
Contiguous and complete assemblies of Blastocystis gut microbiome–associated protists reveal evolutionary diversification to host ecology
- Abigail L. Lind1,2,
- Nathan A. McDonald2,3,
- Elias R. Gerrick4,
- Ami S. Bhatt5,6 and
- Katherine S. Pollard1,7,8
- 1Gladstone Institute for Data Science and Biotechnology, Gladstone Institutes, San Francisco, California 94158, USA;
- 2School of Biological Sciences, Georgia Institute of Technology, Atlanta, Georgia 30332, USA;
- 3Department of Biology, Stanford University, Stanford, California 94305, USA;
- 4Department of Microbiology, University of Chicago, Chicago, Illinois 60637, USA;
- 5Department of Genetics, Stanford University, Stanford, California 94305, USA;
- 6Department of Medicine (Hematology, Blood and Marrow Transplantation), Stanford University, Stanford, California 94304, USA;
- 7Department of Epidemiology and Biostatistics, University of California, San Francisco, California 94158, USA;
- 8Chan Zuckerberg Biohub SF, San Francisco, California 94158, USA
Abstract
Blastocystis, an obligate host-associated protist, is the most common microbial eukaryote in the human gut, and is widely distributed across vertebrate hosts. The evolutionary transition of Blastocystis from its free-living stramenopile ancestors to a radiation of host-associated organisms is poorly understood. To explore this, we cultured and sequenced eight strains representing the significant phylogenetic diversity of the genus using long-read, short-read, and Hi-C DNA sequencing, alongside gene annotation and RNA sequencing. Comparative genomic analyses reveal significant variation in gene content and genome structure across Blastocystis. Notably, three strains from herbivorous tortoises, phylogenetically distant from human subtypes, have markedly larger genomes with longer introns and intergenic regions, and retain canonical stop codons absent in the human-associated strains. Despite these genetic differences, all eight isolates exhibit gene losses linked to the reduced cellular complexity of Blastocystis, including losses of cilia and flagella genes, microtubule motor genes, and signal transduction genes. Isolates from herbivorous tortoises contain higher numbers of plant carbohydrate-metabolizing enzymes, suggesting that, like gut bacteria, these protists ferment plant material in the host gut. We find evidence that some of these carbohydrate-metabolizing enzymes were horizontally acquired from bacteria, indicating that horizontal gene transfer is an ongoing process in Blastocystis that has contributed to host-related adaptation. Together, these results highlight substantial genetic and metabolic diversity within the Blastocystis genus, indicating that different lineages of Blastocystis have varied ecological roles in the host gut.
The vertebrate gut is host to a diverse ecosystem of bacteria, archaea, viruses, and microbial eukaryotes. This diverse microbial community has broad impacts on the physiology and development of the host. Gut microbes can metabolize compounds that are not digestible by the host, providing access to previously inaccessible nutrients (Yadav et al. 2018). Gut microbes also influence the host's immune system and impact host health through their metabolic byproducts (Sommer and Bäckhed 2013). The microbiome's composition, which is influenced by diet, physiology, and the host immune system, varies substantially across vertebrate animals (Godoy-Vitorino et al. 2012; David et al. 2014; Thorman et al. 2023).
Compared to bacteria, substantially less is understood about microbial eukaryote impacts on host physiology and the gut ecosystem, despite the prevalence and diversity of gut microbial eukaryotes across vertebrates (Nash et al. 2017; del Campo et al. 2020). The protist Blastocystis is the most prevalent eukaryotic microorganism in the human gut, found in 10%–50% of industrialized populations and at higher prevalence in non-industrialized communities (Scanlan and Marchesi 2008; Scanlan and Stensvold 2013; Scanlan et al. 2014; Khaled et al. 2020; Asnicar et al. 2021; Jinatham et al. 2021). Beyond humans, Blastocystis colonizes a wide range of animals including other mammals, birds, reptiles, fish, amphibians, and insects (Boreham and Stenzel 1993; Yoshikawa et al. 2016; Gantois et al. 2020). Blastocystis is a genetically diverse genus, with strains adapting to varied gut environments across different host species (Stensvold et al. 2007). Its presence is associated with positive health outcomes in humans, including lower levels of gut inflammation and metabolic health (Nieves-Ramírez et al. 2018; Asnicar et al. 2021; Piperni et al. 2024). However, very little is understood about how Blastocystis contributes to the gut environment and interacts with other gut microbiota.
Blastocystis belongs to the Opalinata lineage of stramenopiles, a group of protists that are obligate animal gut commensals (Kostka 2017). Unlike most stramenopiles, Blastocystis is non-motile, lacks flagella or surface hairs, and grows predominantly as a round cell with a large central vacuole (Zierdt 1988). The Blastocystis genus contains numerous genetically distinct groups categorized into subtypes based on polymorphism in the 18S rRNA gene. Thus far, 40 distinct subtypes have been proposed, and this number is continuing to expand (Stensvold and Clark 2020; Santin et al. 2024). To date, strains from three axenic lab-cultured subtypes (ST1, ST4, and ST7), all of which are subtypes observed in humans, have been sequenced, annotated, and deposited in public databases (Denoeud et al. 2011; Gentekaki et al. 2017). Although these subtypes span a narrow range of the Blastocystis phylogenetic diversity, they have very different genomes and gene contents (Gentekaki et al. 2017). These genomes have either gapped scaffolds (ST7) or highly fragmented non-contiguous genome assemblies (ST1, ST4), limiting the ability to determine synteny between subtypes, and two genomes were annotated without consideration for the unique Blastocystis phenomenon where a portion of genes lack DNA-encoded termination codons. Thus, we lack highly complete and well-annotated genomes necessary for establishing cases of gene loss, horizontal transfer, and sequence evolution so that functional repertoires can be associated with strain diversification.
Like most host-associated eukaryotes, Blastocystis grows best in the presence of other microbiota. Establishing robust axenic cultures of intestinal protists including Blastocystis is laborious and often either fails to remove all microbial contaminants or results in the death of the protist (Zierdt and Williams 1974; Tan 2008). However, removing sources of microbial contamination is important for eukaryotic genome assembly via short-read sequencing (Gough et al. 2020). As a result, genome sequencing for Blastocystis and other intestinal protists has been limited to axenic cultures (Keeling et al. 2014; Gough et al. 2020). To overcome this barrier, we establish high- quality and contamination-free genomes from xenic cultures of Blastocystis grown together with other microbes present in the original sample by using long-read Nanopore and Hi-C sequencing technologies coupled with short reads and metagenomic assembly techniques to establish genomes of Blastocystis strains that are grown together with other microbes. The longer read length of Nanopore sequencing helps span repetitive regions, resulting in greater contig length and fewer misassembles, and has been successfully applied to assembling bacterial metagenomes (Moss et al. 2020). Hi-C sequencing, which captures spatial interactions between DNA fragments, can further improve genome assembly by correcting misassemblies and scaffolding contigs (Šimková et al. 2024). By combining these technologies with metagenomic assembly techniques, we were able to generate nearly complete Blastocystis genomes without the need for axenic culturing. This approach allows us to investigate the full genetic diversity of Blastocystis, even in the presence of other microbes, and to assemble highly contiguous genomes that were previously difficult to obtain.
