
A Hitchhiker's Guide to long-read genomic analysis
- 1Human Genome Sequencing Center, Baylor College of Medicine, Houston, Texas 77030, USA;
- 2Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas 77030, USA;
- 3Department of Computer Science, Rice University, Houston, Texas 77005, USA
-
↵4 These authors contributed equally to this work.
Abstract
Over the past decade, long-read sequencing has evolved into a pivotal technology for uncovering the hidden and complex regions of the genome. Significant cost efficiency, scalability, and accuracy advancements have driven this evolution. Concurrently, novel analytical methods have emerged to harness the full potential of long reads. These advancements have enabled milestones such as the first fully completed human genome, enhanced identification and understanding of complex genomic variants, and deeper insights into the interplay between epigenetics and genomic variation. This mini-review provides a comprehensive overview of the latest developments in long-read DNA sequencing analysis, encompassing reference-based and de novo assembly approaches. We explore the entire workflow, from initial data processing to variant calling and annotation, focusing on how these methods improve our ability to interpret a wide array of genomic variants. Additionally, we discuss the current challenges, limitations, and future directions in the field, offering a detailed examination of the state-of-the-art bioinformatics methods for long-read sequencing.
Short-read sequencing revolutionized genomics by providing a fast and cost-effective method for sequencing entire genomes, establishing it as a cornerstone of modern genomic research (Heather and Chain 2016; Foox et al. 2021). The emergence of long-read sequencing, producing reads of ∼10 kbp–4 Mbp, has enabled unprecedented insights into previously inaccessible genome regions, such as repetitive sequences (Sulovari et al. 2019; Nurk et al. 2022; Chaisson et al. 2023; Olson et al. 2023; Mahmoud et al. 2024b). In addition, long-read sequencing enabled the simultaneous assessment of genomic and epigenomic changes within complex regions (Logsdon et al. 2020; Mahmoud et al. 2021; Vollger et al. 2025). Nevertheless, long-read sequencing requires specialized analysis techniques to unlock its full potential, often requiring in-depth knowledge of rapidly evolving bioinformatic methods.
Two leading long-read sequencing technologies currently dominate the market and have significantly impacted the genomics field (Fig. 1A): Pacific Biosciences (PacBio) HiFi and Oxford Nanopore Technologies (ONT) sequencing. While both technologies produce continuous long reads, they present significant differences in accuracy, price point, read-length profiles, sample requirements, and the amount of DNA, where ONT requires less DNA than PacBio HiFi. DNA quality is crucial for the success of a long-read sequencing run (Oehler et al. 2023), and both PacBio and ONT have multiple extraction and preparation protocols for different organisms. The optimal choice of technology is contingent upon the specific application, with factors such as the complexity of variant calling or the desired level of assembly influencing the decision.
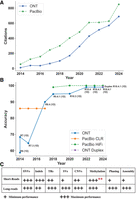
Long-read accuracy, citation trends over time, and comparison to short reads. (A) Citations of PacBio and Oxford Nanopore Technologies (ONT) long-read sequencing publications from 2014 to the present demonstrate their growing impact in the field. We collected citations from PubMed and excluded review articles. (B) This figure presents the evolution of long-read sequencing accuracy over time for ONT (Ashton et al. 2015; Goodwin et al. 2015; Laver et al. 2015; Suzuki et al. 2017; Ferguson et al. 2022; Ni et al. 2023b; Sanderson et al. 2024) and PacBio (Wenger et al. 2019; Amarasinghe et al. 2020; Logsdon et al. 2020; Oxford Nanopore Technologies 2020), illustrating their progress toward achieving >99% accuracy. For ONT, the analysis focuses exclusively on the 1D technology, with the 2014 R7 (1D) and the Duplex data points representing the median value, while the remaining points represent the mean values. We excluded ONT's 2D and 1D2 technologies because they ceased production in 2016. The plot distinguishes between PacBio's continuous long read (CLR) and high fidelity (HiFi) technologies. (C) Comparison between short reads and long reads in variant calling accuracy, methylation calling, and genome assembly (Oehler et al. 2023; Ni et al. 2023a; Dolzhenko et al. 2024; Espinosa et al. 2024; Kosugi and Terao 2024; Höps et al. 2025), where one plus represents the minimum performance and three pluses represent the maximum performance. ** Short reads required biochemical treatment and were used as the benchmark for methylation.
Initially, the adoption of long-read sequencing technologies suffered from both high error rates, at times up to 30%, and high costs, which have so far been significantly reduced over time (Fig. 1B). Currently, PacBio HiFi and ONT generate highly accurate long reads, exceeding 99% accuracy, expanding the applicability of long-read sequencing across diverse genomic studies (Wenger et al. 2019; Koren et al. 2024). Both platforms are capable of DNA and cDNA sequencing and detecting DNA methylation. At the same time, ONT offers additional functionalities such as adaptive sampling and direct RNA-seq (including epigenetic modifications). We recommend that readers interested in more details about the two platforms read the review by Logsdon et al. (2020).
