
Fast and space-efficient taxonomic classification of long reads with hierarchical interleaved XOR filters
- 1Data Analytics and Computational Statistics, Hasso Plattner Institute, Digital Engineering Faculty, University of Potsdam, 14482 Potsdam, Germany;
- 2Phylogenomics Unit, Center for Artificial Intelligence in Public Health Research, Robert Koch Institute, 15745 Wildau, Germany;
- 3Department of Mathematics and Computer Science, Free University of Berlin, 14195 Berlin, Germany
Abstract
Metagenomic long-read sequencing is gaining popularity for various applications, including pathogen detection and microbiome studies. To analyze the large data created in those studies, software tools need to taxonomically classify the sequenced molecules and estimate the relative abundances of organisms in the sequenced sample. Because of the exponential growth of reference genome databases, the current taxonomic classification methods have large computational requirements. This issue motivated us to develop a new data structure for fast and memory-efficient querying of long reads. Here, we present Taxor as a new tool for long-read metagenomic classification using a hierarchical interleaved XOR filter data structure for indexing and querying large reference genome sets. Taxor implements several k-mer-based approaches, such as syncmers, for pseudoalignment to classify reads and an expectation-maximization algorithm for metagenomic profiling. Our results show that Taxor outperforms state-of-the-art tools regarding precision while having a similar recall for long-read taxonomic classification. Most notably, Taxor reduces the memory requirements and index size by >50% and is among the fastest tools regarding query times. This enables real-time metagenomics analysis with large reference databases on a small laptop in the field.
Identifying organisms in an environmental or clinical sample is a fundamental task in many metagenomic sequencing projects. This includes the detection of pathogens in samples with a large host background (Andrusch et al. 2018), as well as studying the composition of microbial communities composed of bacteria, archaea, viruses, and fungi (Doytchinov and Dimov 2022). Over the past years, many tools have been developed that classify short and long sequencing reads by comparing their nucleotide sequences with a predefined set of references (Kim et al. 2016; Dilthey et al. 2019; Wood et al. 2019). Although each tool uses a different classification strategy, they all try to resolve the species present in the sample and determine their relative abundances (Lindner and Renard 2013; Fischer et al. 2017).
Among the different read classification strategies, alignment-based approaches were the first used for taxonomic profiling. Tools like SLIMM (Dadi et al. 2017), DUDes (Piro et al. 2016), or PathoScope (Hong et al. 2014) use the results of common read mappers like Bowtie 2 (Langmead and Salzberg 2012) and bin the sequencing reads across the different reference genomes. Although these methods have high accuracy, their computational performance decreases tremendously when using entire public databases such as NCBI RefSeq or the Genome Taxonomy Database (GTDB) as reference data sets. Thus, high-performance computing clusters are needed to run these tools in a reasonable amount of time and fulfill their memory requirements. In contrast, marker-based approaches such as MetaPhlAn2 (Truong et al. 2015) and mOTUs2 (Milanese et al. 2019) identify bacterial and archaeal species based on collections of marker genes. However, this approach is not feasible for viruses because they have no universally conserved genes. More recent taxonomic classification strategies rely on machine-learning approaches. Tools such as DeepMicrobes (Liang et al. 2020) and BERTax (Mock et al. 2022) show promising results for classifying reads on higher taxonomic levels but perform poorly at the genus and species levels. Most state-of-the-art taxonomic profilers, like Kraken2 (Wood et al. 2019), Ganon (Piro et al. 2020), and KMCP (Shen et al. 2023), use k-mer-based methods for read classification. Initially, these methods count the exact matches of substrings of length k among the different reference sequences in the database and use further statistical analysis to assign reads to references. These profiling tools mainly differ in the indexing of the reference set and/or the k-mer selection method used to calculate the similarity between read and reference sequences.
What all taxonomic classifiers have in common is that they struggle with the ever-increasing amount of reference genomes. Databases such as NCBI RefSeq (O'Leary et al. 2016) and GTDB (Parks et al. 2022) already comprise hundreds of thousands of microbial reference assemblies belonging to 62,000 bacterial species (GTDB release 207) and 12,000 viral species (RefSeq release 211) and are constantly increasing. This poses a major computational challenge to the profilers in terms of memory usage, index construction, and query time.
Several approaches for the efficient indexing and querying of large collections of reference sequence sets have been developed over the past few years to overcome these issues. The popular Kraken2 (Wood et al. 2019) classifier uses minimizers and introduces a probabilistic, compact hash table to reduce the size of the index. On the other hand, color-aggregative methods like Bifrost (Holley and Melsted 2020) or Mantis (Pandey et al. 2018) use compacted de Brujn graphs or counting quotient filters for indexing and querying k-mers. Those methods have the disadvantage that they index each reference separately using data structures for approximate membership queries, for example, Bloom filters (Bloom 1970). In contrast, sequence bloom tree (SBT) (Solomon and Kingsford 2018; Sun et al. 2018; Harris and Medvedev 2020) approaches exploit the k-mer redundancy in data sets with extremely uneven abundance patterns such as those from RNA-seq experiments to compare large sequence data sets. However, these approaches are not optimal for k-mer sets with a high number of unique k-mers, such as microbial genomes. Tools like bitsliced genomic signature index (BIGSI) (Bradley et al. 2019), compact bit-sliced signature index (COBS) (Bingmann et al. 2019), and KMCP (Shen et al. 2023), which are based on Bloom filter matrices, are promising much better results for taxonomic profiling of large sequencing data sets. Interleaved Bloom filters (IBFs), which are very similar to Bloom filter matrices, combine several Bloom filters (one per reference) in an interleaved fashion while allowing the query of all Bloom filters at once (Dadi et al. 2018). The IBF data structure has been used by the taxonomic classifier Ganon (Piro et al. 2020) and was recently enhanced by introducing the hierarchical interleaved Bloom filter (HIBF) in a tool called Raptor (Mehringer et al. 2023).