Here, we applied this hybrid sequencing strategy to eight Blastocystis strains, five isolated from humans and three isolated from the herbivorous tortoise Testudo horsfeldii. These isolates, five of which are xenic, containing other microbes present in the original sample, and three of which are axenic, span the phylogenetic diversity of the Blastocystis genus, enabling us to investigate Blastocystis evolution and determine how Blastocystis has adapted to thrive in animal hosts with different diets, physiologies, and resident microbiota. Our highly contiguous and complete genomes allowed us to investigate patterns of genome evolution within and between subtypes and to compare the genus to related protists. This work demonstrates how high-quality genome assemblies can be created from xenic protist cultures by combining deep long-read sequencing with metagenomic assembly techniques, enabling rigorous comparative genomic analyses.
Results
Capturing Blastocystis diversity
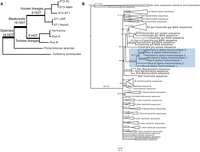
To investigate the evolution of the Blastocystis genus, from its initial divergence through its later diversification, we sequenced eight diverse Blastocystis strains. Strains were cultured anaerobically on egg media overlaid with a balanced salt solution and 25% heat-inactivated horse serum. Five of these strains correspond to three different previously defined “subtypes” (genetically similar groups historically identified in mammalian and avian species [Stensvold and Clark 2020]). These five strains were originally cultured from human fecal samples and were obtained from the American Type Culture Collection. These human isolated strains correspond to three of the four most prevalent Blastocystis “subtypes” (Beghini et al. 2017), genetically similar groups historically identified in mammalian and avian species (Stensvold and Clark 2020): two subtype (ST) 1 (Nandll, JDR), two ST3 (DL, NMH), and one ST4 (BT1). The remaining three strains were isolated from companion animal tortoises: two from different individuals of the domestic Russian tortoise species Testudo horsfeldii (Rus-B, Rus-S), and one from a Hermann's tortoise Testudo hermanni (Hermann's). We performed comparisons of the 18S rRNA gene of these four strains to other Blastocystis strains, finding that the tortoise isolates were distantly related to the human isolates and do not belong to a described mammalian or avian subtype (Fig. 1A; Supplemental Table S1). The closest related named subtypes to the tortoise isolates are ST15, which has been described in non-human primates, and ST28, which has been described in birds (Fig. 1A; Alfellani et al. 2013; Maloney and Santin 2021). Analysis of all eight cultures by light microscopy revealed predominantly vacuolar Blastocystis cells, containing a large central vacuole and a thin peripheral ring of cytoplasm. Although Blastocystis have been observed to adopt a variety of morphologies, vacuolar morphologies dominate cultured samples (Fig. 1B).
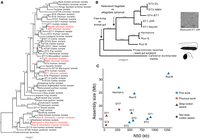
Phylogenetic relationships and genome assembly characteristics of Blastocystis. (A) 18S rRNA maximum likelihood (ML) phylogeny of Blastocystis subtypes, with Proteromonas lacertae as an outgroup. Support values are 1000 replicates of ultrafast bootstrap and SH-aLRT tests (Guindon et al. 2010; Hoang et al. 2018). Strains in red were sequenced in this study. (B) 13-gene concatenation ML phylogeny for all strains and species with sequenced genomes used in this study. Important evolutionary transitions are highlighted. Images represent cellular morphology of Cafeteria burkhardae, Proteromonas lacertae, and Blastocystis. (C) Assembly statistics of Blastocystis strains sequenced in this work compared with previously sequenced and annotated genomes. Stop-codon aware and not stop codon aware designations refer to gene annotation methodologies that take into consideration that Blastocystis genes can lack stop codons or do not consider this, respectively. The ST7 N50 is a gapped scaffold N50 generated with Sanger sequencing scaffolding.
To explore how the Blastocystis genus has emerged and diversified, we compared the Blastocystis genomes generated in this work to those of two other sequenced stramenopiles: Cafeteria burkhardae, a free-living heterotrophic marine flagellate (Hackl et al. 2020), and the recently sequenced Proteromonas lacertae, a reptile gut commensal closely related to Blastocystis (Záhonová et al. 2023). These organisms are the closest relatives to Blastocystis that have sequenced genomes. Genome comparisons allowed us to reconstruct three evolutionary transitions that have occurred during stramenopile evolution: the transition from a free-living aquatic lifestyle to an anaerobic lifestyle inside the animal gut (comparing Cafeteria to Blastocystis/Proteromonas), the change in cell morphology from heterokont flagellate to aflagellate, spherical cells (comparing Cafeteria/Proteromonas to Blastocystis), and adaptations to different animal hosts (comparing Blastocystis tortoise to Blastocystis human) (Fig. 1B).
Contiguous and complete Blastocystis genomes
We prepared high-quality, high molecular weight DNA, as well as RNA, from the eight Blastocystis cultures. DNA was sequenced with Nanopore long-read technology. Short-read DNA and RNA sequencing was also performed for assembly polishing and gene annotation. Hi-C data were additionally generated to help resolve scaffolds for one strain (ST4-BT1), although they resulted in only modest improvements to the final assembly. Non-eukaryotic sequence was detected via multiple methods and excluded from final assemblies (see Methods, “Genome assembly and decontamination”). We additionally validated that our genomes were not contaminated by bacterial sequence by determining that genes with high sequence similarity to bacterial genes arose from polyadenylated eukaryotic transcripts (Supplemental Table S2) (see Methods, “Gene contamination and horizontal gene transfer screens”).
This combination of sequencing technologies enabled us to generate highly contiguous genomes for all six subtypes. The N50 of these assemblies ranged from 289 kb to 1.3 Mb, achieving a much higher assembly quality than currently available Blastocystis genome data sets in NCBI (median: 37 kb) (Table 1; Fig. 1C). We used the BUSCO pipeline to estimate genome completeness based on presence and absence of single-copy universal eukaryotic genes (Manni et al. 2021). BUSCO completeness estimates ranged from 42% to 61.2%, similar to BUSCO completeness results of previously sequenced Blastocystis isolates and related species (Table 1; Fig. 1C). These values likely underestimate the true completeness, because BUSCO is known to have poorer performance in protists relative to plants, animals, and fungi (Saary et al. 2020). Nonetheless, the consistent recovery of a similar fraction of BUSCO genes across stramenopile genomes indicates comparable completeness. It is therefore striking that the sequenced Blastocystis strains showed substantial variability in genome size, from 14.0 to 32.2 mb. Strains in the same subtype exhibited similar lengths: ST3 isolates were 14.0 and 15.3 Mb, and ST1 isolates were 17.2 and 16.0 Mb in size. In contrast, the genomes of the three tortoise isolates were ∼1.5 to 2 times larger than human isolates, ranging from 26.0 Mb to 33.3 Mb.
