
Analytical validation of germline small variant detection using long-read HiFi genome sequencing
Abstract
Long-read sequencing has the capacity to interrogate difficult genomic regions and phase variants; however, short-read sequencing is more commonly implemented for clinical testing. Given the advances in long-read HiFi sequencing chemistry and variant calling, we analytically validated this technology for small variant detection (single nucleotide variants, insertions/deletions; SNVs/indels; <50 bp). HiFi genome sequencing was performed on DNA from reference materials and clinical specimen types, and accuracy results were compared to short-read genome sequencing data. HiFi genome sequencing recall and precision across Genome in a Bottle (GIAB)-defined non-difficult and difficult genomic regions (high confidence) for SNVs are >99.9% and >99.7%, respectively, and for indels are >99.8% and >99.1%, respectively. Moreover, HiFi genome sequencing outperforms short-read genome sequencing on overall SNV/indel F1-score accuracy at all paired sequencing depths, which are further stratified across 100 total GIAB-defined genomic regions for a comprehensive evaluation of performance. Of note, HiFi genome sequencing F1-scores for SNVs and indels surpass 99% at ∼15× and ∼25×, respectively. In addition, high confidence small variant concordance across all HiFi genome sequencing reproducibility assessments (two specimens, three independent sequencing data sets) are >99.8% for SNVs and >98.6% for indels, and average high confidence small variant concordance between paired blood, saliva, and swab specimens are all >99.8%. Taken together, these data underscore that long-read HiFi genome sequencing detection of SNVs and indels is very accurate and robust, which supports the implementation of this technology for clinical diagnostic testing.
Genome sequencing has evolved from a widely used research platform to a comprehensive clinical test at selected medical centers and laboratories (Belkadi et al. 2015; Costain et al. 2020), with the capacity to sequentially interrogate regions of the genome based on new evidence and/or clinical indication (Rehm 2017; Costain et al. 2018; Bick et al. 2019; Yang et al. 2024). Short-read sequencing is the most commonly implemented platform for genome sequencing; however, long-read sequencing is rapidly emerging as an alternative platform with notable benefits over short-read sequencing (Logsdon et al. 2020; Cohen et al. 2022; Conlin et al. 2022). For example, long-read genome sequencing has improved interrogation of clinically significant regions, including structural variants, repeat expansions, homologous gene families, and the HLA region, as well as the inherent benefit of variant phasing (Ardui et al. 2018; Ameur et al. 2019). The two primary long-read sequencing chemistries currently available are single molecule real-time (SMRT; HiFi) and nanopore sequencing, which recently have been employed by the Telomere-to-Telomere (T2T) Consortium to more comprehensively characterize the CHM13 human reference assembly (Nurk et al. 2022).
Long-read SMRT sequencing has been shown to generate highly accurate high-fidelity (HiFi) read lengths of ∼10–25 kb using the Sequel II platform (Pacific Biosciences [PacBio]) (Wenger et al. 2019; Hon et al. 2020). More recently, long-read HiFi genome sequencing has been used to expand the small variant benchmarks in the commonly leveraged Genome in a Bottle (GIAB) Consortium reference material samples to include difficult-to-map regions and segmental duplications that are inherently challenging for short reads (Wagner et al. 2022a). In addition, comparisons of sequencing platforms and variant calling strategies have recently been reported by the PrecisionFDA Truth Challenge V2, which found long-read HiFi genome sequencing to outperform both short-read and long-read nanopore sequencing with genome-wide variant calling accuracy (Olson et al. 2022).
Although long-read HiFi genome sequencing has improved accuracy and haploblock phasing performance compared to short-read sequencing, its adoption into clinical genetic testing laboratories is only now emerging. Of note, resources for validating new clinical sequencing assays are available from the College of American Pathologists (CAP), Association for Molecular Pathology (AMP) (Aziz et al. 2015; Roy et al. 2018), and related professional consortia (Gargis et al. 2012; Matthijs et al. 2016; Santani et al. 2017, 2019), as well as benchmarking reference materials from the GIAB/National Institute of Standards and Technology (NIST) consortium (Zook et al. 2019), and the Global Alliance for Genomics and Health Benchmarking (GA4GH) Team (Krusche et al. 2019). Therefore, to facilitate the implementation of clinical long-read HiFi genome sequencing, our initial effort was centered on a robust analytical validation of germline small variant (SNV/indel) detection using current best practices and benchmarking resources.
