
The additional diagnostic yield of long-read sequencing in undiagnosed rare diseases
Abstract
Long-read sequencing (LRS) is a promising technology positioned to study the significant proportion of rare diseases (RDs) that remain undiagnosed as it addresses many of the limitations of short-read sequencing, detecting and clarifying additional disease-associated variants that may be missed by the current standard diagnostic workflow for RDs. Some key areas where additional diagnostic yields may be realized include: (1) detection and resolution of structural variants (SVs); (2) detection and characterization of tandem repeat expansions; (3) coverage of regions of high sequence similarity; (4) variant phasing; (5) the use of de novo genome assemblies for reference-based or graph genome variant detection; and (6) epigenetic and transcriptomic evaluations. Examples from over 50 studies support that the main areas of added diagnostic yield currently lie in SV detection and characterization, repeat expansion assessment, and phasing (with or without DNA methylation information). Several emerging studies applying LRS in cohorts of undiagnosed RDs also demonstrate that LRS can boost diagnostic yields following negative standard-of-care clinical testing and provide an added yield of 7%–17% following negative short-read genome sequencing. With this evidence of improved diagnostic yield, we discuss the incorporation of LRS into the diagnostic care pathway for undiagnosed RDs, including current challenges and considerations, with the ultimate goal of ending the diagnostic odyssey for countless individuals with RDs.
Rare diseases (RDs) encompass a diverse group of disorders that are individually rare in the population yet represent a significant burden to global health. RDs are conditions that affect fewer than 1 in 2500 individuals (Ferreira 2019), however, with several thousand recognized RDs, they collectively affect up to 1.5%–6.2% of the population globally (Ferreira 2019; Nguengang Wakap et al. 2020). About 70% of RDs are childhood-onset (Nguengang Wakap et al. 2020) and up to 65% are associated with a reduced life span, with about a quarter being potentially life-limiting by 5 years of age (Ferreira 2019). It is estimated that ∼70% of RDs have an underlying genetic etiology (Nguengang Wakap et al. 2020). Currently, the Online Mendelian Inheritance in Man database reports over 6400 phenotypes for which a molecular basis is known and over 4500 genes with phenotype-causing variants (https://www.omim.org/statistics/geneMap), with more disease-gene associations continually discovered. With such large numbers, rarity, and heterogeneity between and within RDs, molecular tools for diagnoses have been of utmost importance. Identifying the molecular cause of RDs provides affected individuals and their families with improved access to support services and potential treatments, information on prognosis and management of the disorder, options for further testing for family planning, and ends often long diagnostic odysseys. Despite the clear importance of reaching a diagnosis, more than half of RDs may remain undiagnosed following standard-of-care clinical genetic testing (Shashi et al. 2014).
Current genetic diagnostic workflows for RDs incorporate the patient's clinical presentation and the suspected mechanism of their rare genetic disease and may use various techniques to achieve a molecular diagnosis (Conlin et al. 2022; Kernohan and Boycott 2024). Over the last decade, short-read sequencing (SRS) of fragments of DNA 50–300 bp has been increasingly used in clinical settings for RD diagnosis, providing sequencing of targeted regions, the protein-coding exome (SR-ES), or nearly the entire genome (SR-GS). SRS enables a high-throughput method to accurately assess sequence variants (single nucleotide variants [SNVs] or insertions/deletions <50 bp), with the added ability to detect certain copy number variants and some structural variants (SVs, genomic alterations >50 bp in size). Since its more widespread adoption, genome-wide SRS, primarily SR-ES, has emerged as an effective first-tier test for indications such as neurodevelopmental disorders (NDDs) or multiple congenital anomalies (Srivastava et al. 2019; Manickam et al. 2021), with a diagnostic yield of ∼30%–35% depending on the indication and previous testing history (Clark et al. 2018; Splinter et al. 2018; Shickh et al. 2021; Chung et al. 2023; Hartley et al. 2024). While this has been a transformative technology, this still leaves nearly two-thirds of patients undiagnosed following SR-ES. Expanding beyond the protein-coding portion of the genome using SR-GS improves the assessment of noncoding sequence variants and detection of copy number variants and SVs; however, to date, the evidence for incremental diagnostic yield over SR-ES has been limited and may only be up to 10% (Ewans et al. 2022).
Several factors may contribute to the high rate of undiagnosed RDs following genome-wide SRS, including interpretation challenges for variants of uncertain significance (VUSs), variants residing in novel disease genes, and complex genetic and/or environmental causes of disease. Additionally, inherent limitations to SRS technology may be a significant contributor. Given the nature of the short reads, SRS struggles in alignment of nonunique sequences such as regions of the genome that are highly repetitive or have high sequence similarity, and in the detection and characterization of SVs (Fig. 1A–C). The short reads are also difficult to use to piece together haplotypes or generate de novo assemblies, which make read-based variant phasing a challenge and limit the reconstruction of complex genomic rearrangements (CGRs) or use of reference-free methods for variant discovery (Fig. 1D,E). Additionally, the polymerase chain reaction (PCR) amplification step in SR-ES can introduce biases and have trouble amplifying regions that are GC-rich. SRS also does not allow for concurrent detection of modifications to the native DNA strand, such as methylation (Fig. 1F). Overall, these limitations may leave many disease-causing variants undiscoverable or uninterpretable in individuals with undiagnosed RDs following SRS. To address many of these limitations, long-read sequencing (LRS) technologies were developed, enabling the genome-wide sequencing of native DNA fragments at multiple orders of magnitude larger than those in SR-GS, over 10 kb and up to megabases in size. In this mini-review, we review the existing evidence for LRS to increase diagnostic yields in undiagnosed RDs by highlighting variant types and situations in which LRS may be a particularly useful approach, emerging evidence from the application of LRS in cohorts of undiagnosed RDs, and considerations for its incorporation into clinical diagnostic pathways for RDs.