Genome assembly statistics for all genomes used in this study
Previous genome sequencing studies have raised questions about the ploidy of Blastocystis and found evidence of segmentally duplicated blocks of genes. With the highly contiguous genomes and multiple isolates of individual subtypes here, we investigated questions of ploidy and conservation of synteny both within Blastocystis subtypes and across the genus. k-mer frequency-based analysis using GenomeScope (Ranallo-Benavidez et al. 2020) demonstrated a strong diploid signal in all Blastocystis isolates, with limited evidence for aneuploidy or polyploidy. We found that strains within ST3 and within ST1 were highly syntenic (Supplemental Fig. S1). However, synteny between subtypes was much lower, broadly following the relatedness between strains based on 18S sequences.
We used a hybrid method of de novo gene prediction and RNA transcript evidence to annotate Blastocystis genes (see Methods, “Genome assembly and decontamination”). Because a substantial fraction of genes in Blastocystis ST1 and ST7 do not have DNA-encoded stop codons (Klimeš et al. 2014; Gentekaki et al. 2017) (discussed below), open reading frame-based gene predictions from DNA sequence are inaccurate. Leveraging our RNA sequencing data, we were able to generate a comprehensive gene annotation for each Blastocystis genome that was able to accurately call genes lacking stop codons (see Methods, “Gene annotation”). Similar approaches were taken for one previously sequenced ST1 genome (Gentekaki et al. 2017), but published Blastocystis genomes for ST4 and ST7 were not annotated with these approaches, leading to incorrect gene annotations. For the Blastocystis strains sequenced in this study, the number of genes per Blastocystis genome sequenced in this work ranged from 6154 to 8547 (Table 1). Protein amino acid identity (AAI) across 753 single-copy orthologs was almost identical between strains of the same subtype (99%–100% AAI for both ST1 and ST3) and decreased to 65%–75% between human Blastocystis isolates from different subtypes and to 40%–42% between human and tortoise isolates (Supplemental Fig. S2). Tortoise isolates had lower pairwise AAI than human isolates, with the most distantly related strains in the tortoise-associated clade having a protein identity of 56% AAI for the same 753 orthologs, suggesting substantial genetic distance within this group.
Genomic diversity within Blastocystis
When we compared sequenced Blastocystis genomes, we found substantial variation in the genomes between human Blastocystis isolates and the tortoise-associated clade, including genome size. Whereas the tortoise Blastocystis isolate genomes are ∼1.5×−2.4× larger than the human isolate genomes, this difference is not reflected in protein-coding gene content, suggesting that non-genic regions are driving the difference in genome size. We sought to determine why the genomes of Blastocystis isolates in the tortoise-associated clade were larger than the genomes of the Blastocystis human isolates.
We found little difference in transposable element presence across Blastocystis, which ranges from 4.2% to 6.7% of the genome in the human isolates and 5.6% to 7.5% of the genome in the tortoise isolates. However, we did observe an increase in low-complexity regions in the tortoise isolates, comprising 0.02%–1.4% of the human isolate genomes and 11.8%–12.7% of the tortoise isolate genomes (Supplemental Table S3). This increase in low complexity regions does not explain the difference in genome size; if these regions are removed, the tortoise isolates remain 1.3×–2.1× larger than the human isolates. These low-complexity regions may be related to the overall lower GC content of the tortoise isolates, which ranges from 21% to 23% in tortoise isolates to 39% to 55% in human isolates (Table 1).
As repetitive elements did not explain differences in genome size, we investigated differences in other genomic features. We found that the almost all introns in human ST1, ST3, and ST4 Blastocystis are exactly 30 bp long, in agreement with previous estimates of intron size in Blastocystis ST1 and ST7 (Denoeud et al. 2011; Gentekaki et al. 2017). However, introns in the tortoise isolates are longer and more variable in length (Fig. 2A). We additionally found that the intergenic spaces between genes are much longer in the tortoise Blastocystis isolate than in human isolates (Fig. 2B). The median intergenic length in the tortoise isolates ranges from 645 to 719 bp, whereas the median intergenic length in the human isolates ranges from 137 to 186 bp. Intergenic lengths in the human isolates can frequently be extremely small, only spanning tens of base pairs (Fig. 2B). Together, the total amount of DNA spanned by introns and intergenic regions is 13.9–16.5 Mb in the tortoise Blastocystis isolates, whereas these regions span only 3.7–5.7 Mb in human Blastocystis isolates. Considered together, the larger introns and intergenic distance in the tortoise isolates explain the difference in their genome sizes relative to the human isolate.
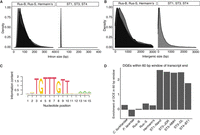
Genomic features differ across Blastocystis strains. (A) Intron lengths and (B) intergenic lengths in tortoise isolates (left) and human isolates (right). (C) Sequence motif enriched adjacent to transcript ends in ST1, ST3, and ST4. Motif generated with STREME (Bailey 2021). This motif is referred to as “downstream gene element” (DGE). (D) Enrichment (log[observed/expected]) of transcripts with downstream conserved element within 30 bp upstream of or downstream from transcript end, normalized by expected DGE occurrence given GC content.
Loss of stop codons is not universal in Blastocystis
In addition to genome size and gene density, we found surprising differences in how genes are encoded in the genome across Blastocystis. In Blastocystis ST1 and ST7, a fraction of protein coding genes (15% for ST7 and 26% for ST1) lack canonical DNA-encoded termination codons (Klimeš et al. 2014; Gentekaki et al. 2017). A proposed mechanism for translation of these genes is that the translational machinery instead uses a U or a UG sequence in mRNA, along with one or two adenines from the poly(A) tail, to generate UAA and UGA on-the-fly stop codons (Klimeš et al. 2014). Additionally, these genomes have a highly conserved motif (downstream gene element, or DGE) roughly 5 bp downstream from transcription stop sites of most genes. These sequences are hypothesized to direct 3′ mRNA cleavage and polyadenylation factors to the pre-mRNA (Klimeš et al. 2014; Gentekaki et al. 2017). In a scenario where the DGE is sufficient for 3′ mRNA cleavage, the loss of DNA-encoded stop codons is driven by relaxation of the selection pressure maintaining them.
We investigated all Blastocystis isolates for the DGE motif, finding enrichment downstream of protein coding genes in human-isolated Blastocystis strains but not downstream from tortoise-isolated Blastocystis strains or other stramenopiles (Fig. 2C,D). We additionally found no evidence of transcripts lacking stop codons in the tortoise-associated Blastocystis clade, suggesting that the unique transcriptional cleavage machinery described in Blastocystis ST1, ST4, and ST7 (Klimeš et al. 2014; Gentekaki et al. 2017) evolved after the divergence of the tortoise- and the human-associated clades sequenced here.