Results
Long-read HiFi genome sequencing small variant accuracy
HiFi genome sequencing accuracy (SNV/indel; <50 bp) was evaluated by sequencing seven GIAB/NIST reference material samples (average 31.1×). Benchmarking was performed using tools and practices recommended by the GA4GH (Krusche et al. 2019), and hap.py version v0.3.15 was used to compare observed results with the published truth set version v4.2.1. Accuracy was measured by recall (i.e., sensitivity) and precision (i.e., positive predictive value), which were stratified by high confidence genomic regions as defined by GIAB/NIST (Zook et al. 2019). SNV/indel detection across the GIAB reference samples was highly accurate, as the average recall and precision for all seven samples were >99.9% for SNVs and >99.8% for indels across the non-difficult genomic regions (Table 1). As expected, recall and precision were slightly lower when interrogating genomic regions with known sequencing challenges (low complexity, low mappability, segmental duplications); however, average recall and precision across all difficult genomic regions were still >98.9% and >98.7% for SNVs and indels, respectively. Of note, in addition to the low complexity, low mappability, and segmental duplication regions highlighted in Table 1, the GIAB “Difficult regions” are defined broadly to include low or high GC content (<25% or >65%), bad promoter regions, false duplications, and other difficult genomic regions (Krusche et al. 2019). The average recall and precision across all genomic regions were >99.7% and >99.1% for SNVs and indels, respectively (Table 1). Among the discordant HiFi small variants with the GIAB truth set, homopolymers were the most common source of error, with ∼75% of indel errors located in or adjacent to a homopolymer run.
Long-read HiFi genome sequencing SNV/indel (<50 bp) accuracy
Long-read HiFi and short-read genome sequencing small variant accuracy
In addition to accuracy benchmarking across the high confidence GIAB/NIST regions (i.e., low complexity, low mappability, segmental duplications, all difficult regions, not in any difficult region, all), HiFi genome sequencing performance was further evaluated across 100 GIAB-defined subregions of the human genome (Zook et al. 2019). F1-scores were generated and stratified by variant type (i.e., SNVs/indels) and results compared to paired analyses with publicly available short-read genome sequencing data (average 40.3×). As illustrated in Figure 1, A–F, HiFi genome sequencing small variant F1-scores were superior to short-read genome sequencing small variant F1-scores across 22 informative genomic subregions within the categories of low mappability, homopolymers, tandem repeats, and GC content, which were most notable for indels of increasing size. In addition, HiFi genome sequencing variant calling accuracy across the recently reported Challenging Medically-Relevant Gene (CMRG) truth set (Wagner et al. 2022b) was also interrogated and compared with short-read genome sequencing accuracy, which identified HiFi's advantage over short-read in detection of >15 bp deletions (87.76% vs. 67.81% recall), >15 bp insertions (84.44% vs. 74.81% recall) but otherwise showed roughly similar performance in the CMRG regions (Supplemental Table S1).
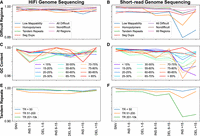
SNV/indel accuracy across GIAB-defined genomic regions. Small variant F1-scores for HiFi genome sequencing (A,C,E) and short-read genome sequencing (B,D,F) across 22 of the 100 interrogated GIAB-defined genomic regions. Results summarized by difficulty (mappability, homopolymers, tandem repeats, segmental duplications, all difficult, not in any difficult, all regions), GC content (<15% to >85%), and tandem repeats (<50 bp, 51–200 bp, 201–10,000 bp), and stratified by variant type (SNV, indels 1–5 bp, 6–15 bp, and ≥16 bp).