Both PacBio and ONT excel in resolving repetitive elements and identifying complex genomic variants, including structural variants (SVs), which have historically posed challenges for short-read approaches (see Fig. 1C; Cameron et al. 2019; English et al. 2024b; Mahmoud et al. 2024b). SVs, defined as genomic alterations of 50 bp or more, encompass deletions, duplications, insertions, inversions, translocations, or a combination thereof (Escaramís et al. 2015; Collins et al. 2020). Furthermore, long-read utility expanded to include haplotype phasing, enabling the study of how genetic variants are inherited together. In addition, long-read sequencing has revolutionized methylation analysis, especially in repetitive regions. Long-read sequencing allows for the simultaneous determination of methylation levels and haplotypes (Gigante et al. 2019), a limited capability in short-read sequencing. This advancement facilitates the identification of differentially methylated regions and enhances our understanding of their potential impact on gene regulation and epigenetic mechanisms. Thus, long read enabled solving epigenetic-related diseases (Xie et al. 2021; Lucas and Novoa 2023), achieving a complete human genome assembly (Nurk et al. 2022), novel insights into repetitive regions (English et al. 2024b), accelerated diagnosis in various diseases (Gorzynski et al. 2022; Akagi et al. 2023; Lau et al. 2023), and comprehensive methylation maps (Gershman et al. 2022).
This mini-review delves into the analysis of long-read sequencing technologies, highlighting how these insights can be replicated and produced for samples of interest. We provide insight into some of the most prevalent long-read analysis approaches and assist in choosing individual analysis tools. This spans different aspects of analysis focusing on DNA, from reference-based mapping to de novo assembly. Although this mini-review mainly focuses on the human genome, many methods described here also apply to other species, while microbiomes have been recently reviewed elsewhere (Agustinho et al. 2024). For readers interested in long-read transcriptomics analyses, we refer them to other works (Calvo-Roitberg et al. 2024; Pardo-Palacios et al. 2024). We cover the alignment-based and de novo assembly-based variant detection, phasing, and annotation range. A comprehensive understanding of these analysis methods is crucial for unlocking the full potential of long-read sequencing in diverse research areas.
Considerations for long-read-based analysis
Long-read sequencing technologies, such as ONT and PacBio, generate raw electrical or optical signals (squiggle) that require specialized basecalling algorithms to convert into nucleotide sequences. ONT's basecaller is continuously updated, with Dorado being the latest development (Table 1). These updates improve basecalling accuracy, now approaching 99% (Fig. 1)—however, frequent updates could present challenges in clinical and multisample projects. Clinical workflows require reproducibility, consistency, and regulatory validation, making frequent revalidation necessary whenever the basecalling software changes. This can delay the integration of advancements into clinical pipelines and affect standardization across laboratories. PacBio's basecaller is integrated into the sequencing machine directly and is not publicly available. PacBio's accuracy is mainly driven by the generation of HiFi reads, where the DNA polymerase reads both forward and reverse strands of the same DNA molecule multiple times in a continuous loop, allowing the software to create a highly accurate consensus sequence from these multiple passes (Wenger et al. 2019). PacBio utilizes the circular consensus sequencing (CCS) method for PacBio platforms to collapse reads and improve their quality. In addition, Google DeepConsensus (Baid et al. 2023) is also available (Table 1). The choice between these approaches depends on the specific project requirements, such as the need for cutting-edge accuracy versus reproducibility and standardization. To ensure data quality, rigorous quality control (QC) is essential. Software like LongQC (Fukasawa et al. 2020) and NanoPack (De Coster et al. 2018) assess read length distribution, base quality, and other metrics, providing crucial insights for downstream analyses.
List of methods for long-read analysis and its function
Following sequencing with either PacBio or ONT, researchers are confronted with a critical decision: using reference-based mapping or de novo assembly (Fig. 2). Reference-based mapping relies on a high-quality reference genome, while de novo assembly often demands longer reads and/or higher coverage (Coster et al. 2021). The choice of either analysis approach often impacts the experimental design itself (Harvey et al. 2023). Other factors influencing this decision include computational resources, DNA quality and quantity, sequencing depth, and the ability of assembly-based approaches to improve existing reference genomes (e.g., for nonmodel organisms). For instance, if only a fragmented and incomplete genome is available as a reference or if the goal is to analyze, e.g., segmental duplications, then a de novo assembly approach might be preferred. Conversely, a reference-based mapping is often more appropriate for projects aiming to interpret and compare variants.
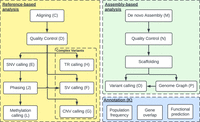
Schematic workflow for long-read-based genomic analysis. The workflow outlines the two approaches to analyzing long-read sequencing data. It details the different routes a researcher can take, either a reference-based approach or an assembly-based approach, and concludes with the types of annotations that can be generated. The letters in parentheses next to each step correspond to their detailed list of tools provided in Table 1.
Below, we discuss and provide insights and methods for the reference-guided alignment and de novo assembly approaches (Fig. 2). In addition, see Table 1 for a complete list of suggested tools for each analysis step.