Gene losses related to cell morphology and signaling unite the Blastocystis genus
Despite the substantial variation in genome diversity for all Blastocystis strains sequenced here, the strains share the typical spherical, non-motile Blastocystis cell morphology lacking flagella. We find that all Blastocystis isolates are united by substantial gene loss related to this morphological change across multiple functional categories. All Blastocystis isolates have lost nearly all cilia- and flagella-related genes (Fig. 3; Supplemental Tables S4, S5). They have also undergone substantial reduction of dynein and kinesin microtubule motor proteins, retaining what is likely the minimal amount of motor proteins required for intracellular transport. This agrees with recent analyses showing reduction in these gene families in human-associated Blastocystis subtypes (Záhonová et al. 2023) and extends that observation by demonstrating that these losses unite the genus. In addition, we find that Blastocystis isolates have lost nucleotide cyclases and many genes related to signal transduction (Fig. 3; Supplemental Tables S4, S5). As cilia are major mediators of cell signaling that use cyclic nucleotides as intermediaries (Johnson and Leroux 2010), the loss of cilia in Blastocystis likely corresponds to the loss of nucleotide cyclases and reduction in signal transduction related genes. Thus, our sequencing of diverse Blastocystis strains provides strong evidence that the flagella-free morphology and associated gene losses evolved in the common ancestor of the Blastocystis genus.
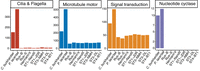
Gene families lost or reduced in all Blastocystis strains. See Supplemental Table S2 for GO categories and InterPro domains used for defining categories.
Lineage-specific genes reveal specialization within the Blastocystis genus
We find multiple differences in gene content in between Blastocystis isolates that likely reflect their ecological niche. All strains in the herbivorous tortoise-associated Blastocystis clade have a larger number of genes involved in plant carbohydrate degradation than the human-associated subtypes. We found five carbohydrate-degrading enzyme families that were either uniquely present in the tortoise isolates or in higher copy numbers, four of which are specific to degrading plant carbohydrates. This includes polygalacturonases, β-D-xylosidases, α-L- arabinofuranosidases, α-L-fucosidases, α-L-rhamnosidases, arabinogalactanases, and β-hexosaminidases (Fig. 4). Polygalacturonase is a pectinase that degrades plant cell walls; it was originally characterized in plants but is also present in phytopathogenic microbes and in bacterial members of ruminant microbiomes (De Lorenzo and Ferrari 2002; Deng et al. 2023). β-D-xylosidases and α-L- arabinofuranosidases are both critical enzymes for degrading xylan, a carbohydrate specific to plant hemicellulose (Yang et al. 2004; Lagaert et al. 2014). The GH154 arabinogalactanase gene family that is present only in the tortoise-associated clade was recently characterized in the gut bacteria Bacteroides thetaiotamicron as acting on plant-derived arabinogalactan glycans (Cartmell et al. 2018). Finally, α-L-rhamnosidases are active on a wide variety of polysaccharide residues produced by plants and bacteria (Guillotin et al. 2019). Together, the presence of multiple plant-specific carbohydrate-digesting enzymes in the herbivorous tortoise-associated clade strongly suggests that, like the bacterial microbiota, Blastocystis utilize dietary plant fiber as a carbon source and participate in the hindgut plant fiber fermentation that is characteristic of herbivorous tortoises (Stevens and Hume 1995).
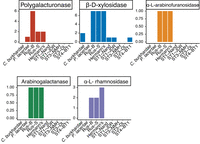
Clade-specific gene family expansions across Blastocystis, identified with InterProScan.
Ongoing horizontal gene transfer from bacteria in the Blastocystis genus
Horizontal gene transfer from bacteria has played an important role in Blastocystis evolution, particularly with respect to survival in an anaerobic environment (Eme et al. 2017). With high-quality genomes of Blastocystis isolates that span the genus, in combination with the genomes of other stramenopiles, we can estimate times of these transfer events. We re-analyzed HGT events into ST1 previously identified in Eme et al. (2017) and additionally screened all sequenced Blastocystis genomes for evidence of additional transferred genes (see Methods, “Gene contamination and horizontal gene transfer screens”).
Previous analysis of Blastocystis ST1, ST4, and ST7 genomes identified 74 horizontal transfer events in Blastocystis ST1 from bacteria (Eme et al. 2017). We were able to confidently identify orthologous genes for 66 of these genes (Supplemental Table S6), of which 29 genes had strong phylogenetic support for horizontal gene transfer from bacteria to eukaryotes, 17 genes lacked strong phylogenetic support for horizontal gene transfer, and the remaining 20 genes were ambiguous. In most ambiguous cases, the Blastocystis genes were either sister to an entirely bacterial tree, there were too few orthologs in the NR database to construct a gene tree, or the placing of the Blastocystis sequences on the phylogeny lacked strong support.
We additionally screened all genomes sequenced for new cases of HGT. To minimize the impact of potential genome contamination, we limited this screen to genes that were expressed in our RNA-seq data, were shared in more than one Blastocystis genome, and were flanked by unambiguously eukaryotic sequence to (see Methods). We found 13 newly identified HGT cases, six which are unique to the tortoise-associated Blastocystis lineage (Fig. 5A).
Horizontal gene transfer across Blastocystis evolution. (A) Estimated time of transfer for 40 horizontally transferred genes in Blastocystis shared between more than one species or strain. These data include previously identified and validated cases originally identified in Eme et al. (2017) and newly identified transfer events. (B) Gene phylogeny of tortoise isolate alpha-rhamnosidase genes and 588 homologous proteins. Support values indicate 1000 replicates of SH-aLRT test and ultrafast bootstraps (UFboot). Branches with support values <95 for UFboot or <80 for SH-aLRT are collapsed.
Two of the six tortoise-unique transfers are glycoside hydrolase genes, alpha-L-rhamnosidase and arabinogalactanase, that are uniquely expanded in this lineage and not present in the human Blastocystis isolates. The remaining transfers are two GNAT family N-acetyltransferases, a cold-shock protein, and a SMI1/KNR4 family protein (Supplemental Table S7).
