicly available RNA-seq data, and a set of consensus and context-specific networks that will aid the scientific community in achieving the full potential of GCN inference in biomedical research.
Results
Manual annotation and clustering of RNA-seq data from recount3 identifies 27 unique tissue contexts
We downloaded uniformly processed RNA-seq samples from humans using the recount3 R package (Wilks et al. 2021) comprised of experiments from three data sources—The Sequence Read Archive (SRA) (Kodama et al. 2012; Katz et al. 2022), Genotype-tissue Expression (GTEx, version 8) (The GTEx Consortium 2020), and The Cancer Genome Atlas (TCGA) (Tomczak et al. 2015)—and selected 1747 projects that included 30 or more samples each. Following quality control (Methods), 95,280 human bulk-RNA sequencing samples remained from 50 GTEx tissues (18,828 samples), 33 TCGA cancer types (11,091 samples), and 884 SRA studies (65,361 samples) (Fig. 1A). The number of genes present following data preprocessing varied by study and is summarized in Supplemental Figure S1. The aggregated data includes samples from a wide array of tissues, cell types, and diseases. Whereas GTEx and TCGA studies included metadata specifying the tissue of origin and disease status for all samples, SRA studies had inconsistent nomenclature. Therefore, to obtain reliable labels for SRA samples, we manually parsed sample descriptions to obtain sample characteristics corresponding to tissue type and disease status for 65,361 SRA samples (Methods). Based on curated annotations, 93.5% of TCGA samples and 30.4% of SRA samples were cancerous. In contrast, all GTEx samples were noncancerous, as expected (Fig. 1B). Tissue labels with the greatest number of samples across all three data sources included blood, central nervous system, breast, skin, and lung (Fig. 1C). SRA included 224 distinct tissue labels derived from manual annotation that was not observed in GTEx or TCGA and reflected a wide range of disease states, including Type I diabetes, Alzheimer's disease, bipolar disorder, arthritis, cancer, and infectious conditions (Fig. 1D). We grouped SRA samples based on their study accession IDs, GTEx samples by tissue, and TCGA samples by cancer code (Methods). To simplify terminology, we defined each group of samples from a data source as a single study. To leverage the extensive biological diversity in the data, we inferred two broad types of networks: consensus and tissue context-specific (context-specific). Our universal consensus network included all samples, regardless of tissue or disease. Our noncancer consensus network included healthy samples and samples with disease status other than cancer. Finally, our cancer consensus network solely included cancerous samples. We restricted our context-specific networks to noncancerous samples grouped by tissue context. We did not examine differential coregulation resulting from noncancer disease and regressed these effects from gene expression. Thus, by including SRA, recount3 provides an unprecedented opportunity to examine unique contexts that were not previously studied in GTEx and TCGA.
Overview of data preprocessing and annotations. (A) Gene expression data was RPKM normalized and log-transformed along with gene-specific and sample-specific filters. Based on the data source, normalized gene expression was processed to merge replicates and exclude miRNA and scRNA-seq samples. (B) Number of samples which were annotated to be noncancerous and cancerous based on available metadata across GTEx, SRA, and TCGA. Sixty-three SRA samples did not have an associated annotation corresponding to cancer status. (C) Top 10 tissue labels by sample size across all three data sources: SRA, GTEx, and TCGA. (D) Top 20 diseases by sample size found in SRA that are not cancer. (E) t-SNE projection of xCell deconvolution scores of 63,193 noncancerous samples colored by the tissue of origin. (F) Increase in the sample size of 27 tissue contexts by using SRA samples compared to GTEx only. SRA studies included seven novel contexts which were not available in GTEx.
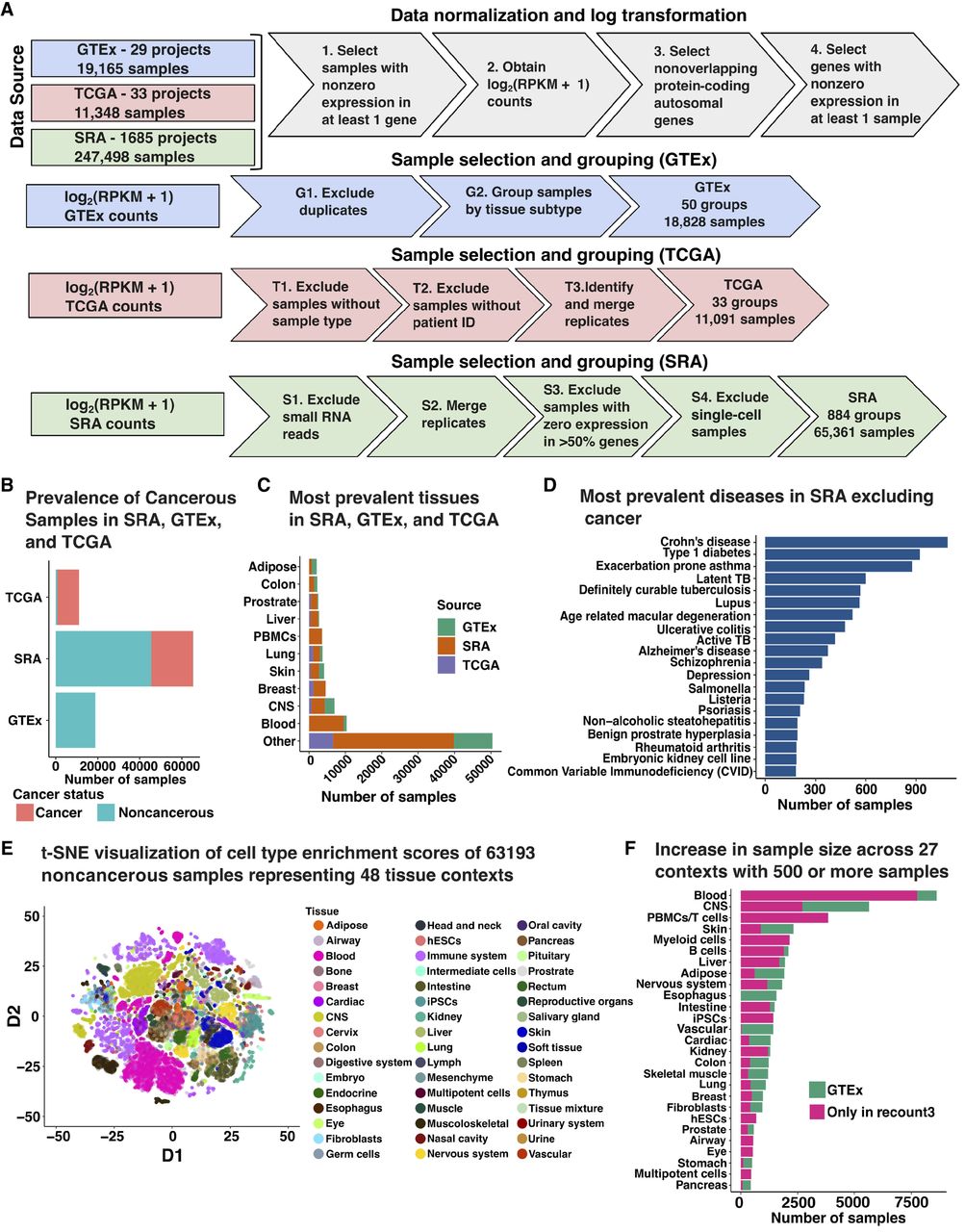
Across the three data sources, SRA, GTEx, and TCGA, we obtained 266 unique manually annotated tissue labels with a median sample size of 31, which was much lower than the number of protein-coding genes. Therefore, we used a study-pooling strategy based on related tissue contexts to increase power (Methods). Specifically, in this work, we define a tissue context as a group of samples with similar cell-type composition, which we infer computationally. Mapping manual annotations to tissue contexts (Supplemental Table S1) generated 48 tissue contexts across 63,193 noncancerous samples for context-specific network analysis. In each context, to ensure that a tissue context represented samples with similar cell-type composition as estimated by xCell (Aran et al. 2017) deconvolution, we learned a joint lower-dimensional t-SNE embedding using cell-type deconvolution scores, and for 25 contexts with more than 500 samples before outlier exclusion, we detected and excluded outliers (Supplemental Fig. S2). For the immune context, we observed that samples displayed extensive heterogeneity in cell-type composition. Thus, we further separated this group into B cells, PBMCs/ T cells, and myeloid cells (Supplemental Fig. S3). Finally, we examined the differences in the empirical covariance estimates obtained when we considered alternate thresholds to exclude outliers and found that, whereas we observed minor differences across covariance estimates obtained by varying the exclusion criterion, these differences were much smaller than the differences in empirical covariance matrices between unrelated contexts. Hence, samples that were found to be outliers by fewer than two methods were selected, to ensure we considered the maximum possible number of samples for each context, while minimizing outliers that may truly reflect different contexts (Supplemental Fig. S4).
In total, we obtained 27 contexts including seven tissue contexts that were not present in GTEx: airway, eye, human embryonic stem cells, induced pluripotent stem cells, multipotent cells, myeloid cells, and PBMCs/T cells (Fig. 1E). The number of samples present in each tissue context following outlier exclusion varied from 8797 (blood) to 485 (multipotent cells) (Fig. 1F). Of the tissue categories present in GTEx, we were able to significantly increase the number of samples for several tissue contexts including blood, central nervous system, skin, and liver (Fig. 1F). These harmonized samples have increased context resolution (i.e., include novel contexts which were not examined in GTEx) and increased sample size, which can be used to improve the inference of consensus and context-specific networks.
Data aggregation improves the inference of consensus and context-specific GCNs
The median study-specific sample size across the three data sources was 44 for SRA, 309 for TCGA, and 285 for GTEx. Further, 766 of 884 SRA studies had a sample size <100. PC-based data correction has been used within a single study to reduce potential false-positive gene regulatory associations (Leek et al. 2012; Parsana et al. 2019), but best practices for applying PC-based data correction in the context of aggregating multiple studies to infer GCNs have not been fully examined. First, we sought to determine whether data correction should be performed jointly across all samples, across samples belonging to a specific tissue context and multiple studies, or across samples from a specific tissue context and a specific study. We observed that PCs recapitulate different sample characteristics depending on the level at which data aggregation is performed. PCs calculated across all samples were driven predominantly by tissue labels followed by technical confounders (e.g., study and data source) (Supplemental Figs. S5A–D, S6A). PCs which were calculated across samples belonging to a single tissue context (blood) but multiple studies were predominantly driven by study and data source. Further, accounting for tissue heterogeneity enabled us to better model cancer status and disease annotations using PCs (Supplemental Figs. S5E–H, S6B). Finally, when we limited samples to a specific tissue context and a specific study (GTEx-skeletal muscle), we found that the top PCs were significantly associated with age, cause of death, and technical batch, consistent with the findings of Parsana et al. (2019) (Supplemental Figs. S5I–L, S6C). Thus, regressing PCs computed by accounting for tissue and cross-study heterogeneity from expression is integral to excluding technical effects and unwanted biological signals.
We examined the effect of PC-based data correction when inferring GCNs by aggregating data across diverse sources by comparing PC-based correction applied to either aggregate data, or individual studies followed by aggregation, where the number of PCs regressed is based on the permutation method described by Buja and Eyuboglu (1992) and Leek et al. (2012). Additionally, we compared the consequences of aggregating empirical covariance matrices inferred from PC-corrected data by either treating all studies irrespective of sample size equally (unweighted aggregation), or upweighting the covariance estimates from studies with a greater sample size (weighted aggregation). Thus, we used four paradigms: (1) Aggregating data before PC-based data correction followed by estimation of empirical covariance from residual expression; (2) PC-based data correction applied to individual studies followed by aggregation of residual expression and joint estimation of empirical covariance; (3) unweighted aggregation of covariance matrices inferred from each study separately after study-specific PC-based correction; and (4) weighted aggregation of covariance matrices computed from individual studies following study-specific PC-based data correction, where the weights were the ratio of the study sample size to the total number of samples used in network reconstruction (Fig. 2A; Methods).