Reference-guided analysis
Aligning reads to a reference genome (i.e., mapping) identifies the likely origin of a sequence read within the reference and helps identify sequence variations relative to a reference genome. These genomic variations encompass a spectrum of sizes, from single-nucleotide variants (SNVs) and short insertions or deletions (indels, <50 bp) to SVs (≥50 bp). Thus, identifying the proper reference genome for long-read alignment is critical as it might impact downstream analyses (Majidian et al. 2023). For example, while GRCh37 and GRCh38 provide more annotation, T2T-CHM13 (newest release v2.0) has been shown to reduce artifacts in the analysis (Aganezov et al. 2022), and a fixed GRCh38 reduces false positive calls in collapsed or falsely duplicated regions of the genome (Behera et al. 2023; Mahmoud et al. 2024a). Moreover, including alternative contigs within reference genomes has been shown to impact alignment precision negatively, necessitating careful consideration during the reference selection process (https://lh3.github.io/2017/11/13/which-human-reference-genome-to-use).
Interestingly, the increased read length and different error characteristics of long-read sequencing pose significant challenges for accurate alignment (Sedlazeck et al. 2018b). Novel computational approaches are continuously developed to improve the efficiency and accuracy of mapping long reads to reference genomes. Furthermore, mapping is an essential step since it is the basis of all subsequent analyses, and hence, an error in this stage will impact the overall results substantially. In the past, multiple aligners have been developed for long reads, such as BLASR (Chaisson and Tesler 2012) and NGMLR (Sedlazeck et al. 2018b), which have been so far overtaken by minimap2 (Table 1; Li 2018). Although minimap2 has become the most widely adopted aligner for long reads, it still struggles to map reads accurately within repetitive genomic regions (5%–10% of the human genome) and regions affected by rearrangements (e.g., inversions) (Ding et al. 2024). Newer aligners, including Winnowmap2 (Jain et al. 2022) and VACmap (Ding et al. 2024), demonstrated improved accuracy in challenging genomic regions compared to minimap2 (Table 1). It has been recently shown that the combination of aligners can achieve improvements over single aligners in speed and accuracy. Vulcan was developed by aligning reads quickly with minimap2 and then reprocessing the ones with abnormally high edit distances using different aligners (Fu et al. 2021).
Several proposed aligners utilize the raw base signal instead of relying on the base caller. For example, cwDTW (Han et al. 2018) employs a dynamic time-warping (DTW) algorithm to measure DNA similarity but is computationally intensive and sensitive to noise (Shih et al. 2022). While tools like Sigmap (Zhang et al. 2021) aim to address these challenges, their performance can suffer when aligning large genomes (Firtina et al. 2024), and the difficulty of processing large FAST5/POD5 files limits their broader adoption. The alignment process typically produces a tab-separated output file in SAM or compressed BAM formats, which includes detailed information (e.g., mapping location and alignment differences) on a per-alignment fragment basis (Li et al. 2009). It is important to note that a read can be aligned in one or multiple fragments (i.e., split reads), while each part of the read is typically aligned only once. Additional information, such as edit distance or methylation tags, is stored with the alignments.
After alignment, rigorous QC is essential to evaluate its performance. Metrics such as percentage of aligned reads, alignment identity, and average base quality are commonly investigated. These assessments are facilitated by tools like SAMtools (Danecek et al. 2021) and NanoPlot (De Coster and Rademakers 2023). In this context, SAMtools can be used to calculate alignment metrics such as the percentage of mapped reads, the presence of split-read alignments, or high numbers of soft-clipped bases. For a typical human genome realignment, one might expect >90% of reads to align successfully, with only a small proportion (e.g., ∼10%) showing split-read alignments. These metrics help identify potential issues, such as low-quality data or misalignments, ensuring reliable downstream analyses.
Identification of genetic and epigenetic alterations from long-read sequencing data
After an alignment has passed the quality assessment, variant calling is the next step in the analysis. Detecting genomic alterations fosters our understanding of genomic differences between individuals and thus may also give insights into diseases or other important phenotypes. In general, genomic alterations can be classified into four different groups based on their type and size:
-
i) SNVs and insertions/deletions (indels) are often defined as smaller than 50 bp. SNVs and indels are the most studied variant type in genetics studies due to their abundance in coding regions and clear relationship to protein changes, with ∼4–5 million variants expected per human genome (The 1000 Genomes Project Consortium 2015). Tools that detect SNVs and indels are broadly categorized into two major approaches: traditional statistical methods and machine learning-based techniques. An example of a statistical method is Longshot, which utilizes the Pair-Hidden Markov Model (Edge and Bansal 2019). While statistics-based tools offer faster runtimes, machine learning methods like DeepVariant (Poplin et al. 2018) and Clair (Zheng et al. 2022) are now preferred for both short- and long-read variant calling due to their higher accuracy. However, these machine learning models require careful selection as they are technology-specific, optimized for particular flow cell generations, and primarily trained on human genome data.