Many k-mer-based classifiers use Bloom filters for approximate membership queries of k-mers in large reference data sets. Their popularity is based on their flexibility, low memory requirements, and fast query times. However, there is a small probability that a k-mer is incorrectly reported as being present in a reference sequence, called a false positive. Some tools let the user define the false-positive rate and adapt the Bloom filter size and/or the number of hash functions to that value. Although Bloom filters have a low memory footprint, they use 44% more memory than the theoretical lower bound, even when applied in an optimal manner (Carter et al. 1978). Therefore, several advanced probabilistic filters like cuckoo filters (Fan et al. 2014; Mitzenmacher et al. 2020) have been developed over the past few years. In particular, XOR filters have been proposed as an alternative to Bloom filters, using only 23% more memory than the theoretical lower bound (Graf and Lemire 2022).
Based on the work of Graf and Lemire (2020) and Dadi et al. (2018), we first developed an interleaved XOR filter (IXF), which can be used in the same manner as the IBF implemented as part of the Seqan C++ library (Reinert et al. 2017). Then, we adapted the hierarchical filter structure proposed by Mehringer et al. (2023) and extended our new data structure to a hierarchical interleaved XOR filter (HIXF) to avoid using more space than necessary when references in the database are highly divergent in size. The HIXF data structure is implemented as part of the taxonomic classification tool Taxor, which allows the user to choose different k-mer-based strategies, such as k-mers and minimizers. Because our new tool is specifically designed for long-read metagenomics experiments, we also implemented open canonical syncmers (OCSs) as a k-mer selection approach, which has been shown to be superior to minimizers for error-prone long reads (Edgar 2021; Dutta et al. 2022). In the final step, taxonomic profiling of the query results is performed by utilizing an expectation-maximization (EM) algorithm for abundance estimation and reassignment of classified reads. We compare Taxor to five state-of-the-art short- and long-read taxonomic classification tools on simulated and real mock communities. Our results show that Taxor can tremendously reduce the index size and memory requirements for queries while still being on par with the evaluated tools regarding precision and recall.
Results
We compare Taxor to five state-of-the-art taxonomic profiling tools, Centrifuge (Kim et al. 2016), MetaMaps (Dilthey et al. 2019), Kraken2 (Wood et al. 2019), KMCP (Shen et al. 2023), and Ganon (Piro et al. 2020). To ensure a fair comparison, we excluded deep-learning methods DeepMicrobes (Liang et al. 2020) and BERTax (Mock et al. 2022) from our benchmarking. Once trained on a large reference data set, those tools can tremendously reduce the time and memory requirements for the taxonomic classification of sequencing reads, but training the models requires extensive computational resources, and both tools show poor results on genus- and species-level assignment. We also did not consider the MetaPhlAn2 (Truong et al. 2015) and mOTUs2 (Milanese et al. 2019), which rely on marker genes and are not suitable for species assignments on viral data sets.
Centrifuge is the tool underlying Oxford Nanopore's “what's in my pot” (WIMP) application for real-time species identification (Juul et al. 2015). Although Centrifuge was initially developed to analyze short-read metagenomic samples, MetaMaps specifically addresses the task of strain-level metagenomic assignment of long reads. We decided to use both tools and Kraken2 with default parameter configurations because this results in the best recall and precision in our experiments on simulated data.
Finally, we evaluated KMCP and Ganon, both utilizing different Bloom filter approaches to store sets of selected k-mers from reference genomes for short-read classification. In our benchmarking, we included the latest version of Ganon, which uses the recently described HIBF (Mehringer et al. 2023). For the calculation of taxonomic abundances, we also included Sourmash (Pierce et al. 2019) in the benchmarking. However, we could not include Sourmash in the read classification benchmarking because it does not classify individual metagenomic reads to either a genome or a taxon. We also tried to add MEGAN-LR (Huson et al. 2018), but it did not assign taxonomic information to the reads when using our custom database and crashed when using the whole NCBI nt database after creating a 3.9 Tb minimap2 index. The specific commands to run the six tools are provided in the Supplemental Methods.
Reference databases
Most taxonomic profilers offer prebuilt reference databases but also allow building custom reference databases. Because the choice of the reference database directly affects the outcome of taxonomic profiling, a fair comparison between tools also requires using the same database. This ensures that observed differences in the single-read assignments are attributed solely to the profiling methods. Thus, we downloaded all complete genome sequences and chromosomes of archaea, bacteria, viruses, and fungi from the NCBI RefSeq database (release 217) (O'Leary et al. 2016) using genome_updater 0.5.2 (https://github.com/pirovc/genome_updater). We used only one representative reference genome per species, according to the sorting criteria of genome_updater. This results in 21,003 genomes used to build custom databases for each tool. This custom database comprises 11,579 viral genomes, 8938 bacterial genomes, 403 archaea genomes, and 83 fungi genomes.
We build customized index data structures based on the described reference database for all tools included in our evaluation. For Centrifuge, we created the reference index using the default parameters, providing only the taxonomic information from the NCBI taxonomy and reference sequences in FASTA format. For MetaMaps, we first created a custom database from our downloaded taxonomy using the provided scripts and following the instructions at GitHub (https://github.com/DiltheyLab/MetaMaps). Then, we created the MetaMaps index from the custom database using default parameters. Because the other tools all use pseudoalignment utilizing k-mer-based approaches, we build indexes using a k-mer size of 22, which is a good compromise between high specificity on species-level identification and high sensitivity for error-prone nanopore read classification. For all four approaches, we decided to use k-mer selection schemes that downsample the used k-mer sets to ∼10% of all reference k-mers to reduce memory usage and index size significantly. Specifically, we used ungapped k-mers of size 22 and a window size of 32 for minimizer-based indexes in Kraken2 and Ganon. For KMCP and Taxor, we used a k-mer size of 22 and a syncmer size of 12. We further set the false-positive rate for the Bloom filter–based approaches, namely, Ganon and KMCP, to 0.39% to reflect the same inherent false-positive rate of XOR filters using 8-bit sequences, as implemented in Taxor (Graf and Lemire 2020). The specific commands and instructions to build the reference indexes of all tools are listed in the Supplemental Methods.