The alpha-L-rhamnosidase gene family cleaves alpha-L-rhamnose from a large diversity of molecules and is present across the tree of life. This gene is present in two copies in Rus-B and Rus-S and three copies in Hermann's and is not present in any human Blastocystis isolates or in P. lacertae. Two genes with the alpha-L-rhamnosidase Interpro domain were present in the C. burkhardae genome. We constructed a gene phylogeny of the C. burkhardae genes and the tortoise genes together with 588 homologous genes retrieved with a sequence search of the Blastocystis and C. burkhardae sequences against the clustered NCBI NR database (see Methods). The gene phylogeny was separated into two fully supported clades, one with the C. burkhardae sequences, eukaryotic sequences from protists and rotifers, and some bacterial sequences, and the other with bacterial sequences and the Blastocystis sequences (Fig. 5B). The tortoise sequences grouped together with sequences from gut isolates and gut metagenome-assembled genomes in the Clostridia bacterial class, which contains prevalent members of the animal gut microbiota. Thus, the Blastocystis tortoise lineage likely acquired this carbohydrate-degrading enzyme from Clostridia bacteria present in the gut microbiome, suggesting a gut-specific function.
The other horizontally transferred carbohydrate gene in the tortoise Blastocystis lineage is within the recently described GH154 glycoside hydrolase gene family, characterized in Bacteroides thetaiotamicron as acting on plant-derived arabinogalactan glycans (Cartmell et al. 2018). This gene family is present exclusively in the tortoise Blastocystis isolates and not found in any other Blastocystis isolates or any other stramenopile genomes. A sequence search for the tortoise Blastocystis genes against the clustered NCBI NR database did not return any eukaryotic hits. A gene phylogeny of all bacterial homologs and the tortoise Blastocystis genes shows the tortoise Blastocystis genes group together with bacteria in the Bacteroidota (syn. Bacteroidetes) phylum (Supplemental Fig. S3).
We did not find strong evidence that the other three carbohydrate gene families uniquely present or expanded in the tortoise Blastocystis lineage were horizontally acquired from bacteria. Homologs to the polygalacturonase gene were all eukaryotic. Although the evolutionary history of this gene is likely complex as most homologs were within Streptophyta, it was not horizontally acquired from bacteria. β-D-xylosidase and α-L- arabinofuranosidase both appeared to have complex evolutionary histories but without conclusive evidence of horizontal gene transfer. Specifically, several of the β-D-xylosidase genes in the tortoise lineages had no eukaryotic homologs, but a gene phylogeny of all β-D-xylosidase genes including the human isolates did not yield a well-supported branch of the tortoise genes nested within bacteria. Similarly, the α-L- arabinofuranosidase gene had both bacterial and eukaryotic homologs, but there was not strong support for the placement of the Blastocystis sequences on this phylogeny either with the eukaryotic sequences or with the bacterial sequences.
Together, these results indicate that horizontal gene transfer played a major role in all evolutionary transitions investigated in this work: at the evolutionary transition from free-living to gut symbiont, in the Blastocystis genus broadly, and is an ongoing method of genetic diversification in this organism.
Discussion
The stramenopile protist Blastocystis is the most prevalent protist in the human gut and is widespread throughout vertebrates. Here, we present contiguous and complete genomes of eight Blastocystis strains, five isolated from humans and three from herbivorous tortoises. Long-read sequencing coupled with metagenomic assembly techniques enabled us to assemble three of these genomes from xenic cultures containing microbes present in the original sample, which has been technically infeasible with standard short-read approaches. Comparative genomic analyses between these Blastocystis genomes alongside other closely related stramenopiles revealed evolutionary changes that the Blastocystis genus has undergone to colonize a wide variety of animal guts.
The herbivorous tortoise isolates of Blastocystis diverged substantially from the human isolated Blastocystis and have many genomic features not seen in the human isolates. We found substantial changes in genome size, with human isolates possessing significantly shorter introns and intergenic regions than the tortoise isolates and substantially lower GC content in tortoise isolates. The GC content of the tortoise isolates is substantially lower than the genomes of most sequenced stramenopiles; of the 299 stramenopiles with sequenced genomes deposited in the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genbank/), only three genomes have lower GC content.
It is not clear whether introns and intergenic regions have shrunk in size after the split of the tortoise-human isolates, or if they have lengthened along the tortoise lineage. Genomic streamlining is a frequently observed process in symbiotic and parasitic organisms (McCutcheon and Moran 2012; Leckenby and Hall 2015), and some parasitic eukaryotes have entirely or almost entirely lost introns (Wilihoeft et al. 2001; Wang et al. 2018). The phenomenon of intergenic and intron size shrinking in Blastocystis likely reflects this general process, but the unexpected differences between different host-associated Blastocystis isolates highlights that this is an ongoing evolutionary process that can proceed at different rates.
Despite these substantial differences in genomic streamlining, all Blastocystis isolates sequenced here have undergone loss of the same genes associated with the more complex flagellated cell morphology of other stramenopiles. Blastocystis isolates have lost nearly all cilia- and flagella-related proteins, substantially reduced the number of microtubule motor proteins, and have lost all nucleotide cyclases and many signal transduction-related genes. These changes unite all Blastocystis isolates, demonstrating shared biology across the genus despite large evolutionary distances and differences in genome structure between isolates.
We additionally find evidence consistent with host adaptation within Blastocystis. The Blastocystis strain isolated from herbivorous tortoises contained many carbohydrate-digesting enzymes not present in other Blastocystis isolates (Fig. 4). Two of these gene families were acquired via horizontal transfer from bacteria and, in one case, via horizontal transfer from bacteria that also colonize the animal gut. The presence of plant-specific carbohydrate-digesting enzymes suggests that, like some ciliates and many gut bacteria, Blastocystis may ferment plant carbohydrates, a critical element of energy homeostasis in predominantly plant-eating animals (Stevens and Hume 1995).
Although Blastocystis is widespread across vertebrates, there is limited evidence for co-phylogeny of Blastocystis subtypes with their host (Fig. 1). Instead, the Blastocystis phylogeny more closely resembles that of spore-forming gut bacteria (Moeller et al. 2016), with host switching as a common phenomenon. Because of the lack of strong co-phylogenetic signal, open questions remain about Blastocystis host association, including what the barriers to colonizing different hosts are and how multiple strains or subtypes (commonly termed “mixed infections”) are maintained, and will require a denser sampling of genomes across the Blastocystis tree of life. As human-associated Blastocystis subtypes are not monophyletic, understanding the biology of the protists that impact humans will require considering Blastocystis subtypes that are present in the guts of diverse vertebrate hosts and how these subtypes function in different environments. Finally, the vast differences seen in the phylogenetically distinct tortoise Blastocystis isolates sequenced here compared to previous sequenced human-associated Blastocystis isolates highlight that substantial genetic diversity in this genus has yet to be explored.
Five of the eight Blastocystis strains sequenced in this work were grown in the presence of other microbes. Despite the challenges of generating highly contiguous and contamination-free genomes from mixed culture metagenomic sequencing, we successfully combined advances in deep long-read DNA sequencing and metagenomic assembly techniques to acquire high-quality genomes from xenic cultures. To address the challenge of annotating genes in genomes where stop codons are not universal, we utilized RNA-sequencing and state-of-the-art annotation pipelines. These two technical accomplishments were essential for enabling robust comparative genomic analyses of Blastocystis and its closest sequenced relatives. The strategy established in this study can be extended in future work to the many eukaryotic microbes that cannot be cultured axenically. The resulting genomes will fill critical gaps in our understanding of eukaryotic microbes and the evolution of this domain of life.