-
ii) Tandem repeats (TRs) consist of consecutive copies of DNA motifs, ranging from 1 bp or larger, with variable copy numbers. TRs span around 8% of the human genomes, range from homopolymers to large segmental duplications, and are highly variable between individuals. So far, over 60 diseases have been linked to TRs, with the majority of them being neurodegenerative (English et al. 2024b). The repetitiveness of the TR sequence often requires specialized methods to overcome alignment artifacts. To account for these, multiple methods have been introduced, such as the Gaussian mixed model (Ummat and Bashir 2014; Liu et al. 2017; Dolzhenko et al. 2024), deep learning (De Roeck et al. 2019; Giesselmann et al. 2019; Fang et al. 2022), and a network-based approach (Guo et al. 2018). In addition, specific methods such as StrSpy leverage TRs’ high variability between individuals for forensic applications by targeting only a small set of TRs (Hall et al. 2022). A significant challenge in TR detection lies in the inconsistent definition of repeat units across different tools. While some methods focus on shorter repeats, others target longer, more complex structures, hindering direct comparisons and meta-analyses. Recent tools from PacBio (TRGT [Dolzhenko et al. 2024]) and ONT (Medaka and pathSTR [De Coster et al. 2024]) have been improved and have shown great performance across the entire genome (English et al. 2024b).
-
iii) Structural variants (SVs) are genome alterations longer than 50 bp, typically numbering between 23,000 and 27,000 variants per healthy human genome (Mahmoud et al. 2019). SV encompasses deletions, duplications, insertions, inversions, and translocations, often occurring in repetitive regions. SVs and TRs are inherently interconnected, as many SVs involve alterations in TR regions, such as expansions or contractions of repeat units. Recognizing this overlap is crucial for a comprehensive understanding of genomic variation. By analyzing SVs and TRs together, we can capture both large-scale structural changes and finer-scale repeat dynamics, offering complementary insights into the genome's complexity (Jensen et al. 2024).
Various methods have been developed to call SVs from mapped reads, including Sniffles2 (Smolka et al. 2024), cuteSV (Jiang et al. 2020), and PBSV (see Table 1). These methods operate on the shared principle of detecting discordant mappings and inferring the SV type by consensus or localized assembly. Additionally, AI-based methods like SVision (Lin et al. 2022b), BreakNet (deletions only) (Luo et al. 2021), and MAMnet (Ding and Luo 2022) have been developed. Most SV callers perform similarly, so differences are often based on runtime or additional features. For example, SVision Pro can detect more complex SVs, while Sniffles2 allows rapid comparison of multiple samples. Of note, there are also cancer-specific variant callers that streamline the comparison of tumor and normal samples, such as NanomonSV (Shiraishi et al. 2023), SAVANA (Elrick et al. 2024), and Severus (Keskus et al. 2024). Additionally, using long reads, cohort analysis tools such as SVJedi (Romain and Lemaitre 2023) and Kanpig (Table 1; English et al. 2024a) were developed to genotype SVs.
-
iv) Copy number variants (CNVs) are typically large alterations that span multiple megabases. While smaller CNVs (50 bp–1 Mbp) are often also detected by SV callers, larger CNVs or even chromosome-size alterations are frequently missed. Due to the undefined boundaries between CNVs and SVs and the predominant focus on SVs within the long-read sequencing community, only a few CNV callers for long reads have been developed. The most prominent CNV callers are HiFiCNV (https://github.com/PacificBiosciences/HiFiCNV) for HiFi reads and Spectre (https://github.com/fritzsedlazeck/Spectre), which works on both ONT and HiFi to identify large CNVs (>100 kb). It is essential to highlight that neither tool reports precise breakpoints, making comparison to other variants typically challenging.
-
v) Epigenetic alterations can be detected directly from long-read sequencing data without prior biochemical alterations of the DNA (such as bisulfite sequencing). The presence of 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC, ONT only) at CpG sites is recorded during the basecalling process. Other modifications, such as N6-methyladenine (6mA), can also be detected using specific contexts or methods (Kong et al. 2023; Agustinho et al. 2024). Methylation has been linked to the regulation of promoters, playing a crucial role in tissue-specific gene expression and the regulation of oncogenes in cancer (Wang et al. 2022; Bhootra et al. 2023). Postprocessing of methylation based on raw reads is typically carried out using tools like Modkit and Jasmine for PacBio or the basecaller Dorado for ONT (see Table 1). Fiber-seq (Stergachis et al. 2020), which requires prior handling of methyltransferases to alter the sample, allows the detection of open chromatin to provide additional insights.
Long-read sequencing has been instrumental in recent breakthroughs in understanding complex genomic regions and epigenetic regulation. The Telomere-to-Telomere (T2T) consortium (Nurk et al. 2022) demonstrated the power of these technologies by resolving previously intractable regions of the human genome. In particular, long reads enabled the first complete characterization of human centromeres, revealing their complex satellite DNA organization and epigenetic states, and provided unprecedented resolution of segmental duplications, which had been resistant to accurate assembly using short reads due to their highly repetitive nature (Nurk et al. 2022). These advances have transformed our understanding of genome architecture, showing how segmental duplications contribute to evolutionary innovation and genomic diversity, while also illuminating the structural organization of centromeres and their role in chromosome segregation (Altemose et al. 2022a). This milestone was accompanied by innovative methodologies leveraging long-read capabilities for epigenetic profiling. Novel approaches such as NanoNOMe (Lee et al. 2020), DiMeLo-seq (Altemose et al. 2022b), and Fiber-seq (Stergachis et al. 2020) have further expanded our ability to profile DNA modifications and chromatin.