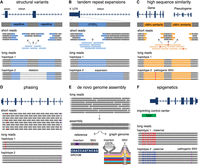
Summary of the utility of LRS over SRS in undiagnosed RDs. (A) LRS has an improved ability to detect SVs compared to SRS, especially in challenging regions for SRS such as repetitive DNA (blue), which often mediates the formation of SVs. (B) LRS enables improved sequencing and alignment of short tandem repeat sequences (blue) compared to SRS, enabling the accurate detection of tandem repeat expansions. Examples of genes with disease-associated short tandem repeat expansions include FMR1 (Fragile X syndrome), HTT (Huntington disease), and several genes associated with cerebellar ataxias (ATXN3, FGF14, etc.). (C) LRS allows for improved mapping and coverage of regions of high sequence similarity in the genome (orange), which are challenging for SRS. This enables the differentiation of sequences between genes and their pseudogenes, and therefore detection of variation in these challenging genes. Examples of disease-associated genes in regions of high sequence similarity include PKD1 (polycystic kidney disease), IKBKG (X-linked immunodeficiencies), and SMN1 (spinal muscular atrophy). (D) LRS enables haplotype phasing over long ranges, which is helpful to confirm compound heterozygosity of variants (red, purple) without the requirement of parental samples for segregation, or in scenarios where one or more variants are de novo. (E) Long reads derived from LRS can be used to build high quality and highly contiguous de novo genome assemblies without requiring alignment to a reference genome. These assemblies can either be compared to a linear reference genome (bottom left) to detect variants (a 4 bp insertion compared to the reference, purple box; and a C > T SNV, red) or they can be compared to de novo assemblies derived from other individuals (bottom right, colored lines indicate assemblies from different individuals) for the generation of graph-genomes that describe genetic variation among individuals (4 bp insertion seen only in one individual; T/C SNV that is more common in the group). (F) Sequencing of native DNA strands in LRS enables concomitant assessment of base modifications, such as differentiating between methylated cytosines (red) and unmethylated cytosines (blue) in cytosine-guanine dinucleotides (CpGs). This can be used in combination with phasing information to investigate imprinted loci that have parent-of-origin-specific DNA methylation patterns. Examples of disease-associated imprinted genes include H19/IGF2 (Silver–Russell syndrome), UBE3A (Angelman syndrome), and PLAGL1 (transient neonatal diabetes mellitus).
Long-read sequencing technologies
LRS technologies can include either “true” LRS or synthetic LRS. In true LRS technologies, long fragments of nucleic acid are directly sequenced. In synthetic methods, such as synthetic long reads (Peters et al. 2012; Li et al. 2015; Bankevich and Pevzner 2016) or linked-reads (Zheng et al. 2016; Marks et al. 2019; Wang et al. 2019; Chen et al. 2020), subfragments of long molecules of nucleic acids are co-barcoded to “link” or tag fragments from the same original long molecule. These short fragments are sequenced by SRS and then synthetically reconstructed into the original long fragment by bioinformatic methods. Synthetic LRS provides improvements over SRS in haplotype phasing, de novo genome assembly, and SV discovery (Bankevich and Pevzner 2016; Zheng et al. 2016; Elyanow et al. 2018; Chaisson et al. 2019; Marks et al. 2019; Wang et al. 2019). However, because the base sequencing unit is still a short read, some limitations of SRS may remain (e.g., poor coverage in low-complexity regions, poor SV reconstruction, GC-biases), and they have historically been inferior to true LRS (Chaisson et al. 2019; Ebbert et al. 2019). As a result, this has likely impacted their more limited application in the field of RDs compared to true LRS. Further development of synthetic LRS technologies is ongoing, with the recently released complete long-read sequencing (CLR) method by Illumina showing particular promise (Gorzynski et al. 2024); however, these are not the primary focus of this mini-review.
In terms of true LRS technologies, two platforms have dominated the market since their introduction: Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT). PacBio high-fidelity (HiFi) sequencing uses a sequencing-by-synthesis method. Double-stranded, high-molecular-weight DNA from a size-selected library of ∼15 kb is first circularized by ligating adaptors to the end of the fragments, then each circular DNA molecule undergoes multiple rounds of sequencing by a polymerase incorporating fluorescently labeled nucleotides to allow for real-time sequence determination. The output of these multiple sequencing passes (subreads) are merged to generate a consensus sequence, a HiFi read, with a very high per-base accuracy and typically between 10 and 30 kb in size (Wenger et al. 2019; Vollger et al. 2020). ONT uses nanopores embedded in electro-resistant membranes through which single-stranded DNA molecules are fed using a motor protein. The disruption of the electric current as bases pass through the nanopore is read in real-time and translated into base sequences using base-calling algorithms. ONT can theoretically be used to sequence fragments of DNA of any size depending on the sample input and library preparation; however, they are typically >10 kb. Library preparation kits for generating ultra-long reads >50 kb are available (https://store.nanoporetech.com/ultra-long-dna-sequencing-kit-v14.html), and studies have demonstrated abilities to generate long reads up to megabases in size (Logsdon et al. 2020). Although initial iterations of both technologies had lower per-base accuracies than common Illumina SRS platforms, these have improved substantially over the years to now be highly competitive with SRS (Logsdon et al. 2020; Damaraju et al. 2024; Kosugi and Terao 2024; Mahmoud et al. 2024). At ∼30× coverage, accuracies (F1 scores) can be up to 98.5%–99.9% for SNVs and 84.9%–99.4% for indels by PacBio HiFi LRS and up to 98.1%–99.7% for SNVs and 69.7%–84.1% for indels by ONT LRS, depending on the chemistry, variant caller, or benchmark sample used (Pei et al. 2021; Harvey et al. 2023; Kolesnikov et al. 2024). Performance for ONT has further improved recently with duplex sequencing using the latest R10.4 chemistry (Kolesnikov et al. 2024). Additional detailed descriptions and comparisons between the two technologies have been summarized elsewhere (Logsdon et al. 2020; Harvey et al. 2023; Mastrorosa et al. 2023; Oehler et al. 2023; van Dijk et al. 2023) and are beyond the scope of this mini-review.