Evaluation data sets
We carried out four experiments to evaluate Taxor, covering multiple metagenome composition scenarios from short- and long-read real data sets as well as sample contamination with eukaryotic DNA. For evaluation on long-read data, we obtained two ONT data sets for the ZymoBIOMICS D6300 microbial community standard (Nicholls et al. 2019) and one Pacific Biosciences (PacBio) HiFi data set for the ZymoBIOMICS cut microbiome standard D6331. The Zymo D6300 standard consists of 10 evenly abundant species, including eight bacteria at 12% sequence abundance and two yeasts at 2% sequence abundance. The first ONT data set comes from a continually updated online resource (https://lomanlab.github.io/mockcommunity/r10.html). We downloaded the R10.3 chemistry data release (February 2020), which was produced from two flowcells on an ONT GridION, resulting in 1.16 million reads (4.64 Gb data). The second ONT data set was obtained from the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/) (PRJEB43406: ERR5396170, released March 2021) and represents the “Q20 chemistry” release for the Zymo D6300 standard (described at https://github.com/Kirk3gaard/2020-05-20_ZymoMock_Q20EA). It was generated using a PromethION, resulting in 5.4 million reads (17.95 Gb data).
The PacBio HiFi data set for the ZymoBIOMICS gut microbiome standard D6331 (obtained from the NCBI BioProject database [https://www.ncbi.nlm.nih.gov/bioproject/] under accession number PRJNA680590: SRX9569057, released November 2020) contains 17 species (including 14 bacteria, one archaeon, and two yeasts) in staggered abundances. Five species occur at 14% sequence abundance, four at 6%, four at 1.5%, and one per 0.1%, 0.01%, 0.001%, and 0.0001% abundance level. There are five strains of Escherichia coli in this community (each at 2.8% sequence abundance), which we treat here as one species at 14% sequence abundance. The PacBio Zymo D6331 data set was generated using the Sequel II system and contains 1.9 million HiFi reads with a median length of 8.1 kb, for a total of 17.99 Gb of data.
To generate a read-level truth set, we use minimap2 (Li 2018) to map the reads against the reference genomes provided by ZymoBIOMICS. All reads that cannot be mapped with minimap2 are excluded, and the primary alignment for each read determines the assumed true placement. This results in two ONT Zymo D6330 evaluation data sets referred as “ZymoR10.3” and “ZymoQ20” and one PacBio HiFi Zymo D6331 evaluation data set referred as “HiFi_D6331.”
As a final comparison to the long-read data sets, we included one short-read sequence data set for the Zymo D6300 community (obtained from the NCBI BioProject database under accession number PRJNA648136: SRX8824472, released July 2020). These data were produced using a NovaSeq 6000, including roughly 100 million 150 bp PE reads. We also generated a read-level truth set, mapping the short reads to the reference genomes provided by ZymoBIOMICS with BWA (Li and Durbin 2009). We refer to this data set as “ILMN_D6300.”
As a negative control, we use pbsim2 with the parameters “–accuracy-mean 0.95,” “–length-min 1000,” and “–hmm_model R103.model” to simulate long-read sequencing data from two eukaryotic genomes not present in the reference database. Specifically, we simulate 685,303 reads from the Aedes aegypti (yellow fever mosquito) genome (GCF_002204515.2) and 142,677 reads from the Toxoplasma gondii ME49 genome (GCF_000006565.2). The two read sets are analyzed independently with Taxor and the other tools.
Evaluation
We evaluated the performance of all six tools using several criteria. We assessed read utilization and classification metrics at the species, genus, and family levels and assessed relative abundance estimates at the species level. First, we evaluated read utilization for each profiling method by calculating the total percentage of reads assigned to specific taxonomic levels. We performed this for the following ranks: class, order, family, genus, and species. Here, we expected methods like Kraken2 and Ganon that use an assignment to the lowest common ancestor to display read assignments across multiple taxonomic levels, whereas methods like Taxor only report the species level. The used evaluation metrics are described in the Supplemental Methods.
For accurate measurement of the read classification metrics for the real mock data sets, we had to control for species synonymies in the taxonomy of the used reference database. To avoid a negative impact on the metrics, we used the sum of cumulative counts for the species and all synonyms as the read count for the taxon. In particular, this included three species in Zymo D6300 (Limosilactobacillus fermentum = Lactobacillus fermentum; Bacillus subtilis = Bacillus spizizenii; Listeria monocytogenes = Listeria sp. LM90SB2) and one species in Zymo D6331 (Limosilactobacillus fermentum = Lactobacillus fermentum), for which we treated the five strains of E. coli contained in this community as one species.
We calculated detection metrics for each data set. To understand the expected performance of each method across different long-read data sets, we averaged the precision, recall, F1, and F0.5 scores at the species level for the three long-read data sets.
Finally, we attempted to obtain relative abundances for each method, acknowledging differences in reporting abundances as described by Sun et al. (2021). In particular, there are clear differences in intended outputs among methods. For example, profiling methods (Sourmash, Ganon, and KMCP) provide taxonomic abundances, whereas classifiers (Centrifuge, Kraken2, and MetaMaps) provide sequence abundances. Because Taxor reports both abundance measurements, we did not transform the reported values of the tools but compared Taxor's output directly to the reported abundance types of the respective tools. We calculated an L1 distance between observed and theoretical abundances for each method as described by Portik et al. (2022). The theoretical abundances were obtained from the manufacturer's specifications based on genomic DNA (sequence abundance) and genome copy (taxonomic abundance). We calculated the L1 distance by summing the absolute error between the theoretical and empirical estimate per species across the three communities. In this calculation, we included the false positives lumped in the “other” category and compared them against a theoretical abundance of zero for this category.