Genome sequencing depth stratification and accuracy
To evaluate HiFi genome sequencing small variant accuracy at different sequencing depths, small variant recall and precision were assessed across a series of sequencing depths downsampled from NA24385 (HG002). As expected, HiFi genome-wide small variant accuracy was reduced at low sequencing depths (Supplemental Table S2); however, HiFi genome sequencing F1-scores for SNVs and indels surpassed 99% at ∼15× and ∼25×, respectively. In comparison, short-read genome sequencing F1-scores for SNVs and indels surpassed 99% at ∼20× and ∼35×, respectively (Fig. 2). At sequencing depths of ∼30×, HiFi genome sequencing F1-scores for SNVs and indels were 99.8% and 99.1%, respectively, and short-read sequencing F1-scores for SNVs and indels were 99.8% and 98.7%, respectively (Fig. 2A–C).
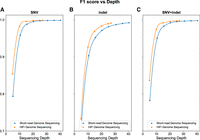
SNV/indel accuracy and depth stratification. Plots of F1-scores across sequencing depths and comparing HiFi genome sequencing and short-read genome sequencing: (A) single nucleotide variants (SNVs); (B) insertions/deletion variants (indels); and (C) SNVs and indels combined.
Long-read HiFi genome sequencing concordance and reproducibility
HiFi genome sequencing library preparation included both manual and automated workflows, and minor updates were introduced to the laboratory procedure to optimize the automated workflow. Workflow and procedure updates were validated by measuring small variant accuracy and/or concordance with reference material and specimen samples as appropriate. As detailed in Supplemental Tables S3 and S4, the accuracy and concordance of the workflow updates (e.g., fragment depletion using the SRE XS and SRE kits) were consistent with paired manual library preparation results, which supported the implementation of these workflow improvements. In addition, a novel validation strategy of two Miro Canvas instruments was accomplished by low-depth NA12878 benchmarking comparisons using a single SMRTcell of data (∼9× each) (Fig. 3A,B), which was supported by consistent quality metrics between manual and automated workflows, and concordant benchmarking results from a subsequent full depth (27.9×) Miro Canvas library preparation (Supplemental Tables S3, S4).
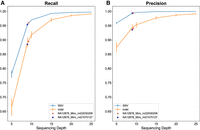
Automated library preparation validation with low depth HiFi genome sequencing. Two Miro Canvas instruments were validated using a single SMRTcell of data (∼9× each) with small variant (SNV/indel) benchmarking of NA12878 and fitting results to reference curves defined by manual library preparation of three GIAB reference samples (NA12878, NA24385, NA24631). Error bars represent standard deviations. Automated library preparation results were considered acceptable if Miro Canvas recall (A) and precision (B) values were equivalent or greater than the average manual preparation reference material accuracy results at comparable depths.
HiFi genome sequencing reproducibility was evaluated by comparing reference sample results to two independent publicly available data sets (NA12878/HG001, NA24385/HG002; see Methods) and measuring F1-score concordance across all three data sets (average 32.9×). Genome-wide SNV/indel non-reference genotype concordance was stratified by genomic context, which ranged from ∼98% to 99.9% (Table 2); however, HiFi genome sequencing reproducibility was reduced when assessed across regions not considered high confidence by GIAB (Supplemental Table S5). Non-reference genotype concordance for the high confidence RefSeq CDS regions across all reproducibility and repeatability assessments were >99.8% and >99.3% for SNVs and indels, respectively (Table 2), indicating that HiFi genome sequencing small variant detection is robust and precise.
Long-read HiFi genome sequencing SNV/indel (<50 bp) reproducibility (GIAB high confidence)
Long-read HiFi genome sequencing specimen validation
Germline specimens were validated by subjecting paired blood, saliva, and swab samples to HiFi genome sequencing and evaluating SNV/indel concordance. As expected, concordance between specimens was reduced for SNVs/indels when evaluating difficult genomic regions, as saliva-based specimens are known to harbor bacterial DNA that interferes with sequencing (Trost et al. 2019; Yao et al. 2020). However, the average paired SNV/indel concordance between all three specimen types were >99% across all high confidence genomic regions (Table 3). Although specimen concordance was reduced when assessed across regions not considered high confidence by GIAB (Supplemental Table S6), these validation results indicate that HiFi genome sequencing of saliva and swab specimens are consistent with blood for germline SNV/indel detection.