Another feature of long-read sequencing is its ability to enable the phasing of variants. Phasing refers to determining whether or not two or more variants co-occur on the same DNA molecule (i.e., haplotype). Long-read-based phasing can detect rare or de novo alleles that population-based phasing methods often miss. Additionally, phasing can reveal inheritance patterns and identify carriers of potentially disease-causing mutations. Phasing is typically conducted by analyzing if variants co-occur on a single read, which is then extended by overlapping reads and statistical clustering of variants, often assuming a diploid model and focusing on heterozygous variants. The primary focus is on SNVs since their frequency across the genome allows them to be phased within a read length. Phased SNVs are then reported in phase blocks (i.e., regions where phasing is consistent) and haplotypes (e.g., HP1, HP2).
There are two main methods for phasing: WhatsHap (Martin et al. 2023) and HapCUT2 (Edge et al. 2017). Other SNV-based phasing methods include LongPhase (Lin et al. 2022a) and HiPhase (Holt et al. 2024), which can incorporate SVs. A novel method, MethPhaser, extends the SNV-based phasing concept by leveraging haplotype-specific methylation signals, which span regions of homozygosity (Fu et al. 2024). Notably, many variant calling methods can leverage phasing information across different variant types (e.g., SNV and SV by Sniffles2). Additionally, when parental data is available, it can serve as a powerful validation tool to assess phasing accuracy by comparing the inferred haplotypes with the expected inheritance patterns. To enable SV phasing, we first need to phase SNVs and then tag the BAM file (e.g., using WhatsHap) before proceeding with additional SV calling.
Another common task after variant identification and phasing (optional) is the comparison of variants across samples. For SNVs, the best practice is to use a genomic variant call format file (gVCF), especially if more than one sample is studied. A gVCF file allows the merging of two or more samples and their variants into a fully genotyped VCF file. This avoids erroneous interpretations of variants whose genotypes are unknown per sample (i.e., ./.). gVCF is a particular type of VCF that contains records for every position or interval in the genome (e.g., read depth), regardless of whether it contains variants. GLnexus (Lin et al. 2018) or BCFtools (Danecek et al. 2021) merge gVCF files across samples. Tools like Sniffles2, Jasmine (Kirsche et al. 2023), and Truvari (English et al. 2024b) are commonly used for multiple sample SV comparisons. Sniffles2 relies on the binary file it creates for each sample to merge SVs. Jasmine uses a minimum spanning forest algorithm to merge SVs but does not call SVs. Truvari can merge and benchmark SVs, including a module named “phab,” which is specific for TR calling (English et al. 2024b).
Variant identification using long-read sequencing has significantly improved over the years and continues to evolve rapidly. This advancement often results in a more comprehensive understanding of the genome. However, annotations are typically required to infer their functional impact on various phenotypes, including diseases, to utilize these variants effectively.
Annotation of variants from long-read sequencing
The next crucial step is variant annotation, which has two primary goals: estimating the potential functional impact of variants and population frequency annotation. Variant annotation does not distinguish between variants identified from long or short reads. However, certain aspects need to be considered. Researchers utilize biological databases and annotation files to predict functional annotation of the identified variant calls. These resources provide crucial information about the variants’ locations within genes, their predicted effects on protein sequence and function, and any known disease associations. Tools such as ANNOVAR (Wang et al. 2010), SnpEff (Cingolani et al. 2012), and Ensembl Variant Effect Predictor (VEP; Hunt et al. 2022) are commonly used, enabling researchers to systematically annotate variants with known functional effects or potential impacts on genes and regulatory elements. ANNOVAR, in particular, queries many databases, including ClinVar (Landrum et al. 2014) for disease associations, dbSNP (Sherry et al. 1999) for known variants, and OMIM (Hamosh et al. 2005) for disease genes. Beyond empirical annotation, VEP also facilitates access to other in silico pathogenicity prediction tools such as PolyPhen-2 (Adzhubei et al. 2013), SIFT (Sim et al. 2012), and CADD (Kircher et al. 2014). These tools generate scores indicative of a variant's potential deleteriousness, ranging from benign to probably damaging. However, their predictions require cautious interpretation as protein context and individual genetics can influence the actual effects.