Comparison of aggregation strategies to optimize network reconstruction. (A) Outline of strategies to compare data correction before and after aggregation and weighted and unweighted aggregation of single tissue/ study covariance matrices included: (1) aggregating data before PC-based data correction followed by estimation of empirical covariance from residual expression (aggregating data, orange); (2) PC-based data correction applied to individual studies followed by aggregation of residual expression and joint estimation of empirical covariance (aggregating data adjusted for confounding, brick red); (3) unweighted aggregation of covariance matrices inferred from each study separately after study-specific PC-based correction (unweighted covariance aggregation, purple); and (4) weighted aggregation of covariance matrices computed from individual studies following study-specific PC-based data correction (weighted covariance aggregation, magenta). (B) Held-out log-likelihood of networks inferred by sequentially aggregating either 10 GTEx studies or 100 SRA studies at a time versus the log of the number of edges (|E|) found in networks obtained by varying the penalization parameter λ (C) F1-score of networks inferred by sequentially aggregating either 10 GTEx studies or 100 SRA studies at a time versus the log of the number of edges (|E|) found in networks obtained by varying the penalization parameter λ when compared to canonical pathways compiled from KEGG, Biocarta, and Pathway Interaction Database. (D) Comparison of held-out log-likelihood corresponding to networks inferred over 49 GTEx studies or 566 SRA studies versus the log of the number of edges (|E|) found in networks obtained by varying the penalization parameter λ using four different aggregation strategies including aggregating data, aggregating data adjusted for confounding, unweighted, and weighted aggregation of covariance matrices. (E) Comparison of F1-scores of obtaining edges corresponding to canonical pathways from KEGG, Biocarta, and Pathway Interaction Database in networks inferred over 49 GTEx studies or 566 SRA studies versus the log of the number of edges (|E|) found in networks obtained by varying the penalization parameter λ using four different aggregation strategies including aggregating data, aggregating data adjusted for confounding, unweighted, and weighted aggregation of covariance matrices. (F) Total number of samples, number of individual studies, and the median sample size of each study which were used in the inference of universal consensus, noncancer consensus, and cancer consensus networks. (G) Comparison of F1-scores of obtaining edges corresponding to canonical pathways in the three consensus networks—universal, noncancer, and cancer—across networks with number of edges (|E|) varying between 5 × 103 and 5 × 106 edges. (H) Log-likelihood of GTEx blood samples based on networks inferred by sequentially aggregating SRA blood studies five at a time for number of edges ranging from 103 to 105 edges. (I) Log-likelihood of GTEx CNS samples based on networks inferred by sequentially aggregating SRA CNS studies five at a time for number of edges ranging from 103 to 105 edges. (J) Odds ratio of finding edges corresponding to tissue-specific protein-protein interactions (PPIs) derived from SNAP in tissue-context-specific networks inferred using all available samples versus only samples found in GTEx for six tissue contexts.
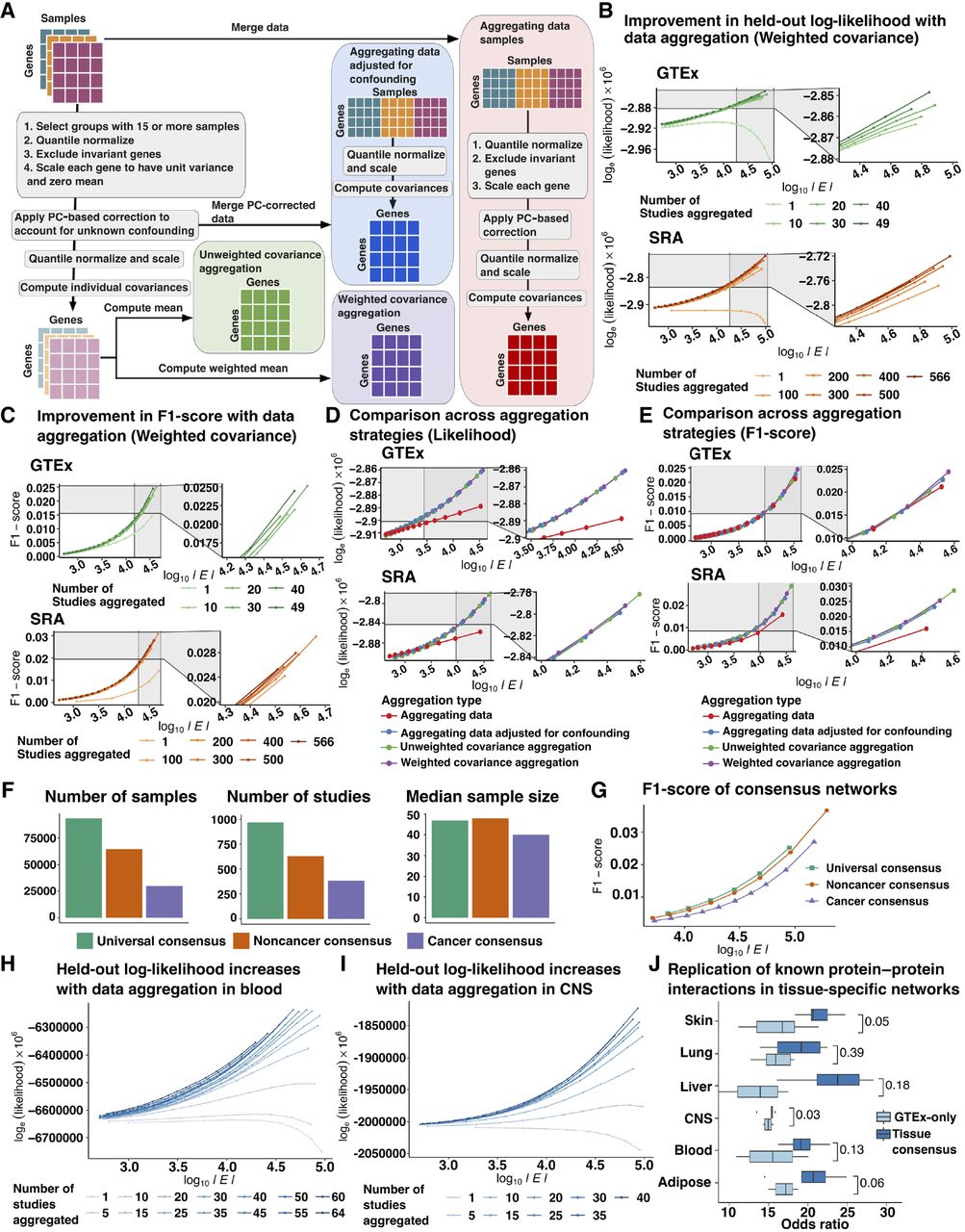
To compare strategies, we split noncancerous samples into two data splits, GTEx and SRA (Supplemental Fig. S7), followed by network inference by graphical lasso on one of the two data splits and evaluation with the held-out split by computing the held-out log-likelihood. Details pertaining to the number of studies, samples, and median PCs regressed for incremental data aggregation are provided in Supplemental Table S2. Additionally, we assessed the recapitulation of known biological pathways by computing the F1-score of finding canonical gene-gene relationships compiled from KEGG (Kanehisa and Goto 2000), Biocarta, and Pathway Interaction Database (Schaefer et al. 2009) obtained using Enrichr (Supplemental Table S3; Chen et al. 2013; Kuleshov et al. 2016). Paradigms in which PC-based data correction preceded aggregation led to a strict increase in held-out log-likelihood and F1-scores of known gene relationships from canonical pathways with the addition of more studies (Fig. 2B,C; Supplemental Figs. S8, S9). Additionally, we found that the low F1-scores were attributed to lower recall while the precision remained high, in accordance with previous studies that utilize graphical lasso to infer GCNs (Parsana et al. 2019). Further, we observed an increase in the precision with data aggregation for a particular value of recall across all strategies which performed PC-based data correction on individual studies (Supplemental Figs. S10, S11). This suggests that data aggregation resulted in GCNs with greater generalizability and recapitulated known biology better when technical confounders were estimated and regressed for individual studies. Because data aggregation led to a decrease in the number of edges found in the network for a particular value of the penalization parameter (Supplemental Fig. S12), we tested whether estimating denser networks would result in higher held-out log-likelihood specifically when the networks were inferred over a greater number of samples but instead observed that a larger number of edges led to overfitting and lower generalizability (Supplemental Fig. S13). Further, the marginal improvement in network reconstruction diminished with the subsequent rounds of aggregation. Although all methods that estimated technical confounders within each study performed similarly and were superior to estimating technical confounders across all samples when evaluated by held-out log-likelihood (Fig. 2D), we found that weighted aggregation of covariance matrices led to a slight improvement in the F1-score of the networks when compared to canonical pathways (Fig. 2E), suggesting that this is the optimal strategy among the alternatives compared.
Although our primary analysis concerns the recapitulation of individual, direct gene-gene edges through graphical lasso, which we expected to be heavily impacted by sample size, we also performed a limited evaluation of the effects of data aggregation with weighted gene coexpression network analysis (WGCNA) (Langfelder and Horvath 2008), a popular method for identifying modules of coexpressed genes. We inferred WGCNA modules by sequentially aggregating GTEx tissues and SRA studies following PC correction (Methods). We discovered that data aggregation led to an improvement in the recapitulation of known gene relationships from KEGG (Kanehisa and Goto 2000), Biocarta, and Pathway Interaction Database (Schaefer et al. 2009) for both GTEx and SRA. However, we note that this improvement is not strictly monotonic, particularly at the highest levels of data aggregation, in contrast to graphical lasso. This may be expected as module detection for WGCNA combines information across multiple gene pairs to identify modules and has been shown to work reasonably well even at small sample sizes. WGCNA module detection simply may not benefit as much from increased sample sizes. Thus, we conclude that PC-correction followed by data aggregation can potentially lead to an improvement in network performance for multiple network inference methods (Supplemental Fig. S14), but the improvement plateaus more quickly when inferring modules with WGCNA than when inferring direct edges with graphical lasso.
Finally, because graphical lasso can potentially identify edges reflecting direct gene-gene interactions, we evaluated the impact of data aggregation by weighted aggregation of covariance matrices, which were estimated from SRA studies following PC correction, on recapitulating known transcription factor (TF)–target gene relationships that were curated by Liska et al. (2022) based on experimental evidence such as ChIP-seq. We observed a modest improvement in the precision of predicting direct gene-gene relationships at higher values of recall with sequential data aggregation. Although we do see improvement, the identification of direct gene interactions remains challenging in the presence of unknown technical and biological confounders (Supplemental Fig. S15A; Kernfeld et al. 2024).
We inferred consensus GCNs across diverse tissues by weighted aggregation of covariance matrices estimated from residual expression and graphical lasso (Methods). In addition to a universal consensus network, which was inferred across 966 studies with sample size greater than or equal to 15, amounting to 95,276 samples across 48 tissue contexts, we constructed a noncancer consensus network and a cancer consensus network. The noncancer consensus network reflects data aggregated across 629 studies and 63,031 samples, and the cancer network reflects 386 studies and 29,967 samples (Fig. 2F). The number of genes which were included in the inference of the three consensus networks is summarized in Supplemental Table S4. We evaluated each consensus network's ability to recapitulate previously reported gene-gene interaction using the F1 score. Across a range of networks with a varying number of edges, we obtained a higher estimate of the F1 score from the universal consensus network, followed by noncancer and cancer consensus networks, which mirrors differences in sample size (Fig. 2G). We observed that the universal consensus and noncancer consensus networks performed better than the cancer consensus network at recapitulating potential TF–target relationships obtained from TFLink (version 1.0) (Liska et al. 2022). However, this could be either due to differences in sample size or the lack of cancer samples in the reference (Supplemental Fig. S15B).