Long-read HiFi genome sequencing SNV/indel (<50 bp) paired specimen concordance (GIAB high confidence)
Discussion
To facilitate the implementation of diagnostic long-read HiFi sequencing, we executed an analytical validation plan that was centered on comprehensively evaluating HiFi genome sequencing for germline SNV/indel detection and specimen types that are used for clinical testing in medical genetics. Results were stratified by variant type and GIAB-defined genomic regions to better inform overall performance, which ultimately determined that HiFi genome sequencing is accurate and robust. The accuracy of germline small variant detection in non-difficult genomic regions across reference materials was >99.9% for both SNVs and indels, and small variant detection accuracy in GIAB-defined difficult regions was >99.5% and >98.8% for SNVs and indels, respectively. These analytical validation analyses underscore the accuracy of long-read HiFi genome sequencing for detecting germline SNV/indels (<50 bp), which supports the implementation of this technology for clinical genetic testing. In addition, quality control (QC) thresholds for clinical long-read HiFi genome sequencing based on CAP requirement MOL.36151 are suggested in Supplemental Table S7; however, these should be considered preliminary recommendations, as clinical laboratories should leverage their own experience and data to define internal QC metrics.
Analytical validation is a critical assessment of any new clinical laboratory test, which is defined by CAP Checklists and other state, federal, and/or professional requirements/recommendations. Test performance specifications include reportable range, accuracy, reproducibility/repeatability, sensitivity/specificity, and other relevant performance characteristics. For long-read HiFi genome sequencing, we adopted the definitions for “Reportable Range” and “Reference Range (Reference Interval)” based on clinical high-throughput sequencing guidelines (Gargis et al. 2012; Santani et al. 2017). However, for a more comprehensive assessment of sequencing performance, reportable range was measured genome-wide but strategically stratified by distinct genomic regions as defined by GIAB/GA4GH. The genomic regions implemented in this validation included high level strata (low complexity, low mappability, segmental duplications, all difficult regions, not in any difficult regions, all high confidence regions, RefSeq CDS regions), as well as the more specific genomic subregions defined by GIAB/NIST and GA4GH (Krusche et al. 2019). These regions were intersected with our SNV/indel performance results, as well as genome sequencing specimen validation data (CAP requirement MOL.31015), as deemed appropriate based on analysis context and intended use.
Sequencing accuracy is a rapidly evolving area that is driven by continual improvements in available chemistries and informatic algorithms developed for calling germline variants. As an integral component of validating clinical sequencing-based platforms (Roy et al. 2018), benchmarking small variant accuracy (i.e., recall, precision, F1) is supported through the GIAB/NIST/GA4GH resources (Majidian et al. 2023; Olson et al. 2023), which recently has been catalyzed by PrecisionFDA challenges that provide more comprehensive evaluations of sequencing-based variant calling (Zook et al. 2019; Olson et al. 2022). Our analytical validation of HiFi genome sequencing is consistent with the most recent PrecisionFDA V2 challenge, which concluded that long-read HiFi sequencing coupled with machine learning-based variant calling tools (Pei et al. 2021; Olson et al. 2022) was superior to short-read genome sequencing using graph-based variant calling.
It is important to note that our reported accuracy results reflect not only the sequencing platforms evaluated but also the variant calling methods used. Given that it was beyond the scope of our study to perform a full comparison of bioinformatics techniques, we selected best-practice tools with high accuracy in challenging genomic regions for each sequencing platform, as demonstrated by PrecisionFDA V2. Our validation also included a detailed evaluation of performance across GIAB-defined genomic stratifications, which highlighted long-read HiFi sequencing accuracy across challenging regions and particularly among indel variants. Of note, our targeted sequencing depth was ≥30×, consistent with recommendations from the Medical Genome Initiative (Marshall et al. 2020), and, as expected, small variant accuracy was reduced at lower depths. However, it is notable that 99% accuracy was surpassed at ∼15–25× for long-read HiFi genome sequencing compared to ∼20–35× for short-read genome sequencing.