While functional annotation is crucial, it cannot be interpreted without considering the allele frequency (AF) of the variant in the population. Population AF is a key factor in ranking variants for their likelihood of being pathogenic (Kobayashi et al. 2017; Gudmundsson et al. 2022). Generally, the likelihood of pathogenicity is positively correlated with the rarity of variants in the population, with few exceptions (Kobayashi et al. 2017). Databases such as gnomAD (Chen et al. 2024) contain annotations about population AF. SVAFotate (Nicholas et al. 2022), AnnotSV (Geoffroy et al. 2018), CADD-SV (Kleinert and Kircher 2022), and gnomAD also enable the annotation of SV and prediction of their deleteriousness. However, these databases are based on short-read sequencing data and often lack comprehensive genome-wide SV annotations (Mahmoud et al. 2024b). A recent study reported that only ∼35% of SV from the HG002 GIAB benchmark could be annotated using gnomAD, while another long read-based annotation achieves 95% (Zheng et al. 2024). Initiatives like the HGSVC, HPRC, and the All of Us Research Program projects are currently developing SV catalogs based on long-read sequencing data of large population groups for AF annotation to address this gap (Gustafson et al. 2024; Mahmoud et al. 2024b).
In conclusion, variant annotation in long-read sequencing workflows is essential for understanding the functional significance of genetic variations, contributing to a more comprehensive understanding of their roles in health and disease.
De novo assembly of long-read data
De novo assembly, the process of reconstructing complete genome sequences from raw sequencing reads without a reference genome, has historically been a significant challenge for short-read sequencing technologies. Due to their inherent length limitations, short reads struggle to span repetitive regions and complex rearrangements in many genomes (Logsdon et al. 2020). These limitations often result in fragmented assemblies riddled with gaps and misassemblies. Long-read sequencing technologies have revolutionized de novo assembly, offering significant advantages over traditional short-read approaches (Espinosa et al. 2024). Several long-read assembly tools have emerged, such as Canu (Koren et al. 2017), Flye (Kolmogorov et al. 2019), Hifiasm (Cheng et al. 2021), and Shasta (Shafin et al. 2020). Hifiasm (Cheng et al. 2021) is currently the most widely used method working on PacBio and ONT, and it provides phased assemblies. Verkko (Rautiainen et al. 2023) is a hybrid assembly pipeline that uses PacBio and/or ONT data with a graph-based approach to producing highly accurate assemblies. Additionally, Verkko could utilize parental Illumina short-read data for phasing. For more details on near-complete genome assembly and assembly algorithms, we encourage the reader to follow the review of Li and Durbin (2024).
In general, genome assemblers first produce continuous sequences built upon overlapping individual reads, effectively tiling them together to create longer contiguous sequences (i.e., contigs) (Sedlazeck et al. 2018a). With modern long-read technologies, particularly PacBio HiFi reads, many assemblers can now produce highly contiguous assemblies directly from the assembly graph, often achieving chromosome-scale contigs without additional scaffolding steps, especially for human genomes (Cheng et al. 2021; Nurk et al. 2022). When needed, scaffolding methods utilize additional information to combine contigs, sometimes with an unresolved sequence in between (i.e., gaps represented with NN). To help create scaffolds, Hi-C, Bionano, or ONT ultra-long reads can provide long-range information extending beyond large repeats (e.g., segmental duplications) and thus joining different contigs into scaffolds. However, scaffolding outside the assembly graph may introduce errors (Nurk et al. 2022) and should be considered carefully based on specific project needs. Given the linking of two or more contigs (i.e., scaffolding), the sequence spanning is often undefined as Hi-C or Bionano does not provide sequence context. Thus, gap-filling strategies can replace these undefined sequences by utilizing unmapped parts of reads originating from the region (Xu et al. 2020; Schmeing and Robinson 2023).
Phasing is another important aspect of genome assembly. Multiple assemblers provide phased assemblies directly from long-read data. These can be extended by Hi-C or Pore-C (Deshpande et al. 2022). Their integration typically leads to larger phase blocks (Garg et al. 2021; Li and Durbin 2024). Alternatively, parental data alongside the proband is another effective approach for whole-genome assembly phasing (Koren et al. 2018).
After generating a preliminary assembly, multiple QC metrics are needed to assess the assembly's completeness and accuracy. Metrics like N50 (i.e., defined as the length of the shortest contig such that the sum of this contig and all larger contigs covers at least 50% of the total assembly length) and the total number and size distribution of contigs provide insights into the assembly continuity. However, these metrics can sometimes be misleading, thus, auN (also known as E-Size) was introduced as a more comprehensive measure (https://lh3.github.io/2020/04/08/a-new-metric-on-assembly-contiguity), calculated as the weighted average of contig lengths across all cumulative coverage thresholds (e.g., N10, N20, N30, …, N100) (Salzberg et al. 2012).
That said, these metrics alone fail to provide deeper insights into the accuracy or completeness of the assembly itself. To address that, benchmarking universal single-copy orthologs (BUSCO) (Manni et al. 2021) analysis is a commonly used approach to evaluate the assembly's content by searching for a set of highly conserved, single-copy orthologous genes. BUSCO reports results in three categories: “Complete” (single-copy or duplicated), “Fragmented,” and “Missing,” providing a quantitative measure of genome assembly quality. In addition to BUSCO, other methods are available for assessing assembly quality. For example, HMM-Flagger (Liao et al. 2023) uses a hidden Markov model to detect misassemblies by analyzing patterns in read mapping, coverage depth, and other alignment signals. Similarly, k-mer-based tools such as Merqury (Rhie et al. 2020) evaluate the assembly by comparing the k-mers from the assembled genome against those derived from unassembled high-accuracy raw sequencing reads, providing insights into the completeness and accuracy of the assembly.