We inferred networks across 27 tissue contexts and examined the impact of data aggregation on context-specific network reconstruction by considering GTEx samples (GTEx only) or by aggregating across samples from all data sources for that tissue context. The number of samples, studies, and median study-specific sample size for each tissue context in either aggregation setting are provided in Supplemental Figure S16. The number of genes over which context-specific networks was inferred is summarized in Supplemental Table S4. As in the consensus network inference, we used weighted aggregation of covariance matrices as the input to network inference by graphical lasso (Methods). To quantify the improvement in network reconstruction with data aggregation, we examined two tissue contexts with the largest sample size: blood and central nervous system (CNS). In both cases, we sequentially aggregated the blood or CNS SRA studies and computed the held-out log-likelihood utilizing context-matched GTEx samples. Similar to the results obtained from our consensus network analyses, we found that data aggregation led to a strict increase in held-out log-likelihood with additional studies and the greatest increase was observed while aggregating the first 20 studies (blood) and 15 studies (CNS) (Methods; Fig. 2H,I). Further, we examined the impact of data aggregation on inferring a context-specific network, specifically, if PC-based data correction sufficiently accounts for interstudy heterogeneity and minimizes the discovery of false positives, while improving the detection of true positive edges, when compared to alternate approaches to minimize false positives such as tuning the penalization parameter to be sufficiently large. Thus, we compared a blood-specific network inferred across all available samples to a blood-specific network inferred from a single study with a large sample-size (GTEx) in recapitulating known gene-gene interactions. We found that increasing the penalization parameter to improve the precision led to lower recall, a limitation that was overcome by data aggregation, which led to higher values of precision even for higher recall values. Finally, we observed that the optimal F1 score obtained by data aggregation exceeded the optimal F1 score obtained by tuning the penalization parameter (Supplemental Fig. S17).
Orthogonally, we verified that our networks recapitulated known context-specific gene relationships by examining the enrichment of previously known tissue-specific protein-protein interactions (PPIs) from SNAP (Leskovec and Sosič 2016) in edges belonging to six tissue context-specific networks (adipose, blood, CNS, liver, lung, and skin) (Methods). Details of the SNAP PPI terms that were mapped to specific tissue-contexts can be found in Supplemental Table S5. Data aggregation and increased sample size in the network significantly increased the estimated odds ratio of tissue-specific PPIs in the liver (median GTEx OR = 7.85, median aggregated OR = 38.85, P = 0.03) and skin (median GTEx OR = 21.83, median aggregated OR = 27.09, P = 0.05), and there was weak enrichment in adipose (median GTEx OR = 18.49, median aggregated OR = 21.14, P = 0.06). In other tissue contexts, we observed a higher median odds ratio of PPI enrichment in networks inferred by data aggregation compared to GTEx-only networks (Fig. 2J). Finally, we observed that context-specific networks obtained from blood, skin, central nervous system, liver, lung, and adipose were able to recapitulate known TF–target relationships from TFLink to varying degrees. The tissue with the highest number of samples, blood, resulted in networks with the best performance in reproducing TF–target relationships as indicated by precision and recall (Supplemental Fig. S15). Thus, we demonstrated that data aggregation led to the improved inference of consensus networks that capture canonical gene interactions and tissue context-specific networks that capture tissue biology by observing better network generalizability and reproduction of known biological processes.
Central network nodes are evolutionarily constrained and include genes that are critical to tissue identity
The biological information captured by a GCN can be evaluated by comparing individual network edges, by examining whether there are edges between genes that are known to interact in a particular cellular pathway (Ha et al. 2015), or by examining the properties of network nodes with a high number of connections or hubs (Pastor-Satorras et al. 2003; Chou et al. 2014; Oh et al. 2015). Because eukaryotic transcriptional networks typically consist of a subset of genes, often transcription factors, that regulate many downstream target genes (Albert 2005), we chose specific values of the penalization parameter λ and number of resulting network edges such that the selected networks are approximately scale-free (Methods; Supplemental Fig. S18; Supplemental Tables S6, S7). We computed different measures of centrality corresponding to each network node and tested for the enrichment of genes involved in GO terms that reflect ubiquitous or context-specific processes (Supplemental Table S8) among network nodes selected with progressively increasing thresholds for degree centrality against a background of all 18,882 protein-coding genes (Methods). Central nodes from all three consensus networks were strongly enriched for genes involved in functions such as microtubule-based process, chromosome organization, and regulation of organelle organization (Fig. 3A; Supplemental Fig. S19). In contrast, we found that central nodes from blood context-specific networks were enriched for genes associated with platelet activation, leukocyte differentiation, and leukocyte chemotaxis to a greater extent than central genes derived from either consensus or a discordant context-specific network (CNS) (Fig. 3B). Further, these trends were reflected across multiple tissue contexts; context-specific networks corresponding to CNS (Fig. 3C), skin, liver, and lung were enriched for genes associated with tissue-matched GO terms (Supplemental Fig. S20). In addition, we examined the enrichment of central genes from tissue-specific gold standards obtained from HumanBase (https://hb.flatironinstitute.org/). We observed that network nodes with a higher degree of centrality were enriched for hub nodes (with a degree in the top 80th percentile) of concordant tissue-specific reference networks, as evidenced by an odds ratio > 1 (Supplemental Fig. S21). Thus, whereas central genes from consensus networks included genes involved in essential cellular processes, context-specific gene relationships are lost with global aggregation and are unlikely to be recovered by increasing the sample size.
Properties of central network nodes of consensus and context-specific networks. (A–C) Enrichment of genes involved in GO processes among network genes selected with increasing thresholds of node degree in three consensus networks: universal (λ = 0.18, 7087 edges); noncancer (λ = 0.18, 7355 edges); and cancer (λ = 0.24, 7552 edges), as well as two context-specific networks: blood (λ = 0.24, 7283 edges); and CNS (λ = 0.28, 8430 edges). Tissue-context-specific networks were inferred only using noncancerous samples. Blood (GTEx) or CNS (GTEx) networks were inferred using samples only found in GTEx whereas blood and CNS networks were inferred using samples from GTEx and SRA. (D) Distribution of the excess overlap of evolutionarily conserved gene sets (Methods) for network nodes binned by the number of neighbors (degree) corresponding to universal consensus networks (λ = 0.14, 0.16, 0.18, 0.20), noncancer consensus networks (λ = 0.14, 0.16, 0.18, 0.20), cancer consensus networks (λ = 0.20, 0.22, 0.24, 0.26), blood networks (λ = 0.18, 0.20, 0.22, 0.24, 0.26), and CNS networks (λ = 0.24, 0.26, 0.28, 0.30, 0.32). Quintile 1 reflects nodes with no neighbors. Nodes with nonzero neighbors are split based on the degree quartile they belong to (Quintiles 2–5). We evaluated the excess overlap of 3104 loss-of-function (LoF) genes with pLI > 0.9, 2853 genes with a Shet > 0.1, 588 genes with a Phi-score > 0.95, and 1440 genes strongly depleted for missense mutations (high missense Z-score). (E) The degree distribution of network nodes that are tissue-specific transcription factors (TFs) in blood (52 TFs), lung (58 TFs), skin (10 TFs), pancreas (16 TFs), cardiac (17 TFs), muscle (7 TFs), CNS (51 TFs), general transcription factors (88 TFs), and protein-coding genes which are not transcription factors in universal consensus (λ = 0.18, 7087 edges), noncancer consensus (λ = 0.18, 7355 edges), skin (λ = 0.26, 7567 edges), skeletal muscle (λ = 0.26, 6254 edges), pancreas (λ = 0.32, 7615 edges), lung (λ = 0.30, 6349 edges), CNS (λ = 0.30, 6316 edges), cardiac (λ = 0.30, 6481 edges), and blood (λ = 0.24, 7283 edges). Pairs with no significance reported were not statistically distinct (P > 0.1). (F) Factor weights were obtained by nonnegative matrix factorization of the presence of hub genes in tissue-specific networks with ∼ 7000 edges. Details of the penalization parameter λ and the number of edges of selected networks for each tissue context are provided in Supplemental Table S7.
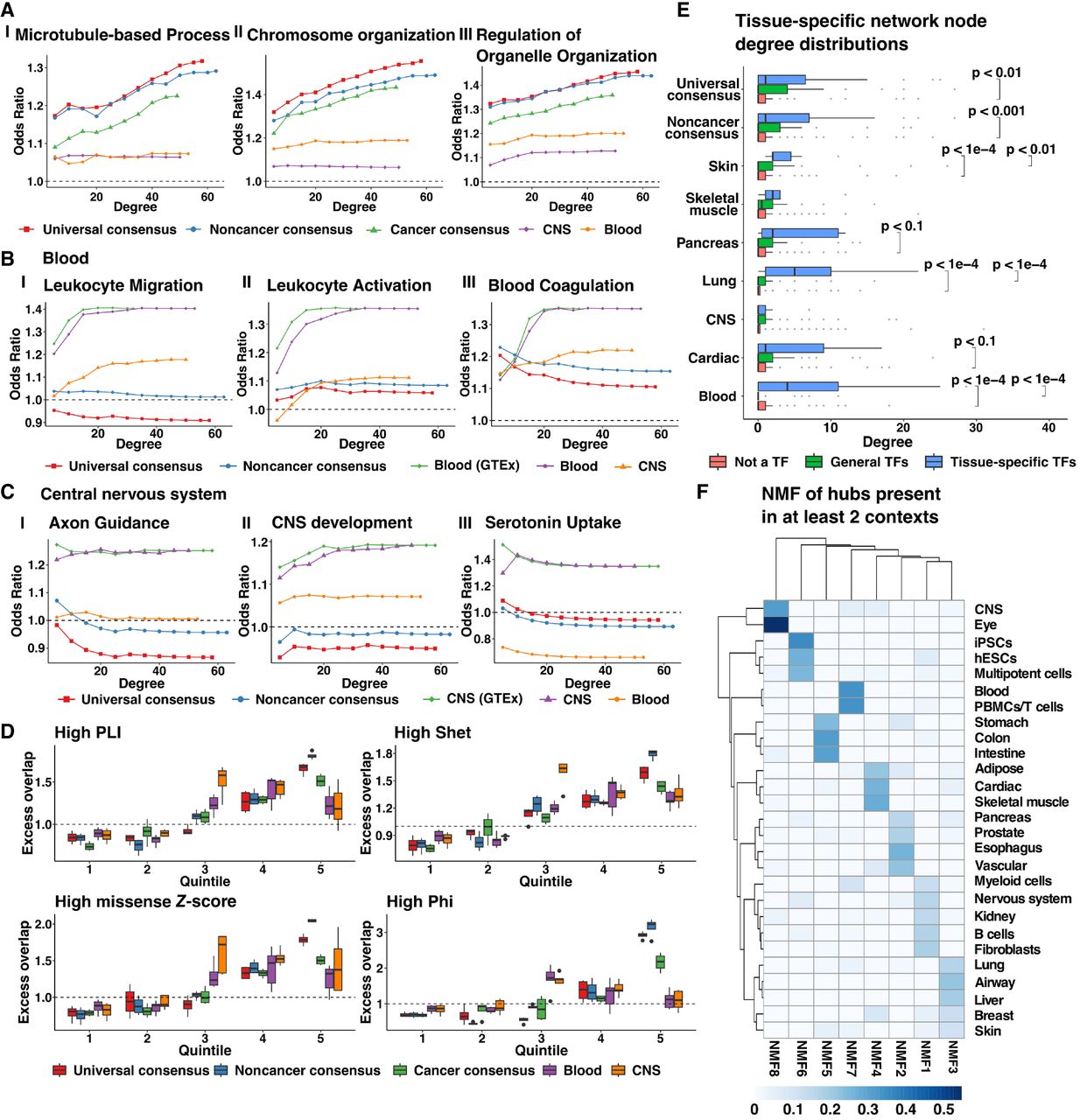
Next, we evaluated whether hub genes were evolutionarily constrained and whether they have known roles in complex traits or diseases (Methods). We first binned network nodes such that genes with a closeness centrality of 0 were assigned to Quintile 1, and those with at least 1 connecting edge were grouped based on estimated quantiles of closeness centrality. We then computed the excess overlap of a particular gene set among network nodes binned by closeness centrality, as the ratio of the fraction of genes from the set found among the network nodes in a particular bin to the fraction of genes from the gene set found among all network nodes (Methods). Consensus network hub genes (Quintile 5) had a significantly higher excess overlap (>1) with evolutionarily constrained gene sets than peripheral nodes (Quintiles 1–3), using metrics including high pLI genes, high shet genes, and high missense Z-score genes. These trends were similar in context-specific networks, with some variation in the strength of the trend across tissues likely driven by sample size differences and differential power in inferring these networks (Fig. 3D; Supplemental Fig. S22). We then examined the enrichment of eQTL-deficient, ClinVar, OMIM, and FDA drug-targeted genes across quintiles of network centrality (Supplemental Figs. S22 and S23; Supplemental Table S9). We observed that peripheral genes (Quintile 1) and central genes (Quintile 5) of consensus networks exhibited an excess overlap > 1 of eQTL-deficient genes (those which lacked significant cis-regulatory variants), whereas genes with intermediate connectivity were depleted of eQTL-deficient genes, whereas the six context-specific networks had widely variable trends. Although central genes from consensus networks were weakly depleted for OMIM and FDA drug-targeted genes, we observed an excess overlap > 1 of hub genes belonging to context-specific networks. This could be explained by our earlier observations that central nodes from consensus networks were involved in essential and nonspecific cellular processes, whereas central nodes from context-specific networks were both specific and critical to tissue identity; thus altering central consensus network hub nodes may have widespread and potentially deleterious off-target effects whereas context-specific hub nodes may identify targetable genes with tissue-specific effects. Finally, we observed that matched tissue-specific transcription factors from Pierson et al. (2015) (Supplemental Table S10) had a significantly greater number of neighbors than 88 general TFs for context-specific networks corresponding to blood (P < 1 × 10−4), lung (P < 1 × 10−4), and skin (P < 0.01), whereas in other tissues, except for CNS, the median degree of tissue-specific TFs was greater than general TFs and nontranscription factors (Methods). In contrast, whereas tissue-specific and general TFs had more neighbors in consensus networks when compared to nontranscription factors, we found no significant differences in the degree distribution of tissue-specific TFs to general TFs in either the universal or noncancer consensus network (Fig. 3E). Therefore, whereas both consensus and context-specific central genes were enriched for genes under high selection pressure, context-specific hub nodes were more likely to be OMIM genes, drug targets, and tissue-specific TFs.