In addition to accuracy, HiFi genome sequencing small variant reproducibility was also interrogated by measuring non-reference genotype concordance between data sets. Concordance across all replicates in high confidence GIAB-defined regions ranged from 99.84% to 99.91% for SNVs and 97.66% to 99.30% for indels, indicating that small variant calling is very robust. Given that exome reproducibility/repeatability is typically higher than that observed with genome sequencing due to the more narrow region interrogated (Linderman et al. 2014), we also stratified our genome results by RefSeq CDS regions, which resulted in highly concordant small variant calling across all replicates in the high confidence regions (all: 99.85%/99.37%; non-difficult: 99.91%/99.73%, for SNVs/indels) and non-high confidence regions (all: 98.22%/93.13%; non-difficult: 99.66%/99.19%, for SNVs/indels).
Of note, the GIAB high confidence regions encompass 81.6% of the autosomal GRCh38 human genome, which translates to ∼2.52 Gb across the seven GIAB reference samples. The remaining 18.4% of non-high confidence autosomal bases (∼567.2 Mb) represent subregions of the genome (and Chromosomes X and Y) that are difficult to benchmark given the uncertainty in the underlying truth set (Zook et al. 2016, 2019, 2020). The concordance results across reference materials and specimen types in our validation study were reduced in the genome-wide analyses (i.e., including non-high confidence regions) compared to the concordance results limited to the high confidence genomic regions, most notably in the GIAB-defined difficult regions (low complexity, low mappability, segmental duplications, etc.). These metrics were considered acceptable, as 40% of the variants in these regions had genotype quality scores of <Q20 compared to <1% of variants in the non-difficult regions before filtering, and the increase in variant numbers was much greater in the difficult regions than the non-difficult regions in the non-high confidence regions (2.51× vs. 1.08×). As such, these thorough reproducibility analyses together indicate that long-read HiFi genome sequencing is highly robust across the high confidence regions of the human genome; however, variants identified in the GIAB non-high confidence difficult regions in a clinical setting would likely require independent confirmation if reportable.
Another critical CAP requirement for test implementation is validating the specific specimen types used for clinical processing (MOL.31015), which, for germline genetic testing, typically includes peripheral blood and/or saliva specimens. To satisfy this requirement, paired specimens were subjected to HiFi genome sequencing and concordance was measured across the genome. Despite known challenges with using oral saliva samples for sequencing due to the presence of competing bacterial DNA (Krusche et al. 2019; Trost et al. 2019), concordance between paired blood, saliva, and assisted saliva (swab) specimens ranged from 99.82% to 99.92% for all SNVs/indels across high confidence genomic regions. However, it is notable that ∼99% of reads aligned to the reference genome for blood and cell line specimens, compared to ∼93% for assisted saliva specimens and 86% for saliva, resulting in lower average depth (blood, cell lines: 33×; assisted saliva: 28×; saliva: 25×). As such, additional sequencing of oral samples to compensate for unmapped reads (as defined by QC thresholds) may be warranted in clinical production. These specimen validation study results indicate that our HiFi genome sequencing procedure and pipeline generates highly comparable results between peripheral blood and DNA isolated from saliva and assisted saliva, which supports their use as acceptable clinical specimens for this test.
Of note, copy number variant (CNV) detection by sequencing is routinely implemented among clinical laboratories (Kadalayil et al. 2015; Rajagopalan et al. 2020), and GIAB/NIST has developed consensus germline structural variant (SV) calls from HG002 (NA24385) (Zook et al. 2020). However, this data set is an integration of 68 callsets from multiple algorithms and four different sequencing technologies, each with their own strengths and weaknesses, and, as a result, it does not currently include robust duplication calls or SVs >100 kb (Whitford et al. 2019). Although long-read HiFi genome sequencing has been shown to be highly effective at CNV/SV detection (Chaisson et al. 2019; Mahmoud et al. 2019; Aganezov et al. 2020), these variants were considered out of scope for this initial analytical validation; however, they are currently being evaluated for a subsequent analytical work product.