Additional diagnostic yield provided by long-read sequencing
Detection and resolution of structural variants
SVs, genomic alterations >50 bp in size including insertions, deletions, duplications, inversions, translocations, and CGRs, account for the largest amount of sequence variation between individual genomes (Sudmant et al. 2015b; Chaisson et al. 2019) and are thus crucial to comprehensively assess in RD diagnosis. It has been long recognized that SVs contribute to genetic disorders (Stankiewicz and Lupski 2010); however, no single previous clinical genomic testing technology has been able to accurately assess SVs across the full spectrum of sizes and genomic context (Conlin et al. 2022; Kernohan and Boycott 2024). Despite the many available tools for SV detection from SR-GS (Kosugi et al. 2019), with significantly longer reads to better span SVs and resolve repetitive and nonunique regions that are enriched for SVs (Sudmant et al. 2015a), LRS consistently outperforms SR-GS in the detection of SVs in a wide range of sizes. In head-to-head comparisons, LRS can detect at least three to five times more SVs than SRS (Huddleston et al. 2017; Chaisson et al. 2019; Ebert et al. 2021). So far, evidence for gains in diagnostic yields by detection of SVs previously missed by SR-GS is primarily in CGRs or SVs involving challenging regions/sequences for SRS. For example, a CGR involving several breakpoints in Chromosomes 7 and 9 with an insertional translocation, inversion, and deletion was identified by PacBio LRS in a proband with a complex NDD that was previously missed by SR-GS analysis (Hiatt et al. 2021). Additionally, examples of insertions involving transposable elements, highly repetitive DNA sequences that can insert themselves into new places in the genome and potentially disrupt coding sequences, splicing, or gene regulation, have been reported. These include SINE-VNTR-Alu (SVA) element insertions in introns of disease genes SMARCB1 and NR5A1 (Sabatella et al. 2021; Del Gobbo et al. 2024) and a LINE-1-mediated insertion that resulted in a single-exon duplication of CDKL5 (Hiatt et al. 2021). There is also ample evidence of LRS successfully identifying disease-causing SVs such as deletions, insertions, inversions, or more complex rearrangements that were missed by previous standard-of-care genetic testing in undiagnosed RD patients (Mizuguchi et al. 2019, 2021; Xie et al. 2020; de la Morena-Barrio et al. 2022; Daida et al. 2023; Damián et al. 2023; Yanagi et al. 2023). As SR-GS was not performed in these examples, the added benefit of LRS over SR-GS was not demonstrated; however, they confirm the utility of LRS as a tool to detect disease-associated SVs following typical diagnostic testing.
Not only can LRS identify SVs that were missed by previous genomic testing in patients with undiagnosed RDs, but it can also clarify known SVs by fine-mapping breakpoints and/or revealing additional SV complexity. In eight individuals with previously identified CGRs, Miller et al. (2021) demonstrated that all rearrangements could be identified by targeted ONT LRS, and additional information about the CGR was gained for all individuals, including the precise resolution of breakpoints, determination of orientation of alterations, or identification of additional events in the CGRs that were not previously detected. This has the potential to boost diagnoses, as highlighted in an affected individual with a previously nondiagnostic balanced translocation t(8;18)(q22;q21) where ONT LRS revealed a chromothripsis-like CGR at the translocation site involving 19 rearranged fragments, including a deletion impacting disease-associated genes RAD21 and EXT1 that explained the patient's presentation (Lei et al. 2020). Additionally, in a study assessing SR-GS for the diagnosis of NDDs, LRS was necessary to fully resolve complex SVs in four patients, supporting its specific added yield over SRS (Sanchis-Juan et al. 2023). Additional examples of LRS revealing greater complexity of SVs and/or fine-mapping breakpoints to clarify and confirm the pathogenicity of variants highlight the utility of this added information to upgrade previously nondiagnostic variants and provide answers to individuals with undiagnosed RDs (Dutta et al. 2019; Schieffer et al. 2021; Sund et al. 2024). This may be particularly useful in reconstructing and mapping breakpoints of CGRs that occur in challenging regions such as segmental duplications or repetitive elements that commonly mediate complex SV formation (Schuy et al. 2022). This was recently demonstrated in a study by Grochowski et al. (2024), in which LRS aided to resolve and fine-map breakpoints of complex duplication–triplication/inverted-duplications associated with MECP2 duplication syndrome that are mediated by nearby inverted low-copy repeats.
Detection and characterization of tandem repeat expansions and contractions
Tandem repeat expansions or contractions are another key class of SV for which LRS can aid in the diagnosis and discovery of genetic causes of undiagnosed RDs. Tandem repeats are regions in the genome in which sequences of DNA are repeated in numerous copies next to one another, often distinguished as short tandem repeats (STRs), repeated motifs of 1–6 bp, or variable number tandem repeats (VNTRs) with repeated motifs of >7 bp. There are an estimated over 1.7 million tandem repeat loci in the human genome, together accounting for ∼8% of our genome (English et al. 2024). Tandem repeats are highly mutable and prone to expansion or contraction due to DNA replication errors. STR expansions in particular contribute to numerous RDs, especially adult-onset neurological disorders including several forms of spinocerebellar ataxia, Huntington disease, and amyotrophic lateral sclerosis (Chintalaphani et al. 2021; Depienne and Mandel 2021). Given their highly repetitive nature and sizes that are often orders of magnitude larger than can be captured in a single SRS read, STR expansions have been notoriously challenging to assess by SRS technologies. In clinical settings, these require targeted approaches including Southern blot or repeat-primed PCR for molecular diagnosis. LRS, therefore, holds promise for the accurate sizing and characterization of sequence composition and epigenetic modifications of STR expansions on a genome-wide scale.