We examined similarities and differences between network architectures of the noncancer consensus and cancer consensus networks based on shared and distinct hub genes, because central genes are likely to be more relevant to network functionality (Pastor-Satorras et al. 2003), and the identification of hubs has led to the discovery of genes involved in cancer (Chou et al. 2014; Oh et al. 2015), tissue regeneration (Rodius et al. 2016), and other diseases (Keller et al. 2008; Presson et al. 2008). Specifically, we found 296 shared hubs between cancer (λ = 0.24, 7552 edges) and noncancer (λ = 0.18, 7355 edges) consensus networks and 312 hubs which were specific to the cancer consensus network (Supplemental Table S11). Cancer-specific hub genes were enriched for pathways such as ncRNA metabolic processing (GO:0034660, P = 5.2 × 10−2) which is believed to play a role in metabolic reprogramming in cancer, DNA damage response (GO:0006974, P = 1.17 × 10−12) which has been posited to play a role in cancer cell survival in nonoptimal conditions, and response to ionizing radiation (GO:0010212, P = 6.1 × 10−4). Further, we found that cancer-specific hub genes were enriched for a plethora of DNA repair and replication pathways, including double-strand break repair via homologous recombination (GO:0000724, P = 6.36 × 10−6), recombinational repair (GO:0000725, P = 1.35 × 10−6), interstrand cross-link repair (GO:0036297, P = 5.48 × 10−3), and double-strand break repair via break-induced replication (GO:0000727, P = 1.36 × 10−3) (Supplemental File 1).
To examine whether network properties were shared between related tissues, we analyzed the overlap of hub genes by focusing on those identified as hubs in at least two tissue contexts (N = 1956). Details of the penalization parameter selected for tissue-context specific networks used in this analysis are summarized in Supplemental Table S12. Grouping tissue contexts based on hub genes using nonnegative matrix factorization with eight latent factors (Methods) led to the grouping of related tissues such as blood and PBMCs/T cells (Factor 7) (Fig. 3F; Supplemental Table S13). As expected, we found that hub genes that led to the grouping of blood and PBMCs/T cells were enriched for defense response (GO:0006952, P = 2.7–24) and cytokine production (GO:0001816, P = 9.9 × 10−7) (Supplemental File 2). Further, Factor 6, which led to the grouping of hESCs, iPSCs, and multipotent cells, comprised hub genes which were enriched for gastrulation (GO:0007369, P = 5.9 × 10−4) and circulatory system development (GO:0072359, P = 6.2 × 10−3) (Supplemental File 3).
Genes with high network centrality are proximal to variants enriched for complex trait heritability
Previous work by Kim et al. (2019) reported that network topology annotations did not contribute to heritability once the LDSC baseline model (Finucane et al. 2015) was included. We examined whether data aggregation would increase the utility of network features for heritability analysis independently of baseline functional annotations. For each network, we calculated centrality annotations by computing six different centrality measures, including, degree, maximum weight, strength, closeness, betweenness, and PageRank. The correlation between the different centrality measures is summarized for select networks in Supplemental Figure S24. We meta-analyzed estimates of heritability enrichment, the ratio of the proportion of heritability explained by SNPs belonging to an annotation to the proportion of SNPs in the annotation, and τ*, an estimate of the heritability of SNPs unique to the annotation (Finucane et al. 2015), using a random effects model to obtain a summary of effect sizes estimated for a set of 42 independent traits considered by Kim et al. (2019) (Methods; Supplemental Table S14). We estimated both heritability enrichment and τ* by either conditioning an annotation corresponding to whether a variant was located in a 100-kb window of all protein-coding genes (all-genes annotation), or conditioning on 97 functional annotations such as known enhancer and promoter regions which are included in the baseline-LD model and the all-genes annotation (all-genes + baseline). Similar to the results found by Kim et al. (2019), we observed a significant estimate τ* corresponding to our consensus network-derived annotations when conditioning on just the all-genes annotation. However, when conditioning on the baseline-LD model, the τ* observed for consensus network-derived annotations were no longer significant (Fig. 4A). Whereas we found no significant differences in enrichment across a varying number of edges present in the network when conditioned on the all-genes annotation, we observed that τ* decreased as the number of network edges decreased. There were no significant differences in either enrichment or τ* with the number of edges found in the network when conditioning on the baseline-LD annotations (Supplemental Fig. S25). Further, our observations were not dependent on the traits studied and remained consistent when we applied s-LDSC to 219 UKBB traits (Supplemental Table S15; Supplemental Fig. S26).
Heritability enrichment of network annotations. Mean and standard deviation of heritability enrichment and the coefficient τ*, an estimate of the heritability of SNPs unique to the annotation. All genes: whether a variant was located in a 100-kb window of all protein-coding genes (translucent), all genes + baseline: all-genes annotation in addition to 97 functional annotations such as known enhancer and promoter regions (opaque). (A) Meta-analysis of 42 independent traits for six centrality measures obtained from the universal consensus network and noncancer consensus networks corresponding to values of the penalization parameter λ between 0.14 and 0.20. (B) Meta-analysis of nine blood-related traits, including Crohn's disease, ulcerative colitis, rheumatoid arthritis, allergy eczema, eosinophil count, red blood cell count, white blood cell count, red blood cell width, and platelet count for network annotations from blood GTEx (λ = 0.24–0.32), Blood consensus (λ = 0.18–0.26), and universal consensus network (λ = 0.14–0.20). (C) Meta-analysis of nine CNS-related traits, including Alzheimer's disease, epilepsy, Parkinson's disease, bipolar disorder, smoking cessation, waist-hip-ratio adjusted BMI, schizophrenia, major depressive disorder, and number of alcoholic drinks per week, for network annotations corresponding to six centrality measures derived from CNS GTEx (λ = 0.26–0.32), CNS (λ = 0.20–0.28), and universal consensus network (λ = 0.14–0.20).
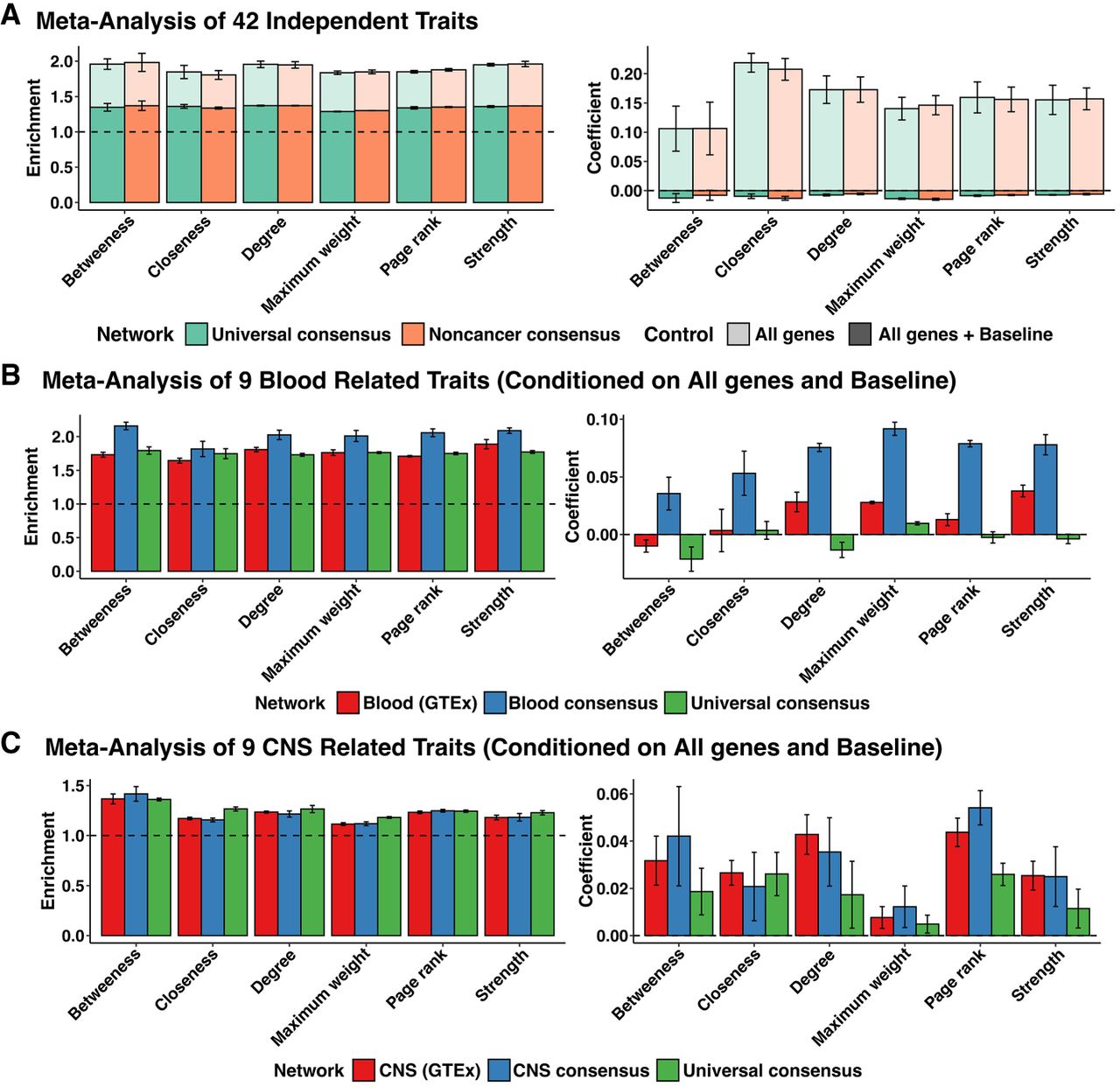
A possible explanation for the lack of heritability enrichment signal unique to network annotations is the redundancy between the baseline LD annotations and network topology annotations. Therefore, we hypothesized that context-specific data aggregation could prioritize variants enriched for heritability of concordant traits independent of baseline annotations. We applied s-LDSC to network centrality annotations derived from networks inferred only from GTEx blood samples (blood GTEx), networks inferred by aggregating recount3 blood samples (blood), and, as a control, networks inferred by aggregating all samples (universal consensus), for a subset of nine blood-related traits from the 42 independent traits (Crohn's disease, rheumatoid arthritis, ulcerative colitis, eosinophil count, platelet count, red blood cell count, red blood cell width, white blood cell count, and eczema) (Supplemental Table S16). As with the consensus networks, tissue-specific networks displayed similar trends in heritability estimates with the number of edges found in the network (Supplemental Figs. S27, S28). When we conditioned on the baseline-LD annotations, we observed that annotations derived from the blood consensus networks had a significant τ* across all centrality annotations, whereas blood GTEx networks had a significant τ* for strength, degree, maximum weight, and PageRank (Fig. 4B). In contrast, we did not observe a significant τ* corresponding to annotations derived from the universal consensus network. We examined the generalizability of our results by conducting a similar experiment in CNS samples, another tissue with a large sample size. We applied s-LDSC to annotations derived from CNS networks inferred from GTEx samples (CNS GTEx), CNS networks inferred by aggregating samples from recount3 (CNS consensus), and the universal consensus networks for CNS-related traits which included waist-hip ratio-adjusted BMI from the earlier set of 42 traits, as well as seven traits from the Psychiatric Genomics Consortium (Alzheimer's, epilepsy, Parkinson's, bipolar disorder, smoking cessation, schizophrenia, major depressive disorder, and number of alcoholic drinks per week) (Supplemental Table S17). We found that annotations derived from both universal consensus and CNS-specific networks led to significant nonzero τ* when conditioning on the baseline-LD model. Although we note that significant nonzero τ* was observed for the consensus networks for the chosen set of CNS traits in contrast to the 42 independent traits, possibly due to power, study quality, and other attributes of the GWAS, we found that annotations from tissue-specific networks led to significantly higher estimates of τ* and outperformed consensus networks (Fig. 4C) for all centrality measures except closeness. Further, for betweenness, maximum weight, and PageRank centrality, CNS consensus networks outperformed CNS GTEx networks, similar to the results in blood, demonstrating context-specific data aggregation results in network annotations that are enriched for trait heritability across tissue contexts. Across both sets of blood- and CNS-related traits, we found that PageRank centrality-derived annotations, which captured both the number of connections that a node has in addition to the centrality of its neighbors to determine the importance of a connection, performed consistently well. We conclude that context-specific aggregation results in the identification of the central genes of the network, which are enriched for the heritability of concordant traits, and an increased sample size leads to a greater heritability enrichment signal.