Classification performance on long-read data
We evaluate Taxor on real long-read data using three sets of metagenomic sequencing data, two ONT data sets for the ZymoBIOMICS D6300 community standard, and one PacBio HiFi data set for the ZymoBIOMICS gut microbiome standard D6331. The read classification performance evaluation results are shown in Figure 1 and Supplemental Table S1. Our results show that Taxor has a high precision on the species level of 0.97 and 0.94 on the ZymoR10.3 and HiFi_D6331 data sets. For both data sets, the precision increases to 0.98 on the genus level and 0.99 on the family level. For the second ONT data set (ZymoQ20), Taxor's species-level precision is slightly lower at 0.91 and increases to 0.92 (genus) and 0.93 (family) on higher taxonomic levels. Taxor further shows a high recall between 0.95 and 0.97 on all taxonomic levels for the ZymoQ20 and HiFi_D6331 data sets. Only for the ZymoR10.3 data set, Taxor's recall has a value of 0.89 on all taxonomic levels. Our new tool has a F1-score between 0.93 and 0.96 and a F0.5-score between 0.92 and 0.95 on the species level across all three data sets.
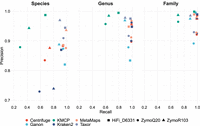
Read classification at the species, genus, and family levels for all three long-read data sets. Precision and recall are shown for all six compared tools on three taxonomic levels. Taxor and KMCP show the highest precisions across all three data sets and all levels. Taxor's recall is comparable to MetaMaps and Ganon on the species level, whereas KMCP consistently has the lowest recall of all tools. Centrifuge and Kraken2 have low species-level recall for the two ONT data sets while performing well on the HiFi data set.
Consistent patterns emerge when comparing Taxor to the other metagenomics read classification tools. Across all three data sets, Taxor and KMCP show the highest precision on the species level, whereas MetaMaps, Ganon, and Taxor have the highest average recall values (see Fig. 2A). When looking at F-scores, we see that Taxor and MetaMaps have the same species-level F1-score across all three data sets, but Taxor outperforms MetaMaps concerning the average F0.5-score when precision is prioritized over recall (Fig. 2B,C).

Average scores on species level across the three long-read mock community data sets. (A) Average precision and recall of the six compared tools on the species level for the two ONT and the HiFi mock community data sets. MetaMaps, Taxor, and Ganon have the highest average recall, whereas Taxor and KMCP outperform the other tools in terms of precision. KMCP is the only tool with an average recall below 0.5 across the three data sets. (B) Average species-level F1-score of the six tools across the three real mock communities. Taxor and MetaMaps show the highest scores, whereas KMCP has the lowest average F1-score. (C) Average species-level F0.5-score of the six tools across the three real mock communities. Our new tool, Taxor, outperforms the other methods when precision is prioritized over recall.
In summary, Taxor and MetaMaps perform best on the species level across the three real data sets, making them the best choice for long-read metagenomic classification. MetaMaps was specifically designed for long-read data sets, and thus, its parameters for read mapping and the following taxonomic classification are tuned for this application. In contrast, Centrifuge, KMCP, and Kraken2 were designed for short-read taxonomic classification. These tools expect fewer sequencing errors, which explains their performance in this evaluation. However, Ganon, another short-read taxonomic classifier, consistently shows good recall and precision on long-read data sets. This may reflect an appropriate parameter choice for long-read classification or a precise refinement of read classifications during the profiling step. Taxor's performance can be explained by using cutoff values for its syncmer-based classification, which were inferred from simulated long-read data sets and the read classification refinement based on taxonomic abundances during the profiling phase.
Effect of host contamination
The effect of contamination with eukaryotic host DNA is an important concern in metagenomics. Thus, we assess the effect of large out-of-database genomes on the read classification performance of all tools. Therefore, we simulated reads from two eukaryotic genomes (A. aegypti and T. gondii), neither of which is present in the reference database. Taxor has a low false-positive rate for both read sets and correctly leaves the large majority of reads unclassified (≥99.99%) on all taxonomic levels. We also note low false-positive rates for KMCP (0% for both data sets) and Ganon (0.17% for A. aegypti and 0.05% for T. gondii). The three tools slightly outperform MetaMaps, having misclassification rates between 1.71% (Aedes) and 2.87% (Toxoplasma). In contrast, Kraken2 and Centrifuge show high false-positive rates, with Kraken2 reporting only 5.67% (Aedes) and 8.84% (Toxoplasma) of reads as unclassified and Centrifuge reporting 22.90% (Aedes) and 26.74% (Toxoplasma) of reads as unclassified. Detailed results for these experiments are provided in Supplemental Table S2.
Relative abundance estimation on real data
All six evaluated tools report relative species abundance estimations after read classification and metagenomic profiling. For the species abundance estimation, we also included Sourmash as an additional taxonomic profiling tool. In Figure 3, we compare the theoretical relative abundances of species in the mock communities against the relative species abundance reported by the different tools. Taxor is the only tool that reports both types of abundance, sequence abundance and taxonomic abundance. When comparing the taxonomic abundance estimates of Taxor to those of Sourmash, Ganon, and KMCP, we see that none of the three tools can accurately predict taxonomic abundances for all species in the three mock communities (Fig. 3A). However, Taxor has the lowest L1 distance of the four tools across all data sets, demonstrating better relative species abundance estimates than Sourmash, Ganon, and KMCP.
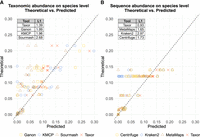
Comparison of theoretical abundances and predicted abundances. For each species in the three long-read real mock communities, predicted abundances by the compared tools are plotted against the theoretical abundances. Points on the dashed line represent a perfect match between predicted and theoretical abundance. (A) Comparison of tools reporting taxonomic abundance. (B) Comparison of tools reporting sequence abundance. Taxor reports both abundance types and outperforms its competitors, having the smallest L1 distance between predicted and theoretical abundance.
Because MetaMaps, Kraken2, and Centrifuge report relative sequence abundances of species instead of relative taxonomic abundances, we compare their profiling results to the theoretical sequence abundances of the mock communities (Fig. 3B). Consistent with the taxonomic abundance evaluation results, none of the investigated tools can accurately predict sequence abundance for the species comprising the three mock communities. As for the taxonomic abundance, Taxor outperforms the other tools by having the smallest L1 distance between the theoretical and predicted species abundances across all data sets. Our investigations indicate that different species with high sequence similarity cause false read classifications and thus also bias the abundance estimations of the tools.