In conclusion, long-read HiFi genome sequencing (≥30×) was analytically validated for germline SNV/indel detection, which supports the implementation of this platform as a robust technology for clinical genetic testing. Of note, practical factors for sequencing platform selection were intentionally excluded from this analytical validation, including cost, labor, and sequencing time, as these variables were not applicable to analytical performance testing. This validation also did not explicitly include “clinical performance characteristics” as defined by CAP (MOL.31590), as these analyses were reserved for subsequent validation of clinically significant germline variants. As such, these analytical validation data provide the infrastructure for long-read HiFi genome sequencing-based detection of germline variation, which supports the use of this innovative technology for clinical diagnostic testing.
Methods
Analytical validation specimens
High molecular weight (HMW) reference material DNA samples were acquired from the Coriell Institute for Medical Research, which included seven benchmarking samples from the GIAB/NIST consortium. Peripheral blood was collected in EDTA vacutainer tubes using standard practices and DNA isolated using the Maxwell RSC Buffy Coat DNA Kit (Promega Corporation) according to manufacturer instructions. Saliva samples were collected using the Oragene Dx OGD-500 kit (DNA Genotek) or the assisted saliva (swab) Oragene Dx OGD-575 kit (DNA Genotek). DNA was isolated from saliva specimens using Maxwell RSC Stabilized Saliva DNA Kit (Promega Corporation) according to manufacturer instructions. All validation samples and sequencing metrics are summarized in Supplemental Tables S7–S10.
Long-read HiFi genome sequencing
Library preparation and long-read HiFi sequencing
Genomic DNA was analyzed with the Femto Pulse Genomic DNA 165 kb kit (Agilent) to confirm an adequate quantity of HMW DNA. Approximately 3–10 μg of DNA was mechanically sheared to 10–20 kb using the Megaruptor Shearing kit (Diagenode), with the DNAFluid+ kit (Diagenode) employed for viscous samples. Library preparation was performed through either a manual or an automated workflow with the SMRTbell Prep kit according to manufacturer instructions, including end repair, A-tailing, adapter ligation, purification with SMRTbell cleanup beads (PacBio), and nuclease treatment.
Manual library preparation included purification of sheared gDNA with SMRTbell cleanup beads, SMRTbell library generation, followed by size selection on Blue Pippin (Sage Science) to remove fragments <10 kb and purification with Ampure PB beads (PacBio). Automated library preparation included small fragment depletion using the Short Read Eliminator (SRE) XS kit (<10 kb) or the SRE kit (<25 kb) (PacBio) as needed prior to shearing, followed by purification with SMRTbell cleanup beads and library preparation using the Miro Canvas (Miroculus). Manual and automated workflow SMRTbell libraries were both quantified by the Qubit dsDNA assay kit (Invitrogen) and bound to sequencing polymerase using the Sequel II Binding kit (PacBio). Long-read HiFi genome sequencing was performed on the Sequel IIe system (PacBio) with a 30 h movie collection time, and each sample was sequenced on three SMRTcells except where otherwise noted.
Publicly available data
To evaluate internal long-read HiFi genome sequencing reproducibility, selected publicly available long-read HiFi genome sequencing data for the NA12878/HG001 and NA24385/HG002 reference materials were acquired from the National Center for Biotechnology Information (NCBI) FTP server (ftp://ftp.ncbi.nlm.nih.gov/giab): PacBio_SequelII_CCS_11kb (NA12878), HudsonAlpha_PacBio_CCS (NA12878) (Zook et al. 2016), PacBio_CCS_15kb_20kb_chemistry2 (NA24385), PacBio_SequelII_CCS_11kb (NA24385).