Methods
Strain cultivation
Blastocystis strains NandII (ATCC 50177), JDR (ATCC 50587), BT1 (ATCC 50608), DL (ATCC 50626), and NMH (ATCC 50754) were obtained from the American Type Culture Collection. The JDR strain was labeled as subtype 3 based on rRNA analysis, but 18S rRNA gene sequencing and subsequent genome sequencing revealed, in fact, that this strain belongs to subtype 1. All strains were isolated from human sources. Cultures were incubated under anaerobic conditions at 37°C on biphasic egg slant media overlaid with Locke's solution supplemented with 25% heat-inactivated horse serum (ATCC Medium 1671). Media was pre-reduced for 24 h before use. ST1 and ST4 strains were maintained in axenic culture, while ST3 and tortoise isolates were grown in xenic culture alongside other microbes present in the original samples. These xenic culture strains proved resistant to axenization.
Blastocystis strains from tortoises were isolated from fresh fecal material that was processed within 8 h of sampling. Fecal material was resuspended in sterile PBS, strained using a 70-µm cell strainer, and centrifuged at 700g for 5 min at room temperature. The pellet was then washed three times with PBS before being resuspended in 80% Percoll. This was overlaid with 40% Percoll and centrifuged at 100g for 10 min at room temperature. Protists were removed from the top of the 40% Percoll layer, washed in PBS, and resuspended in pre-reduced growth medium.
DNA extraction and sequencing
High molecular weight DNA was isolated from Blastocystis cultures with a modified CTAB-based protocol (adapted from McLay 2017) that minimizes polysaccharide contamination. Blastocystis cells were collected from cultures by pelleting with centrifugation at 800g for 5 min and snap frozen at −80°C. Frozen pellets were gently resuspended in two volumes of a high salt CTAB lysis buffer (100 mM Tris pH 8, 1.2 M NaCl, 10 mM EDTA, 4% CTAB, 1% PVP-40), and 200 µg/mL RNase A and 200 µg/mL Proteinase K were added before incubation at 60°C for 90 min. One volume of phenol:chloroform:isoamyl alcohol (25:24:1) was added, and samples were mixed end over end for 15 min. Samples were centrifuged at 17,000g for 10 min, and the upper aqueous layer was recovered. Two additional extractions were performed with pure chloroform, recovering the upper aqueous layer each time. DNA was next precipitated with two volumes of a low salt CTAB precipitation buffer (100 mM Tris pH 8, 10 mM EDTA, 2% CTAB) by end over end rotation at 60°C for 2.5 h. DNA was pelleted by centrifugation at 17,000g for 20 min. DNA pellets were washed twice with 75% ethanol and resuspended in 10 mM Tris pH 8. Genomic DNA was further cleaned with 1.8 volumes of Ampure XP beads according to the manufacturer's protocol (Beckman Coulter). RNA was isolated from Blastocystis cultures with a TRIzol extraction following the manufacturer's protocols (Invitrogen).
Nanopore sequencing libraries for all human isolates and for the Rus-B strain were prepared using the Genomic DNA by Ligation protocol SQK-LSK109 (Oxford Nanopore Technologies) following the manufacturer's instructions, sequenced on a full FLO-MIN106D R9 flow cell on a MinION sequencer, and basecalled with Guppy version 5.0.16. Nanopore sequencing libraries for the Rus-S and Hermann's isolates were prepared using the SQK-LSK110 kit LSK109 (Oxford Nanopore Technologies), sequenced on a full R10.4.1 flow cell, and basecalled with Dorado version 0.5.3 (https://github.com/nanoporetech/dorado).
Short-read Illumina DNA and RNA libraries were prepared using the KAPA HyperPlus PCR-free and KAPA mRNA capture kits (Roche), respectively, following the manufacturer's instructions. RNA integrity and library fragment size distribution were quantified using a Bioanalyzer 2100 (Agilent). Illumina DNA and RNA libraries for all human Blastocystis isolates and for the Rus-S and Hermann's tortoise isolates were sequenced on an Illumina NextSeq 500 (Illumina), and libraries for the Rus-B tortoise isolate were sequenced on an Illumina MiSeq v3.
Genome assembly and decontamination
Basecalled nanopore reads were assembled using Flye version 2.9.2 with the ‐‐meta option enabled (Kolmogorov et al. 2019, 2020). Assembly polishing was performed using four rounds of nanopore-read polishing using racon version 1.4.3 (Vaser et al. 2017), three rounds of Illumina DNA polishing using PolyPolish version 0.5.0 (Wick and Holt 2022), and one round of short-read polishing using POLCA as implemented in MaSuRCA version 4.0.9 (Zimin et al. 2013).
For the ST4 strain BT1, Hi-C data were generated using a Phase Genomics Proximo Hi-C 2.0 kit, and the Phase Genomics Proximo Hi-C genome scaffolding platform was used to create chromosome-scale scaffolds from the hybrid assembled genome. The Hi-C scaffolding resulted only in modest improvements to the original assembly quality; the number of scaffolds reduced from 45 to 34, and the assembly N50 improved to 882 from 876.
We used three different methods to separate eukaryotic, Blastocystis-originating sequence from other microbial sequences for xenic and axenic assemblies. For xenic assemblies, sequences were binned using MetaBAT 2 version 2.17.60 as a binning algorithm, taxonomically annotated using MetaEuk taxtocontig, and additionally annotated as likely eukaryotic or prokaryotic using EukRep (West et al. 2018; Kang et al. 2019; Levy Karin et al. 2020). For binning, short DNA sequencing reads were aligned to the polished assembly using minimap2 version 2.26-r1175 and summarized using the utility jgi_summarize_bam_contig_depths (Li 2018). Metaeuk taxonomy prediction was performed by running version 1f7d7023bdac3d46964ca380bef1b2848685501e, metaeuk easy-predict with mmseqs Swiss-Prot database build and metaeuk taxtocontig with the options “‐‐majority 0.5 ‐‐tax-lineage 1 ‐‐lca-mode 2”. EukRep v0.6.6. was run in “balanced” mode.
Eukaryotic contigs were identified through a union of MetaEuk contigs annotated as eukaryotic, removing any not identified as eukaryotic by EukRep. We found that MetaEuk was not able to identify Blastocystis contigs reliably at lower taxonomic levels than the superkingdom Eukaryota, which we validated by running the pipeline on existing NCBI genomes. Instead, any contigs that were annotated as eukaryotic were considered as potentially Blastocystis in origin. We cross-referenced these eukaryotic contigs with metabat bins, finding bins corresponding to eukaryotic sequence. In two cases, eukaryotic sequences that appeared to be Blastocystis in origin were split across two metabat bins, and these bins were combined. We removed any sequences from these bins that were annotated by MetaEuk as non-eukaryotic. We verified that these final cleaned bins corresponded to Blastocystis by BLASTing the NCBI RefSeq Blastocystis coding gene sequences against these bins. In several cases, we found bins that were annotated as eukaryotic but did not have any hits to Blastocysis coding sequences. These bins were discarded as likely originating from non-Blastocystis eukaryotic organisms in the culture.