Classification performance on short-read data
Finally, we also evaluate Taxor on short-read data using Illumina sequenced metagenomic data of the ZymoBIOMICS D6300 community standard. We compared Taxor's classification results to those of the other tools, except for MetaMaps, which crashed with an error message after setting the minimum read length to 100. The results of this read classification performance evaluation are shown in Supplemental Figure S1 (see Supplemental Methods) and Supplemental Table S1. Although Taxor's intended use is on long-read classification, the results show that Taxor performs comparably well to state-of-the-art short-read taxonomic classifiers. Only Ganon and KMCP show a better recall on the species level, whereas only Ganon outperforms Taxor regarding species-level precision. Although having fewer sequencing errors than long reads, the shorter read lengths of Illumina sequenced reads make it hard to correctly assign them to specific taxa. It also seems that the minimizer-based classification algorithm of Ganon is a better choice for short reads than the syncmer-based k-mer selection used by KMCP and Taxor. We also assume that the choice of the indexed database can lead to improved classification performance, as most tools have difficulties assigning the correct species to a given read if different species with a high sequence similarity are included in the indexed database.
Computational requirements comparison
With the ever-increasing number of genomes in public databases comes the need for faster and more space-efficient methods to facilitate metagenomic read classification and profiling. Thus, we assessed the computational requirements of Taxor for indexing the reference database used in this study. We further included a benchmarking on the whole GTDB database release 214 (Parks et al. 2022) to compare the performance of the tools for a larger database. Then, we also measured the required peak memory usage and runtime for querying the “ZymoR10.3” data set against the built database index. We further compare Taxor's computational requirements against the other five tools used in this study. We used the same reference database for all tools and built and queried the indexes using the specific commands provided in the Supplemental Methods. Because all tools support multithreaded computations, we performed computational benchmarking on an AMD EPYC 7742 cluster node using 30 threads. All times and peak memory usage were measured using the Linux “time” command with the parameter “-v.”
Results of the computational benchmarking are provided in Table 1. For both databases, KMCP is the fastest tool with the lowest peak memory usage when building the index. Ganon is the second fastest tool but has a high memory usage and a large index size on disk. MetaMaps and Centrifuge have the highest computational requirements for building the index of our customized RefSeq database, and both crashed when we tried to build an index of the whole GTDB database. Taxor needs more time to build a database index compared with Kraken2, Ganon2, and KMCP, but it has the second lowest peak memory usage and the smallest index size across all tools. Here, we recognized that Taxor spends most of the time calculating the hierarchical layout and recalculating an IXF if the construction of one of its single XOR filters fails.
Results of computational requirement benchmark
When querying the 426,213 ONT reads of the “ZymoR10.3” data set against the respective database indexes, Taxor, Ganon2, and Kraken2 are the fastest tools, requiring only a few minutes to provide read classification results. These three tools outperform the others, with a six (Centrifuge) to 12 (KMCP) times faster query time on the RefSeq database. MetaMaps is the most resource-hungry tool, needing 335 GB memory and >5 h to query all reads against its index. Finally, we note that Taxor outperforms all evaluated tools regarding peak memory requirements when querying the index. Because of its small index size, Taxor can reduce the peak memory usage up to 50% compared with Centrifuge and KMCP, which both have much higher query times. Here, we also show that KMCP's faster index construction comes at the cost of much slower querying times, which is a disadvantage when the same database is used many times for hundreds of experiments. Compared with Kraken2 and Ganon, which have similar query times, Taxor can reduce the memory footprint by a factor of three (Kraken2) to four (Ganon). These results highlight the combination of fast and space-efficient read classification of Taxor, whereas other tools only provide fast querying or lower memory requirements.
Discussion
The increasing size of public reference genome databases such as NCBI RefSeq makes metagenomic profiling and read classification a computationally challenging task. In particular, reducing memory usage has become an objective of many studies during the past few years. However, state-of-the-art short- and long-read metagenomic classifiers still consume large amounts of memory, which can only be reduced by accepting a higher risk of false classifications. For Bloom filter approaches like Ganon and KMCP, one can, for example, reduce the index size by accepting a higher false-positive rate for the approximate membership queries of k-mers. However, higher false-positive rates can lead to false classifications of viral reads, as proposed by Shen et al. (2023).
This issue motivated us to develop a new data structure that combines low memory requirements, a low false-positive rate, and fast membership queries to facilitate precise metagenomic classification of long reads on large reference sequence databases. Based on the work of Graf and Lemire (2020) and Mehringer et al. (2023), we created a HIXF data structure, which is implemented in the metagenomic classification tool called Taxor. Instead of relying solely on k-mers, Taxor can utilize syncmers as a k-mer selection scheme. Finally, our new tool comes with a profiling step, applying several filter strategies and an EM algorithm that refines the results of the initial read classification.
In this study, we present our work on Taxor and show benchmarking results of a comparison with five commonly used metagenomic profiling tools. Taxor's read classification results are on par with state-of-the-art methods regarding recall while improving precision rates in almost every experiment on real data. Our new tool consistently shows the highest F1- and F0.5-scores on the species level for the three mock communities, indicating the robustness of the findings. We attribute this improvement to using syncmers instead of minimizers, applying several iterative filter steps, and applying our EM algorithm for read classification refinement. Edgar (2021) has shown that syncmers cover the underlying sequence better than minimizers, and both tools in our benchmarking that use syncmers (Taxor and KMCP) perform best regarding precision. Both use either open syncmers (Taxor) or closed syncmers (KMCP), similar filter steps, and a final EM algorithm for the profiling. They mainly differ in the underlying data structure for approximate membership querying and their implementation of the EM algorithm.
Contamination and environmental DNA are important aspects in many metagenomic studies, particularly if their genomes are not in the databases used by the metagenomic profilers. We have shown that Taxor is robust against these out-of-database genomes, minimizing the risk of false classifications of reads in such scenarios. Here, Taxor outperforms the mapping-based method MetaMaps and is on par with the Bloom filter approaches of Ganon and KMCP. The false-positive rate of 0.39%, which we used in this study for the Bloom filter approaches, and our HIXF data structure seem to protect the three tools from falsely classifying reads from out-of-database genomes. Our observations suggest that removing host reads may not be necessary for these tools before the metagenomic classification of long reads in microbiome studies.