Long-read HiFi sequencing bioinformatics pipeline and variant calling
HiFi reads were generated using SMRTLink 10.2 software and the Circular Consensus Sequencing mode. For analyses requiring downsampled data, subsampling was executed with SAMtools v1.18 (Danecek et al. 2021). Alignment and variant calling were performed using a modified version of the PacBio HiFi-human-WGS-WDL pipeline (https://github.com/PacificBiosciences/HiFi-human-WGS-WDL), with the following steps: alignment of HiFi sequencing reads to Genome Reference Consortium Human Build 38 (GRCh38) using pbmm2 v1.7.0 (Hon et al. 2020); small variant calling using DeepVariant v1.4.0 and the PacBio machine learning model included with the software (Poplin et al. 2018); and calculating aligned read depth with mosdepth v0.2.9 (Pedersen and Quinlan 2018). BCFtools v1.20 was used to filter variants, removing all SNV calls with QUAL < 20 (Danecek et al. 2021). Finally, Picard Tools v2.27.4 was used to calculate quality yield metrics (https://broadinstitute.github.io/picard), alignment summary metrics, and variant calling metrics. For analysis of CMRG genes, a modified version of the GRCh38 build was used, wherein false duplications were masked and decoy contigs were added for falsely collapsed duplications (Behera et al. 2023).
Short-read genome sequencing
Publicly available data
To compare long-read HiFi and short-read genome sequencing accuracy, publicly available short-read genome sequencing data for the seven GIAB benchmarking reference materials were acquired from the National Center for Biotechnology Information FTP server (ftp://ftp.ncbi.nlm.nih.gov/giab): NIST_NA12878_HG001_HiSeq_300x, NIST_Illumina_2 × 250 bps (NA24385, NA24143, and NA24149), HG005_NA24631_son_HiSeq_300x, NA24694_Father_HiSeq100x, and NA24695_Mother_HiSeq100x.
Short-read sequencing bioinformatics pipeline and variant calling
For analyses requiring downsampled data, subsampling was executed with SAMtools v1.18 (Danecek et al. 2021). Alignment, germline small variant calling, and calculation of quality control metrics were performed with the Illumina DRAGEN Germline Pipeline v4.2.4 in BaseSpace, using the hg38 alt-masked multi-genome graph reference.
Analytical validation strategy
The HiFi genome sequencing small variant analytical validation plan followed Laboratory Developed Test (LDT) guidelines as defined by the CAP and AMP (Jennings et al. 2009; Aziz et al. 2015; Roy et al. 2018), the American College of Medical Genetics and Genomics (ACMG) (Rehm et al. 2013), high-throughput sequencing recommendations from professional consortia (Gargis et al. 2012; Matthijs et al. 2016; Santani et al. 2017, 2019), and the Clinical Laboratory Evaluation Program at the Wadsworth Center, New York State Department of Healthy (https://www.wadsworth.org/regulatory/clep/clinical-labs/laboratory-standards). The plan was centered on determining the analytical performance characteristics of HiFi genome sequencing for use as a diagnostic technology, as well as defining standard operating procedures (SOPs), quality control/quality assurance procedures, and validating small variant detection and specimen types.
Data access
All GIAB reference material and blood/saliva sample HiFi genome sequencing aligned BAM data sets generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1143955.
Competing interest statement
N.H. is currently an employee of Influx Bio; all other authors declare no conflicts of interest.
Acknowledgments
The authors would like to thank Stanford Health Care, Stanford Children's Health, and Pacific Biosciences for their programmatic support. S.A.S. was supported in part by National Institutes of Health/National Human Genome Research Institute grant U01HG011762.
Author contributions: Project conceptualization: N.H., Y.Y., S.A.S.; data acquisition: N.H., L.L., P.W.T., Z.N.; data analysis, interpretation, and management: N.H., P.W.T., Z.N., C.H., T.P.N., Y.Y., S.A.S.; drafting and revision: N.H., Y.Y., S.A.S.; final approval: N.H., L.L., P.W.T., Z.N., C.H., T.P.N., Y.Y., S.A.S.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278836.123.
- Received December 9, 2023.
- Accepted April 11, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