We found that contigs containing ribosomal RNA genes were often not binned together with protein-coding genes, likely due to their highly repetitive nature and differences in DNA coverage. We searched for contigs containing 18S rRNA genes by BLASTing 18S rRNA genes against the full assembly. Contigs with full length and high percent identity hits to any Blastocystis 18S genes that were also annotated as eukaryotic by MetaEuk were included in final assemblies.
For axenic assemblies, sequences were not binned with MetaBAT 2, but MetaEuk and EukRep were performed as described above to identify potential contaminating sequences. This resulted in two contigs being removed from the BT1 assembly and no contigs being removed from Nand or JDR assemblies.
Genome completeness was determined with sequenced-based and gene-based approaches. For the sequence-based approach, we applied a k-mer frequency-based approach with GenomeScope v2.0 (Ranallo-Benavidez et al. 2020). Gene-based completeness was determined using BUSCO v5.6.1 with the lineage data set eukaryote_odb10 in the genome mode (Manni et al. 2021). Synteny was plotted using odp (Schultz et al. 2023).
Annotating repetitive elements
Repetitive elements in Blastocystis were annotated with a combination of RepeatMasker and RepeatModeler using the Dfam database version 3.8 (Smit et al. 2013; Flynn et al. 2020; Storer et al. 2021). Repetitive elements were predicted from all genomes with RepeatModeler version 2.0.5 with LTRStruct enabled. The predicted transposable elements from all Blastocystis genomes that had a putative predicted annotation were combined, and repetitive elements were tabulated in each genome with a combination of the custom repeat library created with RepeatModeler and the Dfam v3.8 database with RepeatMasker 4.1.7-p1.
Gene annotation
Blastocystis genes occasionally do not encode stop codons, which confounds prediction via open reading frame and requires RNA-seq-based evidence (Klimeš et al. 2014; Gentekaki et al. 2017). Without consideration of lack of stop codons, common gene annotation errors include insertion of erroneous introns and combining adjacent genes. To accurately annotate the Blastocystis genomes, we created a pipeline that used a combination of de novo gene prediction with BRAKER3 and transcriptome assembly, combining all sets of gene models with Mikado (Venturini et al. 2018; Gabriel et al. 2024).
RNA-seq reads were aligned to assemblies with STAR version 2.7.11b (Dobin et al. 2013), with a minimum of three supporting reads required for each junction. Transcript assemblies were generated with StringTie version 2.2.3 (Pertea et al. 2015), which uses RNA-seq alignments against a genome to assemble transcripts. Because Blastocystis has very short intergenic regions, transcript assemblers can occasionally combine multiple transcripts into one. To address this, assembled transcripts were split if an exon had zero RNA-seq coverage. Assembled transcripts <100 bp long were discarded. To generate de novo gene predictions, braker version 3.0.8 was run with OrthoDB11 Eukaryota proteins as protein evidence and the aligned RNA-seq as transcript evidence (Kuznetsov et al. 2022; Gabriel et al. 2024). The Mikado transcript scoring file (available in code repository) was optimized to choose transcripts with strictly RNA-seq-supported introns but to prioritize genes containing stop codons so long as the intron boundaries were strictly supported.
Functional annotation and orthology inference
Proteins from Cafeteria burkhardae BVI (formerly Cafeteria roenbergensis BVI), and predicted proteins from Proteromonas lacertae were used for comparative genomics analyses (Hackl et al. 2020; Záhonová et al. 2023). The genome assembly, transcripts, and proteins for C. burkhardae were downloaded from GenBank under accession GCA_008330645.1. The genome sequence for Proteromonas lacertae was downloaded from GenBank under accession GCA_002245135.1. As no predicted proteins or RNA-seq data were available for P. lacertae, genes were predicted from this genome using the BRAKER3 pipeline, using OrthoDB11 Eukaryota protein sequences as evidence. Proteins from all Blastocystis assemblies, C. burkhardae, and P. lacertae were annotated using a combination of InterProScan version 5.66 (Jones et al. 2014), DeepLoc2 to predict protein localization (Thumuluri et al. 2022), and dbCAN version 4.0.0 to annotate carbohydrate-active enzymes (Zhang et al. 2018). Orthologous sequences were determined using OrthoFinder2 with diamond as the search algorithm and mcl as the clustering algorithm (Li et al. 2003; Buchfink et al. 2015; Emms and Kelly 2019).
Genes related to cilia and flagella, microtubule motor proteins, signal transduction, ion channels, and nucleotide cyclase were identified using either GO categories or Interpro domains corresponding to these categories and are listed in Supplemental Table S4. Carbohydrate-degrading enzymes were identified based on the gene annotation methods mentioned above; specifically, polygalacturonase enzymes were identified with the PANTHER domain PTHR33928, beta-D-xylosidase with the Interpro domain IPR044993, a-L-arabinofuranosidase with the Interpro domain IPR010720, a-L-rhamnosidase with the PANTHER domain PTHR34987, and the GH154 arabinogalactanase with the PANTHER domain PTHR35339.
Identifying motifs adjacent to transcript ends
To identify motifs enriched downstream from transcript ends, we extracted a 60-bp window upstream of and downstream from all annotated transcript ends in all Blastocystis genomes sequenced here, as well as P. lacertae and C. burkhardae. Motifs were identified separately in each genome using STREME version 5.5.7 (Bailey 2021). A motif similar to the one identified by Klimeš et al. (2014) was identified in both ST1, both ST3, and the ST4 human isolate, but no significant and informative motifs were identified in the tortoise-isolated Blastocystis isolates or in the P. lacertae or C. burkhardae genomes. We then combined the sequences from all human isolates and ran STREME on this combined data set to generate a sequence motif (Fig. 2).
To determine enrichment of this motif in all genomes, we searched all transcript ends for matches to a degenerate version of the core region of the sequence motif, TGTTGTT, allowing up to two mismatches. As this motif is inherently an AT-rich sequence, the probability of observing it by chance is a function of the GC content of the sequence that is searched. We calculated the expected probability of seeing the 7-bp motif in each genome given the transcript window GC content and the number of transcripts, and calculated the enrichment of this motif as the log of the observed motif counts divided by the background expectation value (Fig. 2).