Correctly estimating the composition of microbial genomes in metagenomic samples is a main task in many microbiome studies investigating the differential abundances of species between several gut or environmental samples. These differences can be attributed to environmental changes like global warming or anthropogenic pollution. Our benchmarking results show that none of the evaluated tools accurately estimates the abundance of all species in the real mock communities used for evaluation. Although Taxor also has problems estimating the species abundances correctly, its calculated sequence and taxonomic abundances are more consistent with the theoretical abundances of species in the communities. However, these results should be taken with a pinch of salt as various factors, including different DNA extraction methods, can affect the final composition of DNA sequenced for metagenomic samples and can potentially bias relative abundance estimates of the tools (Sui et al. 2020).
The small MinION sequencing devices invented by Oxford Nanopore Technologies (ONT) provide the possibility to sequence a sample at the place of its origin without the need to ship the sample to a laboratory. In such a point-of-care sequencing scenario, computing resources for metagenomic analysis are usually limited. Without a reliable internet connection to perform analysis in the cloud, the tools applied for metagenomic profiling must be as fast and memory efficient as possible while retaining high read classification accuracy. In this context, Taxor represents a significant improvement over state-of-the-art tools, requiring only 50% of the memory and disk space as the best competitor in our benchmarking. Taxor was also among the fastest tools regarding the query time while showing the highest average precision across the three real evaluation data sets. We expect Taxor to be a valuable tool for usage in real-time long-read metagenomic analysis pipelines like minoTour (Munro et al. 2022) or WIMP (Juul et al. 2015), both using Centrifuge for read classification.
Our new HIXF data structure significantly improves the IBF concerning memory consumption and query time. It also reduces the memory requirements compared with the recently published HIBF (Mehringer et al. 2023) when both use the same false-positive rate. However, this comes at the cost of less flexibility and more time needed to build the index. The biggest drawback of the XOR filter is the ability to only index static sets of keys. That means all input data need to be known a priori, and the index cannot be updated after it has been built once. Because the XOR filter is the underlying data structure of our HIXF, this also applies to it. However, we argue that build times for the HIXF are still acceptable and that the lower memory footprint for querying reads compensates for that issue. We even envisage that tools currently relying on Bloom filters or IBFs can benefit from utilizing the HIXF as their index data structure, particularly in time-critical applications like nanopore adaptive sampling (Ulrich et al. 2022).
There are two important directions for the future development of Taxor. First, reducing the computational requirements by enhancing the underlying data structure is worth the effort because the number of genomes in public databases is constantly growing. One possibility would be using binary fuse or ribbon filters instead of the XOR filter in the hierarchical interleave data structure. Recent studies have shown that both filter types are practically smaller than XOR filters (Dillinger and Walzer 2021; Graf and Lemire 2022). However, they have a higher risk of failing to successfully build the index, which results in choosing new hash functions. In an interleaved approach, in which we have to build several single filters using the same hash functions, the failed build process of one filter would require rebuilding all other filters as well. This tremendously increases the runtime of the build process. Thus, further research is needed to improve the build process of those filters to make them suitable for interleaved approaches. Second, estimating the species abundances in the investigated metagenomic samples needs to be improved to reliably use our tool in microbiome studies relying on differential abundance calculations. Here, enhancing the profiling would require further filtering and an improvement of our EM algorithm, which remains an open task for further research studies.
Methods
We have designed and implemented our novel taxonomic profiling tool, Taxor, as a modular workflow that consists of three mandatory steps. First, Taxor computes the k-mer content of the input reference genomes and creates an index for each set of reference genomes. The index is a HIXF, a novel space-efficient data structure for approximate membership queries that we will describe in the following subsections. In the second step, sequencing reads are queried against one or several HIXF index files, resulting in one intermediate file for each index. These intermediate files contain all matches of the reads against the different reference genomes and must be merged before the final profiling step. Three-step filtering of spurious matches is performed before an EM algorithm computes taxonomic abundances and reassigns reads based on the taxonomic profile. We finally provide three output files containing information about the sequence abundances (based solely on nucleotide abundance), taxonomic binning (read to reference assignments), and taxonomic abundances (normalized by genome size).
Interleaved XOR filter
Many tools for taxonomic classification of sequencing reads facilitate approximate membership queries of k-mers of reads against k-mer sets of the reference sequences. A common approach to implement approximate membership queries is using Bloom filters
(Bloom 1970). Dadi et al. (2018) improved this approach by developing an IBF that stores several Bloom filters in one single-bit array that allows querying
all Bloom filters simultaneously. Inspired by their approach, we developed an IXF that combines several XOR filters into one
data structure, enabling simultaneous querying of all XOR filters. As described by Graf and Lemire (2020), and similar to Bloom filters, the XOR filter uses three independent hash functions that return a corresponding position
in the filter for each key (or k-mer). In a Bloom filter, each bit is considered its own array slot, and bits at the positions to which the hash functions
point are set to one. In contrast, in an XOR filter, the bits are grouped together into L-bit sequences, as shown in Supplemental Figure S2 (Supplemental Methods). These L-bit sequences in the XOR filter are set in such a way that a bitwise XOR of the three L-bit sequences, corresponding to positions returned by the hash functions, equals the result of the fingerprint hash function. Although building an XOR filter is almost always successful for larger sets of more than |S| = 107 elements (Botelho et al. 2007), it can fail for smaller sets, which requires rebuilding with other hash functions. Graf and Lemire (2020) have experimentally shown that the estimated probability for the successful building is always greater than 0.8 if the XOR
filter size is set to 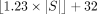 , which makes this size constraint an optimal compromise between space requirement and build time.
, which makes this size constraint an optimal compromise between space requirement and build time.