Discussion
GCNs aid in determining changes in regulatory mechanisms that are key to cellular identity and prioritizing genes that drive phenotypic variability. However, conventional network analyses are often too underpowered to reliably discover gene-gene relationships and are compromised by spurious false-positives and false-negatives that result from limited power, noise, and unobserved technical confounders. We leveraged publicly available RNA-seq data from recount3 and manually curated tissue/cell type annotations to improve the inference of consensus and context-specific GCNs. Utilizing data splits, we demonstrated that accounting for confounders within individual studies followed by weighted aggregation of empirical covariance matrices led to the best improvement in network characteristics with data aggregation across multiple paradigms.
We then inferred three consensus networks (universal, noncancer, and cancer networks) that recapitulated ubiquitous biological processes. Further, we aggregated data belonging to individual tissue contexts to infer 27 tissue context-specific networks that were enriched for matched tissue-specific PPIs and shared similarities across related tissues. All networks and sample annotations are made publicly available as a resource for future studies.
Central genes from both consensus and context-specific networks were enriched for high PLI and high Phi genes, indicating that hub genes are enriched for genes under high selective pressure. Context-specific hub genes were enriched for FDA-approved drug targets and OMIM genes whereas central genes from consensus networks which were inferred over a greater number of samples were depleted for both categories. Thus, context-specific information was lost by global aggregation, cannot be recovered by data aggregation or increased sample sizes, and is important to identifying drug targets and disease mechanisms. Although the central genes of the network as determined by global data aggregation in the consensus network did not explain trait heritability independent of known functional annotations in the baseline-LD model, we found that context-specific data aggregation prioritized variants enriched for concordant trait heritability that did not overlap with previously known functional annotations. Thus, topological properties of genes from context-specific GCNs hold significant promise as a functional annotation for identifying genetic variation that contributes to complex trait heritability.
A commonly used approach to identify genes associated with complex traits is to use colocalization analysis between GWAS and eQTL studies. However, often only about half of the signals colocalize with an eQTL (Mostafavi et al. 2023). Recent work by Mostafavi et al. (2023) demonstrated that genes driving GWAS signals were often genes with complex context-dependent regulatory architecture and were depleted for eQTL variants. This has raised a call in the computational genomics community for orthogonal approaches to identify genes involved in complex traits. We found that annotations derived from context-specific GCNs are informative of trait heritability independent of context-agnostic functional annotations. This suggests that tissue- and context-specific network centrality and other network properties could be used to help prioritize genes near GWAS loci (Zhu et al. 2021) or supplement eQTL data.
One of the major challenges in network inference remains the presence of unobserved technical confounders and undesirable biological signals, which lead to spurious network edges and precludes causality claims. Although PC-based data correction has been extensively utilized to reduce false-positives resulting from confounding, recent work by Cote et al. (2022) suggests that PC-based data correction, and related methods such as PEER (Stegle et al. 2012) and CONFETI (Ju et al. 2017), may overcorrect expression data and remove biological coexpression of potential interest. Correcting or modeling confounders is essential to network accuracy; therefore, tuning parameters such as the number of latent factors to correct as well as exploring alternative methods will continue to be important. Alternate approaches to handle confounding and infer causal regulatory relationships include instrumental variable analysis through the construction of local genetic instruments as outlined by Luijk et al. (2018). However, because central network nodes are evolutionarily constrained and tightly regulated, it can be challenging to construct well-tracking genetic instruments for central genes. Publicly available RNA-seq data including recount3, the extensive annotations we provide, and recent work which illustrated genotype calling using RNA-seq data (Deelen et al. 2015) could improve our ability to detect context-specific cis-regulatory effects, the reconstruction of local genetic instruments, and hence causal regulatory network inference.
Future directions aimed at improving GCN inference could leverage our extensively annotated sample characteristics and data aggregation strategies with complementary strategies, including sharing information between related contexts (Omranian et al. 2016) to increase the effective sample size, introducing constraints or priors corresponding to known regulatory relationships (Hellstern et al. 2021), and using alternate statistical measures of expression similarity that capture nonlinear associations between genes (Margolin et al. 2006). Additionally, heuristic algorithms such as the one proposed by Opgen-Rhein and Strimmer (2007) could be utilized to enrich our current networks with directionality information. Finally, while we studied tissue contexts, we provide annotations of disease status which can be utilized to infer disease-specific GCNs.
Our finding that marginal improvement in network reconstruction decreases with continued data aggregation suggests that simply addressing statistical considerations due to sample size may have limitations for improving GCNs. Including orthogonal sources of information such as gene-enhancer associations inferred from Hi-C data (Babaei et al. 2015), transcription factor binding sites from ChIP-seq data (Zhou et al. 2017), and regulatory information derived from other epigenetic data types, in addition to gene expression quantified by RNA-seq in both bulk and single-cell studies, might result in a more accurate understanding of the shared regulatory architecture between genes. An example of integrating orthogonal information sources is found in recent work by Fu et al. (2025), which leverages paired scATAC-seq and snRNA-seq data across 213 fetal and adult cell types along with prior knowledge of TF motifs, to computationally infer local chromatin environments. The resulting model can then be used to predict gene expression in new cell types and TF regulators of downstream genes. This method and related approaches based on epigenetic data and TF motif information to infer targets of well-characterized TFs can be viewed as complementary to our approach, which is able to capture regulatory and coexpression relationships genome-wide based on large-scale RNA-seq data. Future methods may unify these approaches and incorporate both sources of information. Additionally, experimental protocols such as Perturb-seq (Dixit et al. 2016), which quantifies the transcriptional changes mediated by genetic manipulations on genes, processes, and states, could provide a new avenue for network inference and suggest causal mechanisms and edge directionality (Ota et al. 2025).
In conclusion, the growing availability of publicly shared RNA-seq data presents a valuable opportunity to improve gene GCN inference through data aggregation. Although GCNs offer insights distinct from eQTL analyses and hold promise for uncovering the regulatory mechanisms underlying complex traits, their utility has been limited by challenges in data integration and variability across studies. In this work, we addressed these challenges by developing an approach that accounts for latent confounding and study-specific variability, enabling the construction of context-specific GCNs enriched for complex-trait heritability beyond baseline functional annotations.
Methods
Data preprocessing and quality control
We downloaded uniformly processed human RNA-seq samples using the recount3 R package (version 1.0.7) (Wilks et al. 2021), running on R version 4.0.2 (R Core Team 2020) under Rocky Linux 8.8, and selected 1747 projects that included 30 or more samples each (Fig. 1A). Before normalization, we excluded samples with zero expression across all genes and genes that had zero expression across all samples in a project. We used built-in functions from the recount3 package to compute the RPKM transformed count matrix, selected genes that were protein-coding, autosomal, and unambiguously mapped to the reference genome (Saha and Battle 2018, 2019), and generated the log2(RPKM + 1) count matrix for each project.
Following preliminary processing, we applied a unique data processing pipeline based on the data source. For projects belonging to GTEx, we excluded duplicates (labeled as STUDY_NA) and samples derived from the chronic myelogenous leukemia (CML) cell line. We grouped samples by the tissue of origin to obtain 50 groups and 18,828 samples.
For TCGA, we excluded 67 samples missing sample type and 39 samples lacking patient ID. Replicates were identified as samples sharing both patient ID and sample type, and their gene expression was aggregated using the median. We grouped TCGA samples by cancer code, resulting in 33 groups and 11,091 samples.
For SRA, we first removed samples obtained via size fractionation. Replicates were defined by identical experiment accession numbers and aggregated using the median expression. Further, we excluded 89,101 samples where >50% of genes showed zero expression, which we used as a threshold to eliminate likely microRNA or degraded samples. To further restrict the data set to bulk RNA-seq, we used predicted experiment-type labels from recount3 and excluded samples predicted as single-cell or small RNA-seq. If predicted labels were unavailable, we excluded samples with keywords such as “single cell,” “scRNA,” “snRNA,” or “single nucleus” in the study abstract. Additionally, we retained only those samples whose library selection metadata contained either cDNA or RT-PCR, when available. We also excluded studies which are known scRNA-seq experiments including: SRP096986, SRP135684, SRP166966, SRP200058, and SRP063998. For the remaining studies, we performed a text-based analysis to obtain the study, tissue, organ, biopsy, cell, disease, source and description from the metadata sample_description field. We then manually annotated 10,179 unique combinations of these fields to obtain tissue, cancer status, and disease type. In this manner, we were able to obtain labels for 65,361 samples which we grouped on the basis of the study accession IDs to form 884 SRA studies (Supplemental File 4).