Strain and species phylogenies
The Blastocystis genus phylogeny was constructed using 18S rRNA sequences deposited in GenBank (Supplemental Table S1), along with 18S rRNA sequences extracted from each Blastocystis assembly and from the Proteromonas lacertae genome as an outgroup. Sequences were aligned with MAFFT v7.505 (Katoh and Standley 2013) and a maximum likelihood phylogeny constructed using IQ-TREE version 2.3.5 (Minh et al. 2020) using with the GTR sequence model, 1000 ultrafast bootstrap and SH-aLRT replicates, and rooted on P. lacertae.
To construct a species phylogeny of all species and strains with genomic data used in this study, 13 conserved protein-coding genes identified with BUSCO version 5.6.1 as described above were extracted from Cafeteria burkhardae, Proteromonas lacertae, and all Blastocystis strains sequenced here. Proteins were aligned individually with MAFFT v7.505 and concatenated. Best-fit models for each partition were selected using a relaxed hierarchical clustering algorithm as implemented by ModelFinder in IQ-TREE version 2.3.5 (Chernomor et al. 2016; Kalyaanamoorthy et al. 2017; Minh et al. 2020); 1000 ultrafast bootstrap and SH-aLRT replicates were performed.
Gene contamination and horizontal gene transfer screens
Blastocystis genomes were screened for bacterial contamination and for possible horizontal gene transfer events using a modified version
of an alien index score (Gladyshev et al. 2008; Wisecaver et al. 2016). The alien index score compares the best bit score of a query sequence to a group of related taxa versus to all other unrelated
groups and normalizes this score by the maximum possible bit score of the query sequence. It is determined by the formula
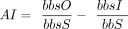 , where bbsO is the bit score of the best hit to a species outside of the group lineage, bbsI is the bit score of the best
hit to a species within the group lineage (skipping self hits), and bbsS is the bit score of the query aligned to itself.
The alien index was calculated for each Blastocystis protein by querying proteins against the NCBI NR database using DIAMOND version 2.1.6 (Buchfink et al. 2015) (see Data access). The self-group was designated as Blastocystis (taxid: 12967), and the group lineage was set as all eukaryotes (taxid: 2759).
, where bbsO is the bit score of the best hit to a species outside of the group lineage, bbsI is the bit score of the best
hit to a species within the group lineage (skipping self hits), and bbsS is the bit score of the query aligned to itself.
The alien index was calculated for each Blastocystis protein by querying proteins against the NCBI NR database using DIAMOND version 2.1.6 (Buchfink et al. 2015) (see Data access). The self-group was designated as Blastocystis (taxid: 12967), and the group lineage was set as all eukaryotes (taxid: 2759).
As an additional control for whether our genomes had high levels of genome contamination, we determined what fraction of genes with high alien index scores were likely transcribed by Blastocystis. Our RNA-seq strategy involved poly(A) purification, which removes non-polyadenylated transcripts. As polyadenylation is unique to eukaryotes, this removes bacterial transcripts. We quantified gene expression using kallisto version v0.48.0 (Bray et al. 2016) and determined whether genes with alien index scores greater than 0.2 were expressed and therefore likely truly integrated into the Blastocystis genomes. The number of genes with alien index scores above 0.2 ranged from 59 to 81 genes across all sequenced genomes, and >85% were expressed in all genomes. For identifying novel cases of HGT, genes with alien index scores >0.2 that were expressed at TPM > 1 and had orthologs in one or more other Blastocystis genomes were further examined for evidence of horizontal gene transfer with gene tree reconstruction. Requiring alien index scores >0.2 enhances the confidence that the identified genes originated in other lineages, although we acknowledge that this is a conservative cutoff and the increased confidence may come at the expense of potentially missing some cases of HGT. We did not use this cutoff for analysis of previously identified genes of HGT and instead performed phylogenetic reconstruction of all previously identified genes (Eme et al. 2017).
Gene tree reconstruction for horizontal transfer
Phylogenetic trees for putative horizontal gene transfer candidates were constructed from all diamond BLASTP hits using the NCBI NR database clustered at 90% sequence identity and 90% sequence length (Buchfink et al. 2015). Sequence hits that were outliers in length were excluded from downstream analyses. Protein sequences were aligned with MAFFT v7.505 (Katoh and Standley 2013), and maximum likelihood phylogenies were constructed using IQ-TREE version 2.3.5 using ModelFinder to determine the best-fit substitution model, and with 1000 ultrafast bootstrap replicates and 1000 replicates of the SH-like approximate likelihood ratio test (Guindon et al. 2010). Branches with SH-aLRT support less than 70 and UF-boot less than 80 were collapsed and trees were midpoint rooted before being examined for horizontal transfer.
Data access
The genome assemblies and gene annotations generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession numbers PRJNA1084774, PRJNA1185195, PRJNA1185180, PRJNA1084796, PRJNA1084798, PRJNA1084795, PRJNA1084799, and PRJNA1084775, and are also available as Supplemental Material. The RNA sequencing generated for Blastocystis strains has been submitted to the NCBI BioProject database under accession number PRJNA1041441. All alignments and phylogenies generated in this work, along with gene annotations for the Proteromonas lacertae gene annotations, are available as Supplemental Material and at Figshare (https://figshare.com/projects/Blastocystis_genomes/186136). Code required to calculate the alien index is available at GitHub (https://github.com/allind/alienindex) and as Supplemental Code 1. All other code required to reproduce this work is available at GitHub (https://github.com/allind/blastannotate) and also as Supplemental Code 2.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank members of the Pollard lab and the Bhatt lab for helpful discussions. We thank Maggie Brown and Southern Grace Reptiles for providing samples for Blastocystis isolation from tortoises. This work was supported by National Heart, Lung, and Blood Institute (NHLBI) grant #R01-HL160862, Chan Zuckberg Biohub SF, and Gladstone Institutes to K.S.P, by the Benioff Center for Microbiome Medicine Trainee Pilot Award, National Institute of Allergy and Infectious Diseases (NIAID) award K22AI173181-02, and Georgia Institute of Technology to A.L.L. A.S.B. is supported by National Institutes of Health grants R01AI143757 and R01AI148623, as well as a Stand Up 2 Cancer grant and a Paul Allen Distinguished Investigator Award.
Author contributions: Conceptualization: A.L.L., A.S.B., and K.S.P.; methodology: A.L.L., N.A.M., E.R.G., A.S.B., and K.S.P.; investigation: A.L.L. and K.S.P.; resources: A.S.B. and K.S.P.; writing (original draft): A.L.L., N.A.M., E.R.G., A.S.B., and K.S.P; writing (review and editing): A.L.L., N.A.M., E.R.G., A.S.B., and K.S.P; visualization: A.L.L.; supervision: K.S.P. and A.S.B.; funding acquisition: A.L.L., K.S.P., and A.S.B.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279080.124.
-
Freely available online through the Genome Research Open Access option.
- Received February 7, 2024.
- Accepted April 10, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