Once the quality of an assembly is established, variant calling can retrieve information concerning sequence differences between the new assembly and another assembly or reference genome. The most commonly used methods for variant calling in assemblies currently include Dipcall (Li et al. 2018), SVIM-asm (Heller and Vingron 2021), and phased assembly variant (PAV) (Ebert et al. 2021). Dipcall can infer SNVs, insertions and deletions (indels), and SVs, though it is limited to detecting only insertions and deletions. In contrast, PAV extends its capabilities by also identifying inversions. SVIM-asm, on the other hand, specializes in detecting a broader range of SVs, including deletions, insertions, tandem and interspersed duplications, and inversions. Variant calling using assembled genomes has advantages over reference-based methods. Assembly-based approaches avoid biases introduced by incomplete or biased reference genomes (Behera et al. 2023). Additionally, assembly-based methods enable the detection of larger insertions, which can still be challenging using long-read mapping-based methods.
Finally, while variant calling can utilize existing reference annotations (e.g., from T2T-CHM13v2 or GRCh), direct functional annotation of a genome assembly can provide additional insights about genes or other functional elements. This is particularly valuable when studying novel sequences or SVs that might affect gene structure or regulation. This process is still highly complicated (Salzberg 2019), often including either a de novo approach over RNA-seq or a liftover approach of a close relative available genome. Multiple methods have been suggested (e.g., liftoff [Shumate and Salzberg 2021]), but most of them require manual curation on top of automated pipelines such as Apollo (Dunn et al. 2019).
In conclusion, long-read sequencing technologies have significantly improved the continuity and accuracy of de novo assembly, paving the way for more accurate and complete genome reconstructions.
Graph genomes for long-read analysis
Reference-based methods rely on mapping reads to a reference genome, assuming that it accurately represents the sample's genetic makeup. However, this single-reference approach is limited by the reference's completeness and accuracy. It often overlooks structural variations and polymorphisms in complex regions such as the human leukocyte antigen (Lai et al. 2024; Zhou et al. 2024), LPA (Behera et al. 2024a), and major histocompatibility complex (Liao et al. 2023) in the human genome. Moreover, a single-reference genome can introduce bias, particularly in nonmodel organisms or genetically diverse populations (Gong et al. 2023; Secomandi et al. 2023; Sun et al. 2025). Graph genome (GG) approaches, or pangenome graphs, address these limitations by representing multiple genomes as graphs (Garrison et al. 2018; Miga and Wang 2021). These graphs provide a more comprehensive representation of genetic diversity and capture variations like insertions, deletions, and SVs as nodes and branches. GGs have proven valuable for studying complex genomes (Paten et al. 2017) and understanding how mobile element insertions impact the epigenome (Groza et al. 2023). Hence, these data structures hold substantial promise for multiple applications, including cancer research and population analysis (Sherman and Salzberg 2020). However, this comes at the cost of increased computational complexity, and variant calls still need to be projected onto a linear reference within the graph for downstream analysis.
GG workflows construct a graph by integrating existing reference genomes with known variants (Garrison et al. 2018). Several methods capitalize on the availability of highly accurate and comprehensive de novo genome assemblies. These approaches build graphs that capture complete genomic variation, enabling accurate population-level analyses (Eizenga et al. 2020; Nurk et al. 2022). For instance, Minigraph (Li et al. 2020) and Minigraph-Cactus (Hickey et al. 2024) leverage de novo assemblies to construct graph-based genome representations and align reads to existing graphs. Other tools like vg, Giraffe (Sirén et al. 2021), HISAT2 (Kim et al. 2019), or DRAGEN (Behera et al. 2024b) align DNA and RNA short reads to GG that are constructed either based on assemblies or previously identified variants. Alternatively, custom graphs with specific alleles enable the visualization of more complex regions across a population set (Chin et al. 2022, 2023). Furthermore, some methods in SV genotyping are already utilizing regional graphs to identify if variants are present in a BAM file (Chen et al. 2019; Ebler et al. 2022; English et al. 2024a)
Finally, while the graph approach holds great promise in genome analysis, further research is essential to fully evaluate its scalability and effectiveness in identifying variants across both genic and intergenic regions, ensuring their utility in diverse genomic analyses; different papers have discussed this in more depth (Sherman and Salzberg 2020; Abondio et al. 2023, 2024; Liao et al. 2023; Rocha et al. 2024).