We grouped GTEx samples by tissue of origin, SRA samples by study accession ID, and TCGA samples by cancer accession code and have referred to each individual group of samples as a “study” to simplify nomenclature. We did not distinguish on the basis of disease state or cancer status while organizing the data, until we proceeded to compute the inputs to network inference. The number of genes retained after filtering varied by study, particularly in smaller SRA projects with limited sample size. These gene count distributions are summarized in Supplemental Figure S1.
Identifying tissue type and cancer status
Wilks et al. (2021) demonstrated that, for human bulk RNA-seq data, tissue or cell type of origin is the dominant source of gene expression variation, with clear tissue-specific clustering visible in the top four principal components. Building on this, we manually refined annotated labels (Supplemental File 4) and grouped 95,484 samples and 5999 genes (with nonzero variance) using t-SNE dimensionality reduction and clustering. Because cancerous and noncancerous samples did not separate distinctly in the t-SNE space, we used manually annotated labels to restrict the analysis to 63,193 noncancerous samples.
To maximize sample size, we merged similar tissues into broader tissue contexts (Supplemental Table S1), defined as a group of samples with similar cell-type composition. Recognizing that gene expression can also vary due to technical factors such as batch effects and library size, we estimated the relative enrichment of 64 stromal and immune cell types using xCell gene signatures (Aran et al. 2017). Enrichment vectors for each sample were then used for visualization and clustering via t-SNE, allowing us to group samples by shared cellular composition into distinct tissue contexts.
For 24 contexts with more than 500 samples, we removed outliers using six different outlier detection methods (Supplemental Methods: “Outlier calling methods to refine sample selection”; Supplemental Fig. S2). We examined whether selecting outliers based on any metric, including outliers labeled by one, two, or three metrics significantly impacted our estimate of the empirical covariance matrix from the data. We compared the Frobenius norm of the differences in empirical covariance estimated for a particular tissue context with varying stringency on the inclusion criterion to the difference in the empirical covariance matrix estimated across contexts. We observed that the differences in the covariance estimates for varying the exclusion criterion were, in general, much smaller than the differences between two unrelated contexts, and hence we included samples within a tissue-specific context if it was labeled an outlier by two or fewer metrics to maximize the number of samples available for downstream analysis (Supplemental Fig. S4).
Although this works with simpler tissue categories such as blood, skeletal muscle, or colon, where the samples have relatively homogenous cell-type compositions, for the immune system, which comprises samples from the distinct myeloid, lymphoid, and innate immune systems, these outlier metrics failed due to large intersample variation. Therefore, for immune cell types, we first performed k-means clustering with three centroids using the in-built kmeans() function. We annotated the three resulting clusters based on the representation of manually annotated labels in each cluster as either B cells, myeloid cells (including monocytes and macrophages), or PBMCs w/T cells (Supplemental Fig. S3). For each cluster, we performed outlier detection using all six methods outlined above and excluded samples that were found to be outliers in any two methods.
Data correction and aggregation
Principal component (PC)-based correction methods can account for technical and biological artifacts that confound gene expression measurements and reduce false-positives in gene network inference (Parsana et al. 2019). However, these methods have been applied to one experiment and not across multiple experiments from disparate sources. We systematically compared four strategies of data aggregation. In the first approach (aggregating data), expression data were pooled across all studies of interest, and confounders were estimated jointly from the combined data set. This strategy reflects a global correction model in which confounding structure is assumed to be shared across data sets.
In the second approach (aggregating data adjusted for confounding), confounder correction was performed independently within each study before combining the corrected data. This addresses the possibility that latent structure differs across studies. The third and fourth approaches involved aggregating covariance matrices rather than gene expression matrices. In the unweighted aggregation of covariance matrices strategy, we treated each study equally and computed the average of study-level covariance matrices after within-study correction. In contrast, the weighted aggregation of covariance matrices strategy assumed that larger studies yield more reliable estimates, and we weighted each study's contribution by its sample size. Both approaches avoid direct normalization across heterogeneous data sets and instead pool structure at the level of gene–gene covariance. Complete implementation details for all four strategies are provided in the Supplemental Methods: “Detailed procedures for data correction and aggregation strategies.”
Network reconstruction with graphical lasso
Following the computation of aggregate covariance matrices using the strategies outlined in Supplemental Methods: “Detailed procedures for data correction and aggregation strategies,” we inferred gene regulatory relationships using graphical lasso (Friedman et al. 2008). The desired network structure is obtained by identifying the precision matrix, Θ = Σ−1 that maximizes the penalized log-likelihood given by Equation 1, where C is the estimated covariance matrix
Network evaluation to determine the optimal aggregation strategy
To evaluate the impact of different data correction and aggregation strategies on network quality, we applied all four approaches (see Supplemental Methods: “Detailed procedures for data correction and aggregation strategies”) to noncancerous samples from two partitions: GTEx and SRA. GTEx samples were organized by tissue type, excluding tissues with fewer than 15 samples (e.g., kidney medulla), resulting in 49 tissue-level groups. For the SRA noncancer partition, we selected 566 studies with noncancer annotations and at least 15 samples per study.
To assess how increasing sample size affects network performance, we constructed aggregate networks by sequentially adding studies or tissues in order of increasing sample size. For SRA, we evaluated networks at one, 100, 200, 300, 400, 500, and all 566 studies. For GTEx, we tested one, 10, 20, 30, 40, and 49 tissues. Each network was inferred using graphical lasso over a grid of penalization parameters between 0.04 and 1.00. Supplemental Table S2 summarizes the number of samples, studies, median sample size, and principal components regressed at each aggregation level.
To compare network quality across aggregation strategies and sample sizes, we used two evaluation criteria: (1) held-out log-likelihood on independent data sets to measure generalizability; and (2) biological validity based on (a) enrichment for known pathway comembership, and (b) transcription factor–target interactions. Precision, recall, and F1-score were computed for each network using standardized reference sets. Full computational details for all evaluation metrics are provided in the Supplemental Methods: “Detailed evaluation metrics to determine the impact of data aggregation.”
Network inference with WGCNA
To evaluate the effect of data aggregation on module-based gene coexpression networks, we used the WGCNA R package (version 1.72-5) (Langfelder and Horvath 2008). We constructed consensus networks by aggregating samples incrementally across three settings: (1) GTEx tissues in batches of one, three, 10, 30, and all 49 tissues; (2) noncancerous SRA studies in batches of one, 10, 20, 30, 100, 300, and all 566 studies; and (3) SRA studies annotated as blood in batches of one, 20, 40, 60, and 65. For each aggregation level, we applied principal component correction to the individual studies or tissues before combining them. We retained only genes that were present in all data sets being aggregated. After performing standard WGCNA preprocessing, we estimated the adjacency matrix for each individual data set using a power parameter selected to ensure approximate scale-free topology. Consensus networks were then constructed by aggregating topological overlap matrices (TOMs) across studies. Modules were identified by hierarchical clustering followed by dynamic tree cutting from the WGCNA package, and similar modules were merged based on eigengene correlation. For each inferred network, we evaluated functional relevance by computing precision, recall, and F1-score based on known gene-gene interactions obtained by curating pathway information from KEGG, Biocarta, and Pathway Interaction Database from Enrichr and selected those pathways that were annotated as canonical pathways by MSigDB. Full computational procedures and parameter thresholds are provided in the Supplemental Methods: “Detailed procedures for WGCNA-based aggregation and evaluation.”
Inference of consensus and tissue context-specific networks
We inferred consensus networks across samples from disparate tissues and cell types to capture shared biological pathways across contexts. Because weighted covariance aggregation yielded the best performance among the four data correction and aggregation strategies, we grouped SRA samples by study accession ID, GTEx samples by tissue of origin, and TCGA samples by cancer code. We inferred the network structure over a range of penalization parameters from 0.08 to 1.00. We constructed three types of consensus networks (universal, noncancer, and cancer), each defined by different sample inclusion criteria (see Supplemental Methods: “Construction of consensus networks” for details).
We inferred context-specific networks in 27 contexts with 500 or more samples. Details of the number of samples, studies, and median sample size across studies for each context are provided in Supplemental Figure S16. For 20 contexts including adipose, B cells, blood, breast, cardiac, central nervous system, colon, esophagus, fibroblasts, intestine, kidney, liver, lung, nervous system, pancreas, prostate, skeletal muscle, skin, stomach, and vascular, we inferred networks either using GTEx sample only or by aggregating context-specific samples from recount3 which included GTEx. For each context-specific network, we first performed PC-based data correction within each study followed by covariance estimation and aggregation by weighting the covariance matrix with the proportion of study-specific sample size to the total number of samples, as detailed in Supplemental Methods: “Detailed procedures for data correction and aggregation strategies.” We then inferred GCNs using graphical lasso using Equations 1 and 2.
Evaluating the impact of data aggregation on the inference of consensus and context-specific networks
To evaluate whether aggregating samples across studies improves the performance of consensus and context-specific networks, we computed the precision, recall, and F1 score of observing known gene coregulatory relationships annotated in canonical biological pathways compiled across KEGG, Biocarta, and the Pathway Interaction Database, as detailed in Supplemental Methods: “Detailed evaluation metrics to determine the impact of data aggregation.” For consensus networks, we compared F1 scores across the universal, noncancer, and cancer networks over a range of network sizes from 5000 to 500,000 edges.
To test context-specific network performance we focused on the two largest contexts: blood and CNS. For each of these contexts, we sequentially aggregated SRA studies by increasing sample size and inferred networks. We then evaluated the generalizability of the networks inferred across varying numbers of samples by computing the held-out log-likelihood using the corresponding GTEx tissues as held-out test sets (Supplemental Methods: “Computation of held-out log-likelihood for content-specific networks”).
Additionally, for six contexts (adipose, blood, CNS, liver, lung, and skin), we compared contex-specific networks constructed solely from GTEx to those derived through data aggregation by assessing the enrichment of tissue-specific protein-protein interactions from the SNAP database. Odds ratios for enrichment were computed using Fisher's exact test. Additional statistical procedures, including two types of permutation testing and a Wilcoxon rank-sum test for comparing aggregated versus GTEx only networks, are described in the Supplemental Methods: “Determining enrichment of known tissue-specific PPI interactions.”
Sparsity parameter selection for consensus and context-specific networks
For each of the three consensus networks and 27 context-specific networks, we evaluated the degree distribution of inferred networks across a range of penalization parameters. Specifically, we identified scale-free networks as those where the degree distribution followed a power law with an estimated scaling exponent between 2 and 3 and a coefficient of determination (R2) >0.8. Additional methodological details, including model fitting and confidence interval calculations, are provided in the Supplemental Methods: “Detailed description of sparsity parameter selection.” Summary statistics and selected network parameters are reported in Supplemental Tables S6 and S7.
Computing gene centrality measures based on network structure
We computed measures of network connectivity for each gene in the network with the igraph R package (version 1.3.5) (Csardi and Nepusz 2006). We used the absolute value of entries in the precision matrix (Methods: “Network reconstruction with graphical lasso”) to define edge weights between genes and normalized these values by the maximum edge weight across the entire graph. Using these weights, we calculated gene-level centrality using several standard metrics: betweenness, closeness, degree, strength, maximum edge weight, and PageRank. For each gene, we used distance-based weights to compute betweenness and closeness centrality, whereas normalized edge weights were used for the remaining centrality measures. Details of how each centrality metric was computed, including mathematical formulas and algorithms used, are provided in the Supplemental Methods: “Computation of centrality measures.” Correlations between centrality measures across select networks are shown in Supplemental Figure S24.
Enrichment of specific pathways among central genes in consensus and context-specific networks
To assess whether highly connected genes in inferred networks were enriched for known biological functions, we developed a general framework to test for the enrichment of functional gene sets among central genes. For each network, we estimated node degree (see Methods: “Computing gene centrality measures based on network structure”) and assessed functional enrichment at a series of increasing degree thresholds (Supplemental Methods: “Functional enrichment analysis of central genes”). For the consensus networks, we first selected the value of the penalization parameter such that the resulting network had ∼7000 edges. We then determined the odds ratio of finding genes belonging to ubiquitous biological processes among network nodes selected by successively increasing degree thresholds and compared the results to those obtained from blood- and CNS-specific networks with a similar number of edges. Full details of GO terms corresponding to ubiquitous biological processes are provided in Supplemental Table S8.