Our IXF implementation combines several XOR filters (bins) in one single bitvector, using 8-bit sequences for each XOR filter. As for the IBF, we initially set the size of all bins to the size of the largest XOR filter, representing the largest reference sequence. When building the IXF for a set of reference sequences, we compute the k-mer sets for each reference sequence and construct the single XOR filters for each reference according to the algorithm proposed by Graf and Lemire (2020). We used the same hash and fingerprint functions for each XOR filter and combined them in an interleaved fashion, as shown in Supplemental Figure S3 (see Supplemental Methods). When querying a read against the references stored in the IXF, every k-mer (or syncmer) of that read is matched against each XOR filter simultaneously. This generally works in the same way as querying an IBF by first retrieving the three subvectors from using the same hash functions as used for building the IXF. Then, the resulting 8-bit sequence calculated by the fingerprint function is concatenated with itself to the length of the subvectors, and a logical bitwise XOR is applied to the three subvectors and the fingerprint vector, which results in a final 8-bit sequence for each reference.
If this sequence equals zero, a bit in a binning bitvector is set to one, indicating the presence of the k-mer in the corresponding reference. Combining the binning bitvectors of the k-mers to a counting vector finally results in the number of matching k-mers between the read and each reference in the IXF. The example in Supplemental Figure S4 (see Supplemental Methods) visualizes this process.
Hierarchical interleaved XOR filter
The interleaved nature of the IXF has two important limitations. First, the largest XOR filter determines the overall size of the IXF because all single bins (XOR filters) of the IXF must have the same size. This means that the largest reference sequence dictates the size of the XOR filters storing the k-mer contents of the other reference sequences. Consequently, we would waste a substantial amount of space for smaller references if the reference sequences in the IXF have highly divergent sizes. Second, the query speed slows down with an increasing number of XOR filters stored in the IXF. This is, in practice, not a problem for a few hundred to a few thousand references but becomes inefficient when storing many thousands of reference genomes. To overcome this issue, we adapted the approach by Mehringer et al. (2023) to create a HIXF. Here, the idea is to split the k-mer content of larger reference sequences into several smaller k-mer sets while merging the k-mer sets of very small reference sequences into one big set of k-mers. The resulting k-mer sets are stored in a high-level IXF, and for each merged k-mer set, a low-level IXF is stored, holding the k-mer sets of the smaller reference sequences in individual bins (XOR filters). Although splitting the k-mer content of large reference sequences and merging the k-mer content of smaller references avoids wasting space, recursively adding an IXF for each merged k-mer set enables querying the individual k-mer sets of the small reference sequences. Depending on the number and size of the reference sequences and the maximum number of bins allowed for each IXF, we can have many levels of the HIXF, as shown in Supplemental Figure S5 (see Supplemental Methods). To compute the layout of the HIXF, we utilize the dynamic programming (DP) approach from Mehringer et al. (2023) that finds the optimal balance between space consumption and query speed. Here, we first calculate HyperLogLog sketches (Flajolet et al. 2007) to determine the Jaccard distance between each pair of reference sequences (Baker and Langmead 2019).
When querying the HIXF, we first calculate the k-mer content of the given query read and determine the minimum number of matching k-mers with each reference sequence to be considered a hit (see Supplemental Fig. S6 in the Supplemental Methods). We calculate this threshold based on the k-mer selection scheme described in the following subsection. Then, for each k-mer of the read, the membership in all filters of the top-level IXF is determined, and we further count the total number of k-mers that match each bin in the top-level IXF. If the counter for a bin exceeds the calculated threshold, the read is considered a match with the corresponding reference sequences. For the split bins, the k-mer counts have to be accumulated before thresholding, whereas we can directly answer the query for single bins that are neither merged nor split. For merged bins that exceed the threshold, we apply the same procedure recursively on the associated child IXF on the next lower level. This approach allows us to skip querying all lower-level IXFs whose upper-level merged bins do not exceed the given threshold. As a final result, we obtain a list of all reference sequences that exceed the minimum number of matching k-mers with the read under investigation.
k-mer selection and thresholding
In the previous subsections, we introduced our new HIXF data structure describing the usage of membership queries for all k-mers of a given read against all k-mers of a given reference sequence set. We consider a read a hit with a reference sequence if the number of matching k-mers is greater than or equal to a given threshold t. For this k-mer model, we calculate the threshold as described in the Supplemental Methods.
Because using all k-mers of the reference sequences can result in huge index sizes, k-mer selection approaches gained much attraction during the past decade, with minimizers being the most popular down-sampling
approach for metagenomic classification of short reads (Wood et al. 2019; Piro et al. 2020). However, Edgar (2021) recently showed that syncmers are more sensitive for selecting conserved k-mers in biological sequences, and Dutta et al. (2022) could also improve long-read mapping by using OCSs instead of minimizers. Therefore, we implemented OCSs as a down-sampling
strategy for large sets of k-mers to decrease the size of the HIXF index. Open syncmers are sampled in our implementation based on three parameters (k, s, t), where k, s, and t are positive integers and s ≤ k. The method then compares the k − s + 1 consecutive s-mers within a k-mer and selects the k-mer as a syncmer if the smallest s-mer occurs at position t ∈ [0, k − s + 1] within the k-mer. The smallest s-mer is defined by the hash value computed for each s-mer. In our implementation, we use the canonical representation of syncmers, meaning that the lexicographically smallest
syncmer out of its forward- and reverse-complement sequence is always selected. We compute such OCSs by choosing 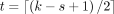 , which has been shown to be the optimal parameter for sequence mapping (Shaw and Yu 2022), given user-specified values for the k-mer and s-mer size.
, which has been shown to be the optimal parameter for sequence mapping (Shaw and Yu 2022), given user-specified values for the k-mer and s-mer size.