Discussion
Long-read sequencing is advancing rapidly, with continuous improvements in read length and accuracy, revolutionizing genomic research (Fig. 1A). Advances in basecalling algorithms and sequencing chemistry have significantly enhanced accuracy, making long-read data more reliable and precise, as evidenced in Figure 1B. These improvements have enabled more accurate detection of SVs, including large insertions, deletions, duplications, and complex rearrangements, which are challenging or impossible to identify with short-read sequencing alone. Additionally, long-read sequencing has dramatically improved de novo assembly capabilities, particularly in regions with complex repetitive elements, where short-read technologies have fallen short. Beyond genome assembly, long-read technologies have expanded their applications to transcriptomics, epigenomics, and metagenomics, providing deeper insights into gene regulation and the genetic diversity within populations. Both PacBio and ONT sequencing technologies support the detection of DNA modifications, such as methylation, while ONT uniquely enables the detection of RNA modifications. This capability provides valuable insights for understanding epigenetic state, chromatin structure, and RNA modifications. Furthermore, developing portable, real-time sequencing devices has opened new possibilities for immediate data analysis, with potential applications in clinical and field settings (Jain et al. 2016; King et al. 2020; Wasswa et al. 2022). These advancements collectively contribute to a more comprehensive understanding of genomic structure and function, with broad implications for personalized medicine (Wojcik et al. 2023) and evolutionary biology (Stergachis et al. 2020).
Despite its revolutionary potential, long-read sequencing still faces several challenges. Compared to short-read technologies, it requires larger DNA input quantities and involves higher per-base sequencing costs. The complexity of data analysis presents ongoing challenges, particularly in accurate alignment, GG creation, and variant calling within complex or duplicated genomic regions. While bioinformatic innovations are helping manage the large data sets generated by this technology, the generation of high-quality libraries and the operation of complex higher-throughput instruments still require specialized expertise and infrastructure. These current limitations notwithstanding, long-read sequencing continues to expand its role in genomics research and clinical applications, particularly in areas where comprehensive structural variation detection and complete genome assembly are crucial.
Nevertheless, efforts are progressing to simplify these processes and reduce DNA input requirements (Heavens et al. 2021). Finally, there is an annotation gap, as current databases predominantly rely on short-read data, which misses around 50% of SVs (Ebert et al. 2021). Integrating long-read data into existing databases or developing new ones tailored for long-read-derived variants, particularly SVs, is essential to fully harness the power of long-read sequencing. This integration will ultimately enhance our understanding of genetic variation and provide a more comprehensive view of genomes. Despite these challenges, the continuous improvements in long-read sequencing technologies promise to unlock new possibilities in genomics research. The future of long-read sequencing is poised for significant advancements to broaden its impact across various research fields (Conesa et al. 2024).
Integrating long-read sequencing with other “omics” technologies, such as proteomics and metabolomics, promises a more comprehensive understanding of biological systems. Additionally, novel single-cell and spatial genomics applications are emerging alongside real-time and in-field sequencing capabilities with platforms like ONT (Izydorczyk et al. 2024), which will enhance field-based genomics, clinical diagnostics, and environmental monitoring. Furthermore, the development of specialized analysis tools, particularly those leveraging machine learning approaches, promises to make long-read data interpretation more efficient and accessible to a broader scientific community (Poplin et al. 2018; Mastoras et al. 2024). Graph genomes will also play a crucial role in addressing reference bias and enabling the exploration of the diversity from complex genomic regions (Miga and Wang 2021). This is particularly valuable for detecting links between genetic markers and diseases, facilitating the genetic study of more prevalent pathologies in different populations. Combining long-read sequencing with CRISPR for targeted sequencing and expanding direct RNA-seq technologies will offer deeper insights into genomic and transcriptomic complexities. The integration of long-read sequencing into clinical diagnostics represents a crucial frontier, with several sequencing centers beginning to validate these platforms for clinical use. Although the rapid evolution of long-read technologies poses challenges for clinical validation, successful diagnostic cases in research settings have demonstrated their potential to resolve previously unsolved cases, particularly those involving complex SVs or repeat expansions (Goenka et al. 2022; Gorzynski et al. 2022).
Long-read sequencing has emerged as a transformative technology in genomics, fundamentally changing our ability to explore complex genomic landscapes. By overcoming key limitations of short-read sequencing, it provides unprecedented insights into genome structure and variation, particularly in challenging regions that have long remained inaccessible. As technical challenges continue to be addressed and new applications emerge, long-read sequencing is poised to revolutionize both basic research and clinical diagnostics, especially in cases where traditional approaches have proven insufficient. The continuous evolution of this technology, coupled with advances in bioinformatics and clinical validation, promises to deepen our understanding of human genetics and accelerate the path toward more precise and personalized medicine.
Competing interest statement
F.J.S. receives research support from Illumina, PacBio, and Oxford Nanopore. The other authors declare no competing interests.
Acknowledgments
The authors would like to thank Androo Markham for constructive discussions. This research was supported in part by the National Institutes of Health (NIH) grants (1U01HG011758-01 and 1UG3NS132105-01), the National Institute of Child Health and Human Development (NICHD) (R01HD106056), and the National Institute of Allergy and Infectious Diseases (1U19AI144297).
Author contributions: All authors contributed to the research and writing of the manuscript.
Footnotes
-
Article and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279975.124.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵








