
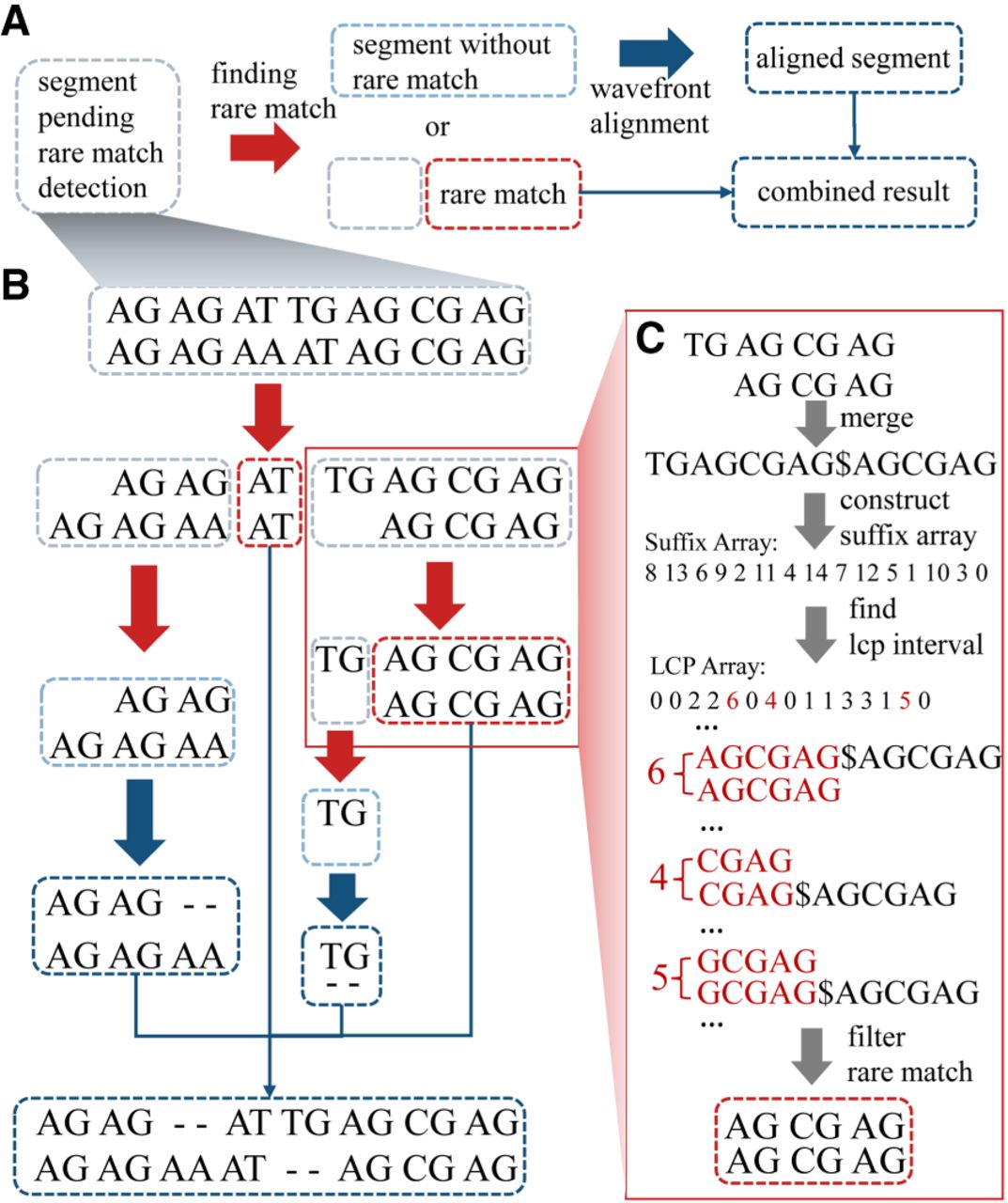
Workflow of RaMA. (A) Recursive logic of RaMA: light gray boxes indicate segments awaiting rare match detection. After the search (red arrow), two scenarios arise: If no rare match is found, the segment turns light blue, undergoes wavefront alignment (blue arrow), and becomes a dark blue aligned segment. If a rare match is identified, it acts as an anchor, splitting the segment while leaving the rest in search mode. The final alignment is formed by combining all detected rare matches and aligned segments. (B) Alignment case: sequences “AG AG AT TG AG CG AG” and “AG CG AG AT AG CG AG,” each containing “AG” as a tandem repeat unit, are aligned using “AT” as a rare match anchor. The sequence is divided; the left part, without further anchors, proceeds to alignment. The right continues to search, finding “AG CG AG” as another anchor. Final results are achieved by merging these segments. (C) Rare match detection: sequences are combined with “$” as a delimiter, then processed to build suffix and LCP arrays. LCP intervals of length 1 are identified, with the longest rare match selected from conflicting results as the final anchor.