Studies benchmarking LRS in patients with a variety of known pathogenic repeat expansions have consistently demonstrated that LRS can effectively recapitulate molecular diagnoses in a single comprehensive assay by identifying pathogenic expansions and providing information on the sequence composition and single base-resolved DNA methylation at STRs (Höijer et al. 2018; Giesselmann et al. 2019; Miyatake et al. 2022; Stevanovski et al. 2022; Erdmann et al. 2023; Dolzhenko et al. 2024). These provide important proof-of-principle for the ability of LRS to detect pathogenic expansions; however, few studies to date have systematically assessed the concordance of sizes of expansions (especially large expansions) detected by LRS compared to standard technologies. Stevanovski et al. (2022) reported an R2= 0.996 for lengths of normal and expanded repeats at the HTT locus, 0.993 for FMR1, and 0.946 for RFC1 using targeted ONT compared to repeat-primed PCR or Southern blot. A similar 100% concordance between repeat sizes from targeted PacBio LRS and PCR fragment analysis at the HTT locus was also observed in 11 patients with Huntington disease (Höijer et al. 2018). Using Cas9-targeted ONT at 10 STR loci associated with ataxia, Erdmann et al. (2023) reported that the majority of loci agreed with PCR fragment analysis within ±3 repeat units for unexpanded or short expansion (<100 bp) alleles in their method validation cohort, within the expected error rate for PCR fragment analysis. In 28 expansion-positive individuals from their cohort of patients with adult-onset ataxia, LRS-predicted sizes were within ±4 repeats of estimates from PCR or were within the range of sizes determined by PCR (Erdmann et al. 2023). In addition to sizing, the accurate assessment of the sequence composition of STRs by LRS can also be helpful. Sequence interruptions or noncanonical repeat motifs may alter the pathogenicity, severity, age of onset, or stability of an expanded repeat, and are, therefore, important for informing the diagnosis and prognosis of RDs caused by STR expansions (Rajan-Babu et al. 2024). This has been demonstrated in sequencing (CTG)n repeat expansions in DMPK associated with myotonic dystrophy type 1, where PacBio LRS enabled accurate detection of sizes, sequence interruptions, and somatic mosaicism in individuals with >1000 repeats, and the finding that CCG interruptions near the 3′ end of the STR are associated with increased somatic stability of the repeat and milder phenotypes (Cumming et al. 2018; Mangin et al. 2021). Additional studies of larger cohorts of patients with various known expansions are necessary to evaluate the accuracy of LRS in comparison to current diagnostic standards and to facilitate its incorporation as a comprehensive molecular tool for STR expansion disorders.
LRS has also been instrumental in the discovery of novel STR expansion disorders in undiagnosed RDs, contributing to the increase in discoveries over the past several years. Some early examples include the discovery of an intronic (TTTCA)n or (TTTTA)n expansion in SAMD12 associated with autosomal dominant (AD) benign familial adult myoclonic epilepsy (Ishiura et al. 2018; Zeng et al. 2019) and a (GGC)n expansion in the 5′ UTR of NOTCH2NLC associated with neuronal intranuclear inclusion disease (Sone et al. 2019; Tian et al. 2019). Since then, LRS has aided in the discovery or further characterization of several novel STR disorders, reviewed elsewhere (Chintalaphani et al. 2021; Depienne and Mandel 2021; Gall-Duncan et al. 2022). This includes recent discoveries of novel expansions in ZFX3 and THAP11 associated with AD forms of spinocerebellar ataxia (Tan et al. 2023; Chen et al. 2024c), and a 27 bp duplication in a polyalanine tract in HOXD13 associated with synpolydactyly 1 (Melas et al. 2022). Additionally, LRS was also necessary to fully characterize the size and sequence composition of the recently discovered intronic (GAA)n expansion in FGF14 associated with AD late-onset spinocerebellar ataxia type 27B (Pellerin et al. 2023; Rafehi et al. 2023). This STR expansion has since proven to account for a considerable proportion of undiagnosed patients with ataxia, particularly in individuals of European descent and especially in French Canadians (Hengel et al. 2023; Novis et al. 2023; Pellerin et al. 2023; Rafehi et al. 2023; Méreaux et al. 2024). The ability to accurately sequence the expanded repeats >1000 bp supported that only pure (GAA)n expansions are associated with disease, as large expansions in unaffected individuals were found to contain different GA-rich motifs than the expanded (GAA)n motif observed in all affected individuals (Pellerin et al. 2023). Given the limited availability of STR genotype data from population cohorts sequenced by LRS and tools for analysis, the discovery of novel STR expansions from genome-wide LRS is challenging. As such, novel STR expansion discoveries have been powered by large pedigrees, often guided by linkage studies identifying candidate regions for targeted sequencing or analysis of LRS data. With recent improvements in tools for STR genotyping and discovery and a growing number of population cohorts, genome-wide analyses will soon be more feasible and will help to power additional discoveries of novel STR expansions underlying undiagnosed RDs.
Variant discovery in regions of high sequence similarity
Genomic regions with high sequence similarity are another problem area for SRS technologies as the difficulty to uniquely map short reads between two or more highly similar regions contributes to regions with poor or no sequencing coverage in SRS. This is relevant for undiagnosed RDs because many disease-associated genes reside in such regions, having either one or more pseudogenes (genomic regions with high sequence similarity to known genes that do not generate functional protein products), high sequence similarity to other functional genes, or multiple regions within the gene itself that are highly similar (Mandelker et al. 2016). Examples include PKD1, HYDIN, IKBKG, and SMN1, all of which have historically required multiple targeted molecular technologies to comprehensively assess disease-associated variation. In a study published in 2019, Ebbert et al. identified 36,794 regions in gene bodies, including 2855 in coding sequences, that they termed as SR-GS “dark regions”: regions with either no/low sequencing coverage or low mapping quality of sequencing reads because of difficulties in adequately assembling or aligning SR-GS reads. Using earlier iterations of LRS technologies, they demonstrated that PacBio and ONT LRS significantly improved coverage in 88% and 95% of the regions in coding sequences, respectively (Ebbert et al. 2019). Furthermore, Wenger et al. (2019) found that 152/193 (79%) of medically relevant genes with at least one exon in an SRS dark region were fully mappable using PacBio CCS. Recently, Sanford Kobayashi et al. (2022) found that PacBio HiFi LRS successfully covered 98% of annotated SRS dark regions genome-wide in their cohort of 30 participants. This facilitated the identification of a pathogenic variant in IKBKG in a participant with a previously undiagnosed immunological disorder, demonstrating the successful use of LRS to uncover new diagnoses in these challenging regions for SRS (Sanford Kobayashi et al. 2022). Additional studies of specific genes also support this utility. Borràs et al. (2017) found that targeted PacBio LRS of PKD1 and PKD2 in a cohort of patients with AD polycystic kidney disease identified all previously known pathogenic variants with high sensitivity and specificity, and also identified additional variants that were missed by previous testing. Additionally, Fleming et al. (2024) demonstrated that ONT LRS with a modified SRS bioinformatic pipeline aided in differentiating variation in HYDIN from its pseudogene HYDIN2 and supported disease-associated variant discovery in a cohort of patients with primary ciliary dyskinesia. Recently, Chen et al. (2023) developed a tool, Paraphase, that accurately differentiates full-length haplotypes in SMN1 and its paralog SMN2 to facilitate variant discovery and diagnosis from PacBio LRS data. This tool can now be applied to 160 long (>10 kb) segmental duplication regions with >99% sequence similarity, encompassing 316 genes, including 11 medically relevant genes (Chen et al. 2024b). With the human genome having over 6000 genes with SRS dark regions (Ebbert et al. 2019), it is likely that yet undiscovered pathogenic variation in these regions may underlie undiagnosed RDs, and LRS holds promise to bring them to light.