We extended this analysis to context-specific networks by selecting GO terms relevant to each context and testing for enrichment among high-degree nodes. For each of six contexts (blood, CNS, skin, lung, liver, and adipose), we selected one network inferred solely from GTEx samples and one inferred from aggregated samples (including GTEx) with ∼7000 edges. For example, in the blood context, we tested enrichment for GO terms such as leukocyte migration, leukocyte activation, and blood coagulation among high-degree nodes. As negative controls, we compared the observed enrichment in the blood-specific networks to that found in an unrelated context-specific network inferred across aggregated samples, as well as the universal and noncancer consensus networks. A complete list of GO terms tested for each context is provided in Supplemental Table S8.
In a separate analysis, we compared inferred context-specific networks to gold standard tissue-specific networks from HumanBase (Greene et al. 2015). For each inferred network, we assigned nodes to degree-based bins and tested whether high-degree genes in our networks were enriched for hub genes (defined as those above the 80th percentile in degree) in the corresponding HumanBase network. Enrichment was tested across bins to assess the degree to which centrality in inferred networks aligns with regulatory importance in an external reference. Detailed procedures for these enrichment analyses are described in Supplemental Methods: “Enrichment of HumanBase hub nodes among central network nodes.”
Excess overlap of genes grouped by centrality measures with known evolutionarily constrained and functionally prioritized gene sets
We grouped genes for each consensus network such that the first bin includes genes with a closeness centrality of 0, the second bin contains nodes with closeness centrality in the lowest 25th percentile, the third bin includes genes with closeness centrality in the 25th–50th percentile, the fourth bin includes genes with closeness centrality in the 50th–75th percentile, and the fifth and final bin includes genes with closeness centrality greater than the 75th percentile.
For each bin, we computed the excess overlap with evolutionarily constrained and functionally prioritized gene sets as defined in Kim et al. (2019). We quantified enrichment by comparing genes in bin i, with each reference gene set j, , relative to the set of all genes in the network Gtot, using the following equations:
Details on the reference gene sets are provided in the Supplemental Methods: “Reference gene sets used for overlap analysis” and in Supplemental Table S9.
Identifying biological processes associated with shared and distinct hub genes from noncancer and cancer consensus networks
In accordance with the scale-free criterion, we selected the noncancer network inferred using a penalization parameter λ = 0.18, resulting in a network with 7355 edges, and the cancer consensus network corresponding to the penalization parameter λ = 0.24 and a network with 7552 edges to compare the biological processes which are represented by shared and distinct network hubs. First, we defined hub genes as network nodes with a closeness centrality in the 90th percentile independently for each consensus network, such that the noncancer hub genes are given by HN and the cancer hub genes are given by HC. We then identified hub genes shared between cancer and noncancer consensus networks , noncancer-specific hub genes , and cancer-specific hub genes as (Supplemental Table S11). We then used the GOTermFinder tool (Boyle et al. 2004) to identify GO terms that are shared by the genes in the sets HS, HNS, and HCS.
Examining the differences in the degree distribution of tissue-specific versus general transcription factors in consensus and context-specific networks
We selected context-specific networks inferred from CNS, blood, cardiac, skin, lung, skeletal muscle, and pancreas samples from recount3 which each had approximately 7000 edges (Supplemental Table S12). Additionally, we selected values of the penalization parameter λ which yielded universal and noncancer consensus networks with ∼ 7000 edges. We referred to Pierson et al. (2015) to obtain a list of tissue-specific and general transcription factors which are provided in Supplemental Table S10. To select a background set of non-TFs, we first obtained the intersection of genes which were included in each network considered. Then, we excluded both general and tissue-specific TFs from this list and randomly selected 100 of these genes. The selected background is provided in Supplemental Table S10. For each network, we compared the degree distribution of tissue-specific and general transcription factors to non-TFs and the degree distribution of tissue-specific to general transcription factors using the Wilcoxon rank-sum test. Across all tests, we adjusted for multiple hypothesis correction using the Holm-Bonferroni method.
Nonnegative matrix factorization to determine shared coregulatory relationships in similar tissues
We selected context-specific networks with ∼7000 edges across 27 contexts for which we inferred GCNs by aggregating samples assigned to the context from recount3 (Supplemental Table S12). Further, the selected networks were in accordance with the scale-free selection criterion detailed in Methods: “Sparsity parameter selection for consensus and context-specific networks.” For each network, we identified hub genes as network nodes with a degree centrality in the 95th percentile. Across the 27 contexts, we found 3682 unique hubs. We subsetted to hubs that are present in at least two contexts, which resulted in 1956 hubs. We then used the R package RcppML (version 0.3.7; https://cran.r-project.org/web/packages/RcppML/index.html) to perform nonnegative matrix factorization to learn eight underlying factors to group similar patterns of hub genes. Specifically, H is a binary matrix of dimensions NHubs × Nc, where Nc is 27 (i.e., the number of contexts). Hi, j = 1 when the ith gene is a hub gene in context j and Hi, j = 0 otherwise. We then obtain matrices W of dimensions NHubs × 8, and C of dimensions Nc × 8 by solving the following optimization function
Finally, we chose a solution after 10 iterations that resulted in the greatest sparsity, that is, smallest values of and . To interpret the resulting context cluster, we examined the genes that contributed the most to the corresponding factor. Specifically, for a given factor, W[:, p], we first obtained genes with loadings in the 80% percentile. Then, for each gene we computed the maximum difference between the loading of the gene on factor p to all other factors and selected genes where this difference is ≥ 5 × 10−5. Thus, we obtained a set of factor-specific hub genes Gp (Supplemental Table S13). We then used the GOTermFinder tool (Boyle et al. 2004) to identify GO terms that were more likely to be present in the set Gp than a background of all protein-coding genes by estimating the odds ratio and P-value by applying the hypergeometric test.
Stratified LD-score regression to quantify the heritability enrichment of variants proximal to central network genes in consensus and context-specific networks
We applied stratified LD-score regression (Finucane et al. 2015) to assess whether SNPs near central genes in inferred networks contributed disproportionately to complex trait heritability. For the following networks—universal consensus, noncancer consensus, blood-specific (inferred from only GTEx data and across aggregated data from recount3), and CNS-specific (inferred from only GTEx data and across aggregated data from recount3)—we obtained annotations corresponding to six different centrality measures, that is, degree, betweenness, closeness, maximum edge weight, strength, and PageRank (Methods: “Computing gene centrality measures based on network structure”) by transforming the centrality scores to lie between 0 and 1. Each SNP was annotated based on the centrality of genes within 100 kb, taking the maximum value when multiple genes were nearby. These annotations were tested for heritability enrichment and standardized effect () using s-LDSC under two models: (1) one including only an all-genes annotation; and (2) another including the all-genes annotation and 97 annotations from the baseline-LD v2.2 model (Villar et al. 2015; Gazal et al. 2017; Marnetto et al. 2018; Hujoel et al. 2019). Trait sets (Supplemental Methods: “Description of trait sets used in sLDSC”) included 42 curated traits (Kim et al. 2019; Supplemental Table S14), 219 UK Biobank traits (Supplemental Table S15), nine blood-related traits (Supplemental Table S16), and nine CNS-related traits (Supplemental Table S17). All networks used in the analysis satisfied the scale-free degree criterion (see Methods: “Sparsity parameter selection for consensus and context-specific networks”). Final enrichment and estimates were summarized using random-effects meta-analysis by using the function meta.summaries() from the R package rmeta (version 3.0; https://cran.r-project.org/web/packages/rmeta/rmeta.pdf). Full procedural details and model specifications are provided in Supplemental Methods: “Stratified LD score regression procedures.”
Data access
Scripts to reproduce the analysis and figures included in this paper are available in the Supplemental Code and can also be accessed at GitHub (https://github.com/prashanthi-ravichandran/recount3_networks). Networks and annotations used in this project can be obtained from Zenodo (https://zenodo.org/records/10480999).
Competing interest statement
A.B. is a consultant for Third Rock Ventures, LLC, a shareholder in Alphabet, Inc., and a founder of CellCipher, Inc.
Acknowledgments
We thank Battle lab members for helpful discussions throughout the course of this work, particularly Joshua Weinstock and Eric Kernfeld for code review and manuscript feedback. A.B. was supported by the National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS) R35GM139580. K.D.H. and A.B. were supported by NIH/NIGMS R01GM121459.
Author contributions: P.R., P.P., and A.B. conceived the project. P.R., P.P., and A.B. designed the analyses. P.R. and P.P. performed the analyses. R.K. and K.D.H. contributed feedback to experimental design and interpretation. P.R., R.K., and A.B. organized and wrote the paper with input from all authors.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280808.125.
[3] Freely available online through the Genome Research Open Access option.
References
- ↵Albert R. 2005. Scale-free networks in cell biology. J Cell Sci 118: 4947–4957. 10.1242/jcs.02714
- ↵Alon U. 2003. Biological networks: the tinkerer as an engineer. Science 301: 1866–1867. 10.1126/science.1089072
- ↵Aran D, Hu Z, Butte AJ. 2017. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 18: 220. 10.1186/s13059-017-1349-1
- ↵Babaei S, Mahfouz A, Hulsman M, Lelieveldt BPF, de Ridder J, Reinders M. 2015. Hi-C chromatin interaction networks predict co-expression in the mouse cortex. PLoS Comput Biol 11: e1004221. 10.1371/journal.pcbi.1004221
- ↵Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. 2004. GO::TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20: 3710–3715. 10.1093/bioinformatics/bth456
- ↵Buja A, Eyuboglu N. 1992. Remarks on parallel analysis. Multivariate Behav Res 27: 509–540. 10.1207/s15327906mbr2704_2
- ↵Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A. 2013. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128. 10.1186/1471-2105-14-128
- ↵Chou W-C, Cheng A-L, Brotto M, Chuang C-Y. 2014. Visual gene-network analysis reveals the cancer gene co-expression in human endometrial cancer. BMC Genomics 15: 300. 10.1186/1471-2164-15-300
- ↵Cote AC, Young HE, Huckins LM. 2022. Comparison of confound adjustment methods in the construction of gene co-expression networks. Genome Biol 23: 44. 10.1186/s13059-022-02606-0
- ↵Csardi G, Nepusz T. 2006. The igraph software package for complex network research. InterJournal, Complex Systems 1695. https://igraph.org.
- ↵Deelen P, Zhernakova DV, de Haan M, van der Sijde M, Bonder MJ, Karjalainen J, van der Velde KJ, Abbott KM, Fu J, Wijmenga C, 2015. Calling genotypes from public RNA-sequencing data enables identification of genetic variants that affect gene-expression levels. Genome Med 7: 30. 10.1186/s13073-015-0152-4
- ↵Diaz LPM, Stumpf MPH. 2022. Gaining confidence in inferred networks. Sci Rep 12: 2394. 10.1038/s41598-022-05402-9
- ↵Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, 2016. Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167: 1853–1866.e17. 10.1016/j.cell.2016.11.038
- ↵Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P-R, Anttila V, Xu H, Zang C, Farh K, 2015. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 47: 1228–1235. 10.1038/ng.3404
- ↵Friedman J, Hastie T, Tibshirani R. 2008. Sparse inverse covariance estimation with the graphical lasso. Biostatistics 9: 432–441. 10.1093/biostatistics/kxm045
- ↵Fu X, Mo S, Buendia A, Laurent AP, Shao A, del Mar Alvarez-Torres M, Yu T, Tan J, Su J, Sagatelian R, 2025. A foundation model of transcription across human cell types. Nature 637: 965–973. 10.1038/s41586-024-08391-z
- ↵Gazal S, Finucane HK, Furlotte NA, Loh P-R, Palamara PF, Liu X, Schoech A, Bulik-Sullivan B, Neale BM, Gusev A, 2017. Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat Genet 49: 1421–1427. 10.1038/ng.3954
- ↵Greene CS, Krishnan A, Wong AK, Ricciotti E, Zelaya RA, Himmelstein DS, Zhang R, Hartmann BM, Zaslavsky E, Sealfon SC, 2015. Understanding multicellular function and disease with human tissue-specific networks. Nat Genet 47: 569–576. 10.1038/ng.3259
- ↵The GTEx Consortium. 2020. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369: 1318–1330. 10.1126/science.aaz1776
- ↵Ha MJ, Baladandayuthapani V, Do K-A. 2015. DINGO: differential network analysis in genomics. Bioinformatics 31: 3413–3420. 10.1093/bioinformatics/btv406
- ↵Hartwell LH, Hopfield JJ, Leibler S, Murray AW. 1999. From molecular to modular cell biology. Nature 402: C47–C52. 10.1038/35011540
- ↵Hastie T, Tibshirani R, Friedman J. 2013. The elements of statistical learning: data mining, inference, and prediction. Springer Science & Business Media, Berlin.