Analogous to the k-mer-based approach, we need to determine a threshold for a read's minimum number of matching syncmers to consider it a hit with a reference sequence. In contrast to k-mers, there is no theoretical derivation for a (1 − α) confidence interval of the number of erroneous syncmers. Thus, we decided to derive the threshold empirically by simulating error-prone nanopore reads from a random sample of 1000 bacterial reference genomes from the GTDB (Parks et al. 2022). We used the Rust implementation of the read simulator Badread (Wick 2019) to simulate nanopore reads with five different read lengths between 1000 and 5000 bp and repeated the simulations for 20 different read accuracy rates (80%, 81%, 82%, …, 99%). Next, we build separate HIXF index files of the 1000 genomes, one for each even k-mer value between 16 and 30. We only allow for even-numbered values for the k-mer size because we use canonical syncmers, with 16 being the practically smallest value to distinguish k-mers from different reference sequences and 30 being the maximum value that allows finding k-mer matches between error-prone reads and a reference sequence. Finally, we separately queried the simulated reads of different read accuracies against the created HIXF index files and calculated the minimum fraction of found syncmers for each read and every combination of read accuracy and k-mer size (Supplemental Table S3). This minimum matching ratio for the different read accuracy and k-mer size combinations is used by Taxor to calculate the threshold for the minimum number of syncmer matches between a given read and the reference sequences stored in the HIXF index.
Taxonomic profiling
The existence of homologous regions of genome sequences across multiple microbial species can lead to high false-positive rates if taxonomic profiling methods exclusively rely on sequence similarity information. However, setting a low sequence similarity threshold is essential for detecting all species in a sample if the sequencing accuracy hardly reaches values of 98%. Therefore, we apply a three-step filtering approach of potential hits between reads and reference sequences before refining the results using an EM algorithm that reassigns reads to references based on the number of k-mer matches and taxonomic abundances of matched references.
Before the filtering, reads are assigned to matched reference genomes if the number of matching k-mers exceeds a certain threshold. Thus, a read can be assigned to many reference genomes in the index. We perform the first filter step on the single read level, determining the best matching reference genome based on the maximum number of k-mer matches (maxkmatch) with the given read. All reference assignments to that read for which the number of matching k-mers is smaller than 0.8 × maxkmatch are considered spurious matches and removed from the results. The second filtering step creates a list of all reference genomes with at least one uniquely mapped read (a read assigned to exactly one reference genome). Consequently, we remove all read-to-reference assignments in the results for which the reference genome has no uniquely mapped read. Finally, we apply the two-stage taxonomy assignment algorithm used in MegaPath (Leung et al. 2020) to reduce suspicious matched references. In short, the algorithm identifies reference genomes with <5% of their matches being uniquely matched reads. If such a reference genome S also shares a certain amount (e.g., 95%) of matches with another reference genome T, all matches of reads to S are reassigned to T.
After filtering, we estimate the relative abundances of all matched references and reassign reads using a standard EM algorithm.
This approach iteratively maximizes the likelihood that a given read r comes from a reference genome g, which also maximizes the likelihood of the relative taxonomic abundances in the whole read set. Let G be the set of genomes in the database, and let πg be the probability that a sequencing read in the sample emanates from database genome g ∈ G. We define the likelihood of the mapped read set R as
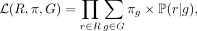 (1)
where ℙ(r|g) is the probability of read r coming from reference genome g. We use mc(r, g), the number of k-mer (or syncmer) matches between read r ∈ R and genome g ∈ G, and define ℙ(r|g) as
(1)
where ℙ(r|g) is the probability of read r coming from reference genome g. We use mc(r, g), the number of k-mer (or syncmer) matches between read r ∈ R and genome g ∈ G, and define ℙ(r|g) as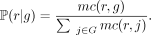 (2)
After initialization of the taxonomic compositions with
(2)
After initialization of the taxonomic compositions with 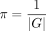 for all g ∈ G, we calculate in each iteration step an updated read assignment for each r ∈ R by
for all g ∈ G, we calculate in each iteration step an updated read assignment for each r ∈ R by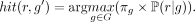 (3)
Based on the reassignment of reads, we update the taxonomic compositions π in each iteration step by accumulating the read
lengths of all reads mapping to a certain genome and normalizing this value by the genome length. Letting len(r) be defined as the length of read r and len(g) be defined as the length of genome g, we can write
(3)
Based on the reassignment of reads, we update the taxonomic compositions π in each iteration step by accumulating the read
lengths of all reads mapping to a certain genome and normalizing this value by the genome length. Letting len(r) be defined as the length of read r and len(g) be defined as the length of genome g, we can write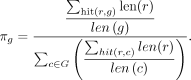 (4)
The numerator in Equation 4 can be interpreted as the depth of coverage on genome g in the sample under investigation. We divide the coverage of g by the sum of all genome coverages to get the relative taxonomic abundance of g in the sample. The single steps of the EM algorithm are repeated until convergence of the likelihood
(4)
The numerator in Equation 4 can be interpreted as the depth of coverage on genome g in the sample under investigation. We divide the coverage of g by the sum of all genome coverages to get the relative taxonomic abundance of g in the sample. The single steps of the EM algorithm are repeated until convergence of the likelihood 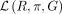 or after a predefined number of iteration steps (default, 10).
or after a predefined number of iteration steps (default, 10).
Software availability
We have made the source code of Taxor available as open-source software at GitLab (https://gitlab.com/dacs-hpi/taxor) and as Supplemental Code 1. For easy installation, we also made Taxor available as a Bioconda package (https://bioconda.github.io/recipes/taxor/README.html#package-taxor). Output files of the compared tools and of the Python and R scripts (R Core Team 2021) used for data analysis are available at https://osf.io/vqkcn/ and as Supplemental Code 2.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Svenja Mehringer, Vitor Piro, Enrico Seiler, and Knut Reinert (FU Berlin) for valuable discussions and comments on hierarchical filters and metagenomic profiling. This work was funded by the German Federal Ministry of Education and Research (BMBF) in the Computational Life Science program (Live-DREAM, 031L0175B) and Global Health program (ZooSeq, 01KI1905D) and has been supported by a grant from the BMBF/German Center for Infection Research (TI 06.904-FP2019 to B.Y.R.).
Author contributions: J.-U.U. developed software, analyzed and interpreted the data, and wrote the manuscript. B.Y.R. conceived and supervised the study, interpreted the data, and wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278623.123.
- Received October 10, 2023.
- Accepted May 23, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