Improved read-based phasing
Variant phasing, in which variants are assigned to either the maternal or paternal chromosome, is often an important step in RD diagnostics. Particularly for individuals with compound heterozygous variants in genes associated with recessive disease, determining whether the variants are in cis (on the same parental chromosome) or in trans (on different parental chromosomes) is crucial for variant interpretation. Phasing directly from SRS reads requires variants to be near enough to one another to either both be captured within a single short-read or paired-end read or have nearby heterozygous variants that can act as proxies. Alternatively, phase can be determined for inherited variants when genotypes from both parents or other informative family members are available, by statistical methods using genotypes from large population data sets to infer phase (typically requiring genome-sequencing data), or by more laborious methodologies that physically separate chromosomes or selectively amplify one allele before sequencing. Statistical phasing methods can be applied to SRS data with good success; however, these methods typically have increased error rates for rare variants that are not commonly feasible in clinical RD diagnostics. Laboratories primarily rely on familial genotyping or direct read-based phasing if possible.
With significantly longer read lengths, LRS outperforms SRS in read-based phasing, increasing phase block N50s (largest haplotype block length such that 50% of all heterozygous sites are contained in haplotype blocks of equal or greater size) by at least 10-fold from ∼1 kb to over 100 kb for PacBio and up to megabases in size for ONT (Choi et al. 2018; Chaisson et al. 2019; Majidian and Sedlazeck 2020). LRS read-based phasing has further improved with the latest sequencing platforms and phasing tools. Using haplotype-aware variant calling with PEPPER-Margin-DeepVariant, highly accurate haplotype blocks with N50s of 0.24 Mb from 35× PacBio HiFi or 2–6 Mb from 25–75× ONT LRS have been achieved (Shafin et al. 2021). This method enabled up to 66% or 93% of annotated genes to be fully captured within a haplotype block using 35× PacBio HiFi or 75× ONT LRS, respectively (Shafin et al. 2021). Incorporating other variant types such as SVs or STRs in addition to small variants further improves phasing, as demonstrated by a new tool HiPhase which generated haplotype block N50s of 0.48 Mb and fully phased 88% of annotated genes from PacBio HiFi data (Holt et al. 2024). When informative family members are unavailable for testing, one or more variants are de novo, or distances between variants are too great than can be phased by standard clinical methodologies, LRS, therefore, has the potential to provide a resolution for RD diagnostics. Several examples of LRS successfully phasing compound heterozygous variants in genes associated with autosomal recessive (AR) disease have been reported. For example, a LINE-1 insertion in exon 7 was confirmed in trans with a maternally inherited coding variant in exon 36 of CC2D2A in two siblings with a clinical diagnosis of Joubert syndrome, even when paternal DNA was unavailable (Yanagi et al. 2023). The readily available phasing information from LRS also improves the analysis and interpretation of singleton data, especially when a candidate gene or region may be targeted for sequencing or for analysis. This was demonstrated by Miller et al. (2022), who used targeted ONT LRS to identify missing second variants in trans in 8 of 9 individuals with a clinical diagnosis of AR Werner syndrome.
De novo genome assembly and pangenome approaches for variant discovery
Another compelling benefit of LRS over SRS is the improved feasibility of generating de novo genome assemblies and using pangenome approaches for variant discovery. Standard reference-based variant detection methods rely on the alignment of sequencing reads to the reference genome and then using bioinformatic tools to identify variants compared to the reference. However, until the most recent CHM13-T2T reference genome (Nurk et al. 2022), previous reference genomes remained incomplete, leaving regions of unknown sequence (gaps) distributed throughout the genome which hinder read alignment and variant discovery (Schneider et al. 2017; Nurk et al. 2022). Additionally, these references are representative consensus calls from only a small number of human genomes, which can introduce biases in the calling of nonreference sequences (Miga and Wang 2021). LRS allows the generation of de novo genome assemblies, where individual genomes are assembled into long contiguous haplotype-resolved sequences directly from the long reads instead of first aligning these to a reference genome. Variant calling from LRS assemblies against the reference genome can improve the precision and recall of SVs and indels compared to standard reference-based variant detection (Ebert et al. 2021; Harvey et al. 2023). Alternatively, these assemblies can be further leveraged to discover variants without relying on the reference at all by comparing variation among multiple assembled genomes as a pangenome graph. This can enhance variant discovery, particularly for SVs (Liao et al. 2023). This approach has not yet been thoroughly explored in the RD space; however, a recent study demonstrated that the generation of a graph pangenome of 574 assemblies from a pediatric RD cohort and 94 control assemblies improved the reproducibility of SVs compared to standard reference-based approaches (Groza et al. 2024). It also improved the prioritization of rare, potentially disease-associated SVs, leading to the discovery of a novel diagnostic SV in KMT2E in a patient with a previously undiagnosed RD (Groza et al. 2024). Several limitations to pangenome approaches such as high computational burden and the need for additional tools tailored for variant discovery mean that the current utility of these methods in RD diagnostics is primarily in improved variant calling accuracy and genotyping, especially for complex SVs or variants in complex loci (Taylor et al. 2024). With the increasing use of LRS and continued developments in pangenome references and analytic tools (Liao et al. 2023; Taylor et al. 2024), this emerging field may prove useful to improve yields in undiagnosed RDs in the future.
Utility beyond the DNA sequence
Because LRS technologies directly sequence unamplified DNA, modifications to DNA bases are preserved and can be detected by both ONT and PacBio LRS either based on unique disruptions to the electrical current or alterations in polymerase kinetics, respectively (Flusberg et al. 2010; Rand et al. 2017). Thus, LRS enables the assessment of epigenetic modifications at a base pair resolution. This has been useful in delineating pathogenic mechanisms of newly discovered RDs such as demonstrating 5-methylcytosine (5mC) hypermethylation of expanded (CGG)n alleles in NOTCH2NLC associated with neuronal intranuclear inclusion disorder (Ishiura et al. 2019). It also has significant clinical utility by supporting molecular diagnoses for RDs in which 5mC is altered. This includes imprinting disorders, where disrupted parent-of-origin-specific DNA methylation patterns at imprinted loci may be detected with the use of haplotype-phased reads and 5mC information (Cheung et al. 2023; Yamada et al. 2023; Bækgaard et al. 2024), or STR expansion disorders such as Fragile X syndrome, Friedreich's ataxia, or myotonic dystrophy type 1 in which pathogenic expanded STRs are hypermethylated (Giesselmann et al. 2019; Stevanovski et al. 2022; Cheung et al. 2023; Erdmann et al. 2023; Dolzhenko et al. 2024). Methods and tools are also being developed for the assessment of genome-wide DNA methylation outliers to aid in improving yields in undiagnosed RDs. In a recent proof-of-principle study, Cheung et al. (2023) analyzed rare 5mC hypermethylation events in PacBio LRS data from a cohort of 276 individuals from 152 families with undiagnosed pediatric RDs and identified hypermethylation associated with a repeat expansion in DIP2B in a patient with global developmental delay. This added information on epigenetic modifications provided along with DNA sequence in LRS, therefore, has the potential to aid in identifying novel candidates and diagnoses in undiagnosed RDs.
Beyond DNA sequencing, the benefits of longer reads in LRS also extend to RNA sequencing (RNA-seq). RNA-seq can increase diagnostic yields in RDs through the assessment of gene expression alterations, splicing changes, and allele-specific expression related to disease-associated DNA variants (Cummings et al. 2017; Kremer et al. 2017; Frésard et al. 2019; Gonorazky et al. 2019; Lee et al. 2020; Murdock et al. 2021; Yépez et al. 2022). However, previous studies have relied on SRS, which has limited ability to reconstruct full-length mRNA transcripts, making it challenging to interpret the exact impact of splice-altering variants or fully resolve novel gene fusions. Long-read RNA-seq in contrast, enables full-length isoform sequencing and quantification. This has powered the discovery of thousands of novel transcript isoforms in human tissues (Glinos et al. 2022). It has also helped clarify the impact of splice-altering VUSs in undiagnosed RDs, such as a homozygous intronic c.600-31T>C in MFN2 that created five novel isoforms that all disrupted the reading frame and resulted in nonsense-mediated mRNA decay (Stergachis et al. 2023) and an intronic c.1079-23T>A in CLPB that created a novel isoform with a new splice site causing the insertion of 7 amino acids in the conserved P-loop of CLPB (Farrow et al. 2023). With the increasing interest in using RNA-seq as a second-tier test to clarify transcriptomic alterations of noncoding SNVs and SVs, it will be important to consider the added strengths of long-read RNA-seq to further boost diagnoses in the future.
Emerging evidence of diagnostic yields in cohorts of undiagnosed RDs
Much of the existing literature applying LRS in undiagnosed RDs to improve diagnostic yields is from individual case reports or proof-of-concept studies sequencing known positive controls, making it difficult to accurately assess the increased diagnostic yield provided by LRS. Increasingly, however, studies are emerging applying LRS to cohorts of individuals with undiagnosed RDs, providing some of the first demonstrations of the true potential added yield of LRS. The exact indications and previous genetic testing in each study are variable and therefore diagnostic yields vary accordingly. Several cohort studies have applied LRS directly following standard clinical testing using SRS gene panels or SR-ES. This includes a cohort of 34 families with various RDs of suspected AR inheritance that remained undiagnosed following SR-ES in which analysis of regions of homozygosity in PacBio LRS data identified diagnostic variants in 13 families, 8 of which (23.5% of total) were not detectable by SR-ES (AlAbdi et al. 2023). Additional yields associated with LRS reported in more specific RD cohorts undiagnosed following SRS panels or SR-ES are highly variable. These include 18% in a cohort of 11 families with antithrombin deficiency with previous negative analyses of SERPINC1 (de la Morena-Barrio et al. 2022), 44% in a cohort of nine families with muscular dystrophy (Bruels et al. 2022), 50% in a cohort of 26 patients with clinical diagnoses of tuberous sclerosis complex (Duan et al. 2024), and up to 100% in a small cohort of five families with undiagnosed hereditary spastic paraplegia (Fukuda et al. 2023). Finally, Miller et al. (2021) demonstrated a 60% yield of targeted ONT LRS at candidate genes to identify a missing pathogenic or likely pathogenic variant in a cohort of 10 individuals with various undiagnosed RDs for which either a single pathogenic variant in an AR disease gene, or no pathogenic variant for a specific suspected AD or X-linked disorder had been identified by previous clinical testing. Unfortunately, these studies do not allow us to assess the yield of LRS over a more comparable SRS technology, SR-GS. Indeed, some of the identified diagnostic variants could have been detected by SR-GS such as some larger deletions and splice-altering intronic SNVs. However, these studies nonetheless support an increased diagnostic yield of LRS following typical standard-of-care testing and may help in the future when determining where to place LRS in the diagnostic care pathway.
Only a few reported studies have used LRS in cohorts of undiagnosed RDs following negative SR-GS. These studies are crucial to determine the additional yield of LRS over currently available SRS technologies. Some of the earliest reports are from small cohorts (<10) of patients with undiagnosed NDDs. While Pauper et al. (2021) did not identify any disease-associated candidates in five probands with undiagnosed NDDs presenting with intellectual disability and other features following trio PacBio LRS, Hiatt et al. (2021) identified a likely diagnostic variant in 2 of 6 (33%) probands also using trio PacBio LRS, including a de novo CGR and a de novo LINE-1-mediated insertion, both impacting known disease genes. Notably, both studies only focused on assessing de novo variants, which commonly underlie NDDs (Sebat et al. 2007; Vissers et al. 2010). Further larger studies in heterogeneous cohorts of undiagnosed RDs have also supported incremental yields over SR-GS. In a cohort of 30 patients from 26 families with undiagnosed pediatric RDs, Sanford Kobayashi et al. (2022) found a modest increased yield by PacBio LRS using a singleton approach with one additional diagnosis that was missed by SR-GS, a known likely pathogenic hemizygous stop-loss variant in IKBKG. Another larger effort from the Genomic Answers for Kids project sequenced 256 affected participants with diverse pediatric disorders, many of which remained undiagnosed following SR-GS (Cohen et al. 2022). Although specific diagnostic yields were not reported, at least five examples of diagnoses made by LRS were provided as proof-of-principle for the technology, including previously unidentified pathogenic repeat expansions, a CGR, and the use of phasing of compound heterozygous variants to support diagnoses (Cohen et al. 2022).
Driven by increased throughput, decreased costs, and improved analysis capabilities of LRS, recent comprehensive genome-wide analyses of LRS data in larger cohorts of undiagnosed RDs have begun to shed more light on the increased diagnostic yields LRS may provide over SR-GS. Two studies of cohorts of ∼100 patients with various undiagnosed RDs suggest a specific increased yield of LRS over SR-GS between 7% and 17% (Hiatt et al. 2024; Steyaert et al. 2024). Building on their 2021 study (Hiatt et al. 2021), Hiatt et al. (2024) applied PacBio LRS in 96 probands with undiagnosed RDs presenting with NDD, multiple congenital anomalies, or a suspected congenital myopathy. They found new disease-relevant or potentially disease-associated variants in 16 probands, noting that 7 of these (7.3% of total) were exclusively identifiable by LRS (Hiatt et al. 2024). Additionally, in a cohort of 232 individuals from 93 families with undiagnosed RDs presenting with neurological, neuromuscular, or epilepsy phenotypes, Steyaert et al. (2024) identified 13 novel diagnoses (13%) and four additional compelling candidate disease-associated SVs (4.3%) using PacBio LRS. In addition to this undiagnosed cohort, they also studied a small cohort of 21 families with rare clinically recognizable unsolved syndromes (Aicardi, Hallermann–Streiff, Gomez–Lopez–Hernandez, Oculo-auriculo-vertebral spectrum disorders), but did not identify candidate genes or loci shared among affected individuals for any of these syndromes (Steyaert et al. 2024).
Together, evidence from these cohort studies is promising for a modest but significant increase in diagnostic yield from LRS over SRS technologies, ultimately providing diagnoses for families with RDs that would not have otherwise been possible. It is also worth noting that most of these studies have focused primarily on variants impacting known disease-associated loci, de novo variants, or large, exon-overlapping SVs. As analysis methods and control cohorts continue to grow and improve, these yields may further increase as the community is better able to harness the full potential of LRS in undiagnosed RDs.
Considerations for incorporating long-read sequencing in the RD diagnostic workflow
Although the surge in studies demonstrating the utility of LRS in undiagnosed RDs shows its promise to improve diagnostic yields, several factors from sample input through to analysis still limit the widespread incorporation of this new technology as part of the RD diagnostic workflow. Firstly, current library preparation procedures require relatively large amounts of high-quality, high-molecular-weight DNA to achieve long-read lengths for genome-wide LRS. This necessitates starting biological materials and DNA extraction protocols that preserve the integrity of these large DNA molecules and may be a limitation when minimal amounts of samples are available or when collection of blood or other invasive samples may not be possible. Secondly, the higher cost and lower throughput compared to SRS have been limitations to its widespread adoption. Although recent developments such as the PacBio Revio and ONT PromethION systems have made significant improvements, bringing material costs per 30× genome down to ∼$720–$1000 USD and sequencing up to 1300 (Revio) to nearly 5000 (PromethION) genomes per year (https://www.pacb.com/revio/; https://nanoporetech.com/products/sequence/promethion), this is still not nearly comparable to the latest developments in SRS technology such as Illumina's NovaSeq X Series which proposes to be able to generate more than 20,000 genomes per year at as low as $200 USD per sample in material costs (https://www.illumina.com/systems/sequencing-platforms/novaseq-x-plus/applications/transition.html). It is worth noting, however, that many additional factors that contribute to the cost of a clinical test (e.g., sample collection and handling, laboratory overhead costs, analysis and reporting) may be more similar between the two methods; therefore, this cost differential between SRS and LRS testing is likely less significant overall than suggested simply by the cost of sequencing. Thirdly, the historically higher costs and lower throughput of LRS also contributed to a limited availability of control data sets for variant allele frequency annotation. Given the greater sensitivity for the detection of SVs and coverage of challenging SRS regions by LRS, databases of allele frequencies derived from population cohorts also sequenced by LRS are essential for rare variant analyses in RDs. Unfortunately, the number of publicly available LRS genomes currently pales in comparison to those from SRS data sets such as gnomAD (Chen et al. 2024a). Promisingly, several recent efforts have begun to make headway. This includes CoLoRSdb (https://colorsdb.org/), a single resource of compiled data from over 1400 PacBio LRS genomes from several cohorts, including the Human Pangenome Reference Consortium (Liao et al. 2023), the Human Genome Structural Variant Consortium (Ebert et al. 2021), and RD cohorts such as the Genomic Answers for Kids project (Cohen et al. 2022). Additionally, a data set of 1019 samples from the 1000 Genomes Project sequenced at intermediate coverage (16.9×) by ONT LRS suitable for SV analysis (Schloissnig et al. 2024) and the first 100 samples sequenced at a minimum 30× depth from the 1000 Genomes Project ONT Sequencing Consortium were recently released (Gustafson et al. 2024). As more human genomes are sequenced by LRS, these allele frequency databases will continue to grow and improve. Indeed, the All of Us initiative recently performed a feasibility study, establishing a method for LRS using PacBio at scale for accurate small variant and SV discovery at the population-level in preparation for its planned population-scale LRS effort (Mahmoud et al. 2024). Data from the first 1000 participants are now available by registered access through their Researcher Workbench (https://www.researchallofus.org/). Finally, a fourth key factor still limiting the incorporation of LRS into RD diagnostic workflows centers around data analysis and infrastructure. While many tools for LRS data analysis exist to support initial base calling, alignment, assembly, phasing, variant calling, and more (Amarasinghe et al. 2020), best practices and standards for bioinformatic and analytical pipelines have yet to be established. These are necessary to ensure consistency of results among and within clinical laboratories. There is also a need to consider the increased infrastructure required for the computation and storage of this genome-wide data. All these limitations are active areas of development and have improved significantly even in the past few years, making incorporating LRS into clinical workflows more and more feasible soon.
As we consider incorporating LRS into clinical testing for RDs, we face a major consideration of where to place it in the RD diagnostic workflow. Until many of the aforementioned limitations have been addressed, the current utility of LRS clinically lies in its application as a second-tier test following nondiagnostic clinical genome-wide SRS (Fig. 2). Focusing LRS in undiagnosed RDs in situations where LRS is especially powerful over SRS may provide the highest yields. For example, in patients with missing second mutations for AR conditions, to clarify and fine-map SVs identified by karyotyping or microarray, when phasing would clarify molecular diagnoses, in undiagnosed RDs suspected to be caused by tandem repeat expansions (e.g., neurodegenerative conditions, AD inheritance, demonstrate anticipation), suspected imprinting disorders, or other RDs for which family history or phenotypic presentation supports a high likelihood of an underlying monogenic disorder (Fig. 2). However, given that LRS can identify nearly the full spectrum of variant types and can assess epigenetic modifications that historically have all required multiple different and often consecutive molecular diagnostic methods, it is also promising to implement as a first-tier test (Fig. 2). Using LRS near the beginning of the RD diagnostic workflow would streamline clinical genetic testing and improve access to comprehensive genetic testing for all individuals with RDs, ultimately reducing the diagnostic odyssey by identifying molecular diagnoses faster and for more individuals than current clinical standards (Conlin et al. 2022; Damaraju et al. 2024). When we also consider the collective costs and burden of multiple different sample collection and handling procedures, specific training required, analyses and reporting, and other overhead costs associated with each individual test in the current standard of cascade testing, using LRS as a single first-tier test could even prove more efficient and cost-effective in certain cases (Damaraju et al. 2024). This has not yet been thoroughly demonstrated, therefore, studies evaluating diagnostic yields, clinical utility, and economic analyses of LRS as a first-tier test compared to standard clinical testing workflows will be crucial. As a starting point, considering LRS as a first-tier test for scenarios where rapid and thorough diagnostics are needed or for presentations in which numerous step-wise and laborious clinical tests could be replaced by a single LRS test would aid in demonstrating this utility (Fig. 2). For example, LRS as a first-tier test in critically ill patients for rapid diagnostics, or as a single test to thoroughly assess all known disease-causing repeat expansions in individuals presenting with ataxia. Additionally, first-tier LRS is compelling for infants presenting with hypotonia, where current standards may include many different tests including karyotyping, microarray, and targeted assessments of several genes depending on clinical suspicion, including challenging genes SMN1 and SMN2 for spinal muscular atrophy, STR expansions in DMPK for myotonic dystrophy, and/or methylation and copy number testing at 15q11.2 for Prader–Willi syndrome (Fig. 2; Sharma et al. 2021). So far, studies have demonstrated that LRS is feasible as a first-tier test for comprehensive assessment of STRs associated with ataxia (Stevanovski et al. 2022) and to provide ultrarapid diagnoses in a pediatric critical care setting (Gorzynski et al. 2022; Zalusky et al. 2024). As LRS costs continue to decrease, throughput increases, and analyses mature, we anticipate a more rapid incorporation of this technology into diagnostic care pathways.
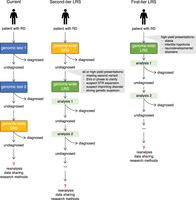
Hypothetical incorporation of LRS into the undiagnosed RD care pathway. (Left) The current care pathway in which patients with RDs may undergo long diagnostic odysseys, receiving numerous different consecutive genomic tests depending on indications, including genome-wide SRS. (Middle) Proposed incorporation of LRS as a second-tier test following nondiagnostic genome-wide SRS testing. Studies support an increased diagnostic yield when incorporating LRS following genome-wide SRS; therefore, this pathway has the potential to reduce the number of patients with undiagnosed RDs. (Right) Proposal for incorporating LRS as a first-tier test in the future, which would be primarily useful for RDs in which LRS first may be most cost-effective. This reduces the step-wise diagnostic pathway but allows for consecutive analyses of LRS data (e.g., coding variation, repeat expansions, SVs, methylation).
Conclusions and future prospects
A key approach to alleviating some of the global burden of RDs is to provide accurate diagnoses. These direct care, management, counseling, and access to resources for families, in addition to improving our understanding of the causes of RDs to improve targeted therapies and disease management. With about two-thirds of RDs remaining undiagnosed following genome-wide SRS, the need for additional strategies to find answers for these remaining families is of high importance. LRS is a key technology to incorporate into the undiagnosed RD care pathway to tackle this challenge. LRS has demonstrated the capability to identify challenging variants for SRS, resolve known VUSs, and provide additional supportive information such as epigenetic alterations to boost diagnostic yields in undiagnosed RDs. LRS is also an attractive technology to provide streamlined comprehensive genomic testing in the future that could improve timeliness to diagnoses and access to comprehensive genomic testing for more individuals with undiagnosed RDs. Additional studies of LRS that clearly define diagnostic utility will further solidify the need for this technology, and studies of outcomes and costs in comparison to standard-of-care testing and/or genome-wide SRS will aid in navigating where LRS fits best in the undiagnosed RD care pathway. Further developments to improve throughput, reduce costs, increase available control/population cohort data for allele frequency annotation, and standardize analysis methods will support the incorporation of this technology in clinical diagnostic laboratories. Given the significant advancements that have been made in the field of LRS to date, we believe the time for more widespread use of LRS to tackle the remaining undiagnosed RDs is here, and we are optimistic that this technology will help take the field closer to the ultimate goal of accurate and timely diagnoses for all RDs.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was performed under the Care4Rare Canada Consortium funded by Genome Canada and the Ontario Genomics Institute (OGI-147), the Canadian Institutes of Health Research, Ontario Research Fund, Genome Alberta, Genome British Columbia, Génome Québec, and Children's Hospital of Eastern Ontario Foundation. G.F.D.G. is supported by a CIHR Fellowship award (MFE-491710) and K.M.B. is supported by a CIHR Foundation Grant (FDN-154279) and a Tier 1 Canada Research Chair in Rare Disease Precision Health.
Footnotes
-
Article published online before print. Article and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279970.124.
-
Freely available online through the Genome Research Open Access option.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