- ↵Hasty J, McMillen D, Collins JJ. 2002. Engineered gene circuits. Nature 420: 224–230. 10.1038/nature01257
- ↵Hellstern M, Ma J, Yue K, Shojaie A. 2021. netgsa: fast computation and interactive visualization for topology-based pathway enrichment analysis. PLoS Comput Biol 17: e1008979. 10.1371/journal.pcbi.1008979
- ↵Huang Y-J, Lu T-P, Hsiao CK. 2020. Application of graphical lasso in estimating network structure in gene set. Ann Transl Med 8: 1556. 10.21037/atm-20-6490
- ↵Hujoel MLA, Gazal S, Hormozdiari F, van de Geijn B, Price AL. 2019. Disease heritability enrichment of regulatory elements is concentrated in elements with ancient sequence age and conserved function across species. Am J Hum Genet 104: 611–624. 10.1016/j.ajhg.2019.02.008
- ↵Ju JH, Shenoy SA, Crystal RG, Mezey JG. 2017. An independent component analysis confounding factor correction framework for identifying broad impact expression quantitative trait loci. PLoS Comput Biol 13: e1005537. 10.1371/journal.pcbi.1005537
- ↵Kanehisa M, Goto S. 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28: 27–30. 10.1093/nar/28.1.27
- ↵Katz K, Shutov O, Lapoint R, Kimelman M, Brister JR, O'Sullivan C. 2022. The Sequence Read Archive: a decade more of explosive growth. Nucleic Acids Res 50: D387–D390. 10.1093/nar/gkab1053
- ↵Keller MP, Choi Y, Wang P, Davis DB, Rabaglia ME, Oler AT, Stapleton DS, Argmann C, Schueler KL, Edwards S, 2008. A gene expression network model of Type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res 18: 706–716. 10.1101/gr.074914.107
- ↵Kernfeld E, Keener R, Battle A, Cahan P. 2024. Transcriptome data are insufficient to control false discoveries in regulatory network inference. Cell Syst 15: 709–724.e13. 10.1016/j.cels.2024.07.006
- ↵Kim SS, Dai C, Hormozdiari F, van de Geijn B, Gazal S, Park Y, O'Connor L, Amariuta T, Loh P-R, Finucane H, 2019. Genes with high network connectivity are enriched for disease heritability. Am J Hum Genet 105: 1302. 10.1016/j.ajhg/2019.03.020
- ↵Kodama Y, Shumway M, Leinonen R, International Nucleotide Sequence Database Collaboration. 2012. The Sequence Read Archive: explosive growth of sequencing data. Nucleic Acids Res 40: D54–D56. 10.1093/nar/gkr854
- ↵Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97. 10.1093/nar/gkw377
- ↵Langfelder P, Horvath S. 2008. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9: 559. 10.1186/1471-2105-9-559
- ↵Leek JT, Evan Johnson W, Parker HS, Jaffe AE, Storey JD. 2012. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28: 882–883. 10.1093/bioinformatics/bts034
- ↵Leskovec J, Sosič R. 2016. SNAP: a general purpose network analysis and graph mining library. ACM Trans Intell Syst Technol 8: 1–20. 10.1145/2898361
- ↵Liska O, Bohár B, Hidas A, Korcsmáros T, Papp B, Fazekas D, Ari E. 2022. TFLink: an integrated gateway to access transcription factor–target gene interactions for multiple species. Database 2022: baac083. 10.1093/database/baac083
- ↵Luijk R, Dekkers KF, van Iterson M, Arindrarto W, Claringbould A, Hop P, Boomsma DI, van Duijn CM, van Greevenbroek MMJ, Veldink JH, 2018. Genome-wide identification of directed gene networks using large-scale population genomics data. Nat Commun 9: 3097. 10.1038/s41467-018-05452-6
- ↵Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Favera RD, Califano A. 2006. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 7(Suppl. 1): S7. 10.1186/1471-2105-7-S1-S7
- ↵Marnetto D, Mantica F, Molineris I, Grassi E, Pesando I, Provero P. 2018. Evolutionary rewiring of human regulatory networks by waves of genome expansion. Am J Hum Genet 102: 207–218. 10.1016/j.ajhg.2017.12.014
- ↵Mostafavi H, Spence JP, Naqvi S, Pritchard JK. 2023. Systematic differences in discovery of genetic effects on gene expression and complex traits. Nat Genet 55: 1866–1875. 10.1038/s41588-023-01529-1
- ↵Narang V, Ramli MA, Singhal A, Kumar P, de Libero G, Poidinger M, Monterola C. 2015. Automated identification of core regulatory genes in human gene regulatory networks. PLoS Comput Biol 11: e1004504. 10.1371/journal.pcbi.1004504
- ↵Oh E-Y, Christensen SM, Ghanta S, Jeong JC, Bucur O, Glass B, Montaser-Kouhsari L, Knoblauch NW, Bertos N, Saleh SMI, 2015. Extensive rewiring of epithelial-stromal co-expression networks in breast cancer. Genome Biol 16: 128. 10.1186/s13059-015-0675-4
- ↵Oltvai ZN, Barabási A-L. 2002. Systems biology. Life's complexity pyramid. Science 298: 763–764. 10.1126/science.1078563
- ↵Omranian N, Eloundou-Mbebi JMO, Mueller-Roeber B, Nikoloski Z. 2016. Gene regulatory network inference using fused LASSO on multiple data sets. Sci Rep 6: 20533. 10.1038/srep20533
- ↵Opgen-Rhein R, Strimmer K. 2007. From correlation to causation networks: a simple approximate learning algorithm and its application to high-dimensional plant gene expression data. BMC Syst Biol 1: 37. 10.1186/1752-0509-1-37
- ↵Ota M, Spence JP, Zeng T, Dann E, Marson A, Pritchard JK. 2025. Causal modeling of gene effects from regulators to programs to traits: integration of genetic associations and Perturb-seq. bioRxiv 10.1101/2025.01.22.634424
- ↵Parsana P, Ruberman C, Jaffe AE, Schatz MC, Battle A, Leek JT. 2019. Addressing confounding artifacts in reconstruction of gene co-expression networks. Genome Biol 20: 94. 10.1186/s13059-019-1700-9
- ↵Pastor-Satorras R, Rubi M, Diaz-Guilera A. 2003. Statistical mechanics of complex networks. Springer Science & Business Media, Berlin.
- ↵Pierson E, GTEx Consortium, Koller D, Battle A, Mostafavi S, Ardlie KG, Getz G, Wright FA, Kellis M, Volpi S, 2015. Sharing and specificity of co-expression networks across 35 human tissues. PLoS Comput Biol 11: e1004220. 10.1371/journal.pcbi.1004220
- ↵Presson AP, Sobel EM, Papp JC, Suarez CJ, Whistler T, Rajeevan MS, Vernon SD, Horvath S. 2008. Integrated weighted gene co-expression network analysis with an application to chronic fatigue syndrome. BMC Syst Biol 2: 95. 10.1186/1752-0509-2-95
- ↵R Core Team. 2020. R: a language and environment for statistical computing (version 4.0.2). R Foundation for Statistical Computing, Vienna. https://www.R-project.org/.
- ↵Rodius S, Fournier A, Götz L, Liechti R, Crespo I, Merz S, Nazarov PV, de Klein N, Jeanty C, González-Rosa JM, 2016. Analysis of the dynamic co-expression network of heart regeneration in the zebrafish. Sci Rep 6: 26822. 10.1038/srep26822
- ↵Saha A, Battle A. 2018. False positives in trans-eQTL and co-expression analyses arising from RNA-sequencing alignment errors. F1000Res 7: 1860. 10.12688/f1000research.17145.1
- ↵Saha A, Battle A. 2019. Pre-computed cross-mappability resources for human genomes (hg19 and GRCh38). figshare. Collection. 10.6084/m9.figshare.c.4297352.v4
- ↵Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, Buetow KH. 2009. PID: the Pathway Interaction Database. Nucleic Acids Res 37: D674–D679. 10.1093/nar/gkn653
- ↵Schlitt T, Palin K, Rung J, Dietmann S, Lappe M, Ukkonen E, Brazma A. 2003. From gene networks to gene function. Genome Res 13: 2568–2576. 10.1101/gr.1111403
- ↵Segal E, Shapira M, Regev A, Pe'er D, Botstein D, Koller D, Friedman N. 2003. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet 34: 166–176. 10.1038/ng1165
- ↵Stegle O, Parts L, Piipari M, Winn J, Durbin R. 2012. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protoc 7: 500–507. 10.1038/nprot.2011.457
- ↵Stuart JM, Segal E, Koller D, Kim SK. 2003. A gene-coexpression network for global discovery of conserved genetic modules. Science 302: 249–255. 10.1126/science.1087447
- ↵Tomczak K, Czerwińska P, Wiznerowicz M. 2015. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol 19: A68–A77. 10.5114/wo.2014.47136
- ↵van Dam S, Võsa U, van der Graaf A, Franke L, de Magalhães JP. 2017. Gene co-expression analysis for functional classification and gene–disease predictions. Brief Bioinform 19: bbw139. 10.1093/bib/bbw139
- ↵Villar D, Berthelot C, Aldridge S, Rayner TF, Lukk M, Pignatelli M, Park TJ, Deaville R, Erichsen JT, Jasinska AJ, 2015. Enhancer evolution across 20 mammalian species. Cell 160: 554–566. 10.1016/j.cell.2015.01.006
- ↵Wang X, Choi D, Roeder K. 2021. Constructing local cell-specific networks from single-cell data. Proc Natl Acad Sci 118: e2113178118. 10.1073/pnas.2113178118
- ↵Wilks C, Zheng SC, Chen FY, Charles R, Solomon B, Ling JP, Imada EL, Zhang D, Joseph L, Leek JT, 2021. recount3: summaries and queries for large-scale RNA-seq expression and splicing. Genome Biol 22: 323. 10.1186/s13059-021-02533-6
- ↵Wolf DM, Lenburg ME, Yau C, Boudreau A, van ‘t Veer LJ. 2014. Gene co-expression modules as clinically relevant hallmarks of breast cancer diversity. PLoS One 9: e88309. 10.1371/journal.pone.0088309
- ↵Zhou K-R, Liu S, Sun W-J, Zheng L-L, Zhou H, Yang J-H, Qu L-H. 2017. ChIPBase v2.0: decoding transcriptional regulatory networks of non-coding RNAs and protein-coding genes from ChIP-seq data. Nucleic Acids Res 45: D43–D50. 10.1093/nar/gkw965
- ↵Zhu X, Duren Z, Wong WH. 2021. Modeling regulatory network topology improves genome-wide analyses of complex human traits. Nat Commun 12: 2851. 10.1038/s41467-021-22588-0