
Toward telomere-to-telomere cat genomes for precision medicine and conservation biology
- 1Department of Veterinary Integrative Biosciences, Texas A&M University, College Station, Texas 77843-4458, USA;
- 2Department of Biology, Texas A&M University, College Station, Texas 77843-4458, USA;
- 3Interdisciplinary Program in Genetics and Genomics, Texas A&M University, College Station, Texas 77843-4458, USA
Abstract
Genomic data from species of the cat family Felidae promise to stimulate veterinary and human medical advances, and clarify the coherence of genome organization. We describe how interspecies hybrids have been instrumental in the genetic analysis of cats, from the first genetic maps to propelling cat genomes toward the T2T standard set by the human genome project. Genotype-to-phenotype mapping in cat models has revealed dozens of health-related genetic variants, the molecular basis for mammalian pigmentation and patterning, and species-specific adaptations. Improved genomic surveillance of natural and captive populations across the cat family tree will increase our understanding of the genetic architecture of traits, population dynamics, and guide a future of genome-enabled biodiversity conservation.
The world's cat species (family Felidae) are among the most charismatic and recognizable flagship symbols of wildlife and animal conservation (Macdonald et al. 2015). This successful vertebrate lineage of apex predators has undergone diversification and extinction over the past 30 million years in response to the emergence and migrations of its prey species across nearly all terrestrial ecosystems, except for parts of Australasia and the polar extremes (Meachen-Samuels and Van Valkenburgh 2009; Price et al. 2012). The size of living cat species ranges two orders of magnitude, from the 1- to 2-kg rusty-spotted cat (Prionailurus rubiginosus) to the more than 300-kg tiger (Panthera tigris) (Wilson and Mittermeier 2009). One of the smallest felids, the nocturnal black-footed cat (Felis nigripes), inhabits the arid deserts of southern Africa and is a close genetic cousin to Felis catus, the domestic cat. This diminutive species is heralded as the deadliest feline, with the highest hunting success rate and appetite to match its enormous metabolism (Wilson and Mittermeier 2009). The largest felid that ever lived, the extinct saber-toothed cat Smilodon populator, bore long and formidable upper canines and weighed as much as 435 kg (Manzuetti et al. 2020). Aside from this large range in body size, the otherwise conserved felid body plan is so remarkably well-honed to predatory efficiency that it has changed only modestly in a few lineages throughout its history. The relative stasis in skeletal morphology masks a tremendous amount of variation in behavior, diet, pelage, and ecological preference.
So, what are the genetic alterations that distinguish a domestic cat from a tiger; make some cats orange, black, spotted, or striped; and make the popular Persian cat breed particularly susceptible to diseases like ringworm? For many of these questions, we now have solid answers owing to a long history of genetic investigation into the domestic cat for veterinary medicine and as an animal model for basic and biomedical research. Over the past 30 years, the feline genomic community has attempted to learn from the lessons of the Human Genome Project (HGP) and apply the latest technological advances to increase our knowledge about the most captivating aspects of feline biology and evolution. The domestic cat is one of the most common household pets worldwide, and it benefits from health care bested only by our own. This intimate familiarity of humans with cats in their living rooms and the many wild cat species displayed in zoos worldwide are characteristics that make cats a valuable vehicle for engaging students and the general public in the modern principles of genomics, conservation, and evolution (O'Brien 2003; Losos 2023). We have learned through comparative genomics that the chromosomes of humans and cats are far more conserved in gene content and gene order than similar comparisons of humans to the premier mammalian genetic model, the mouse, or nearly any other pair of mammal species separated by more than 70 million years of independent evolutionary history (O'Brien and Nash 1982; O'Brien et al. 1997; Murphy et al. 1999, 2000, 2005).
In the era of the democratization of whole-genome sequencing, what genetic secrets remain to be learned about all cats, great and small? How can lessons from the HGP continue to advance veterinary medical and conservation genomic advances for cat species? One of the most compelling findings of the HGP that became obvious with the first complete telomere-to-telomere (T2T) genome assembly in 2022 was that previous human assemblies were woefully incomplete despite nearly 20 years of the most intensive expert genome annotation and curation. Earlier drafts of the human genome were unfinished and missing hundreds of genes and ∼240 Mbp (equivalent to the length of human Chromosome 1) of particularly complex repetitive and functional sequences (Nurk et al. 2022). The major lesson in this biomedical milestone is that we must assume that all non-T2T eukaryotic genomes are missing at least this much (∼5%–10%) sequence information and likely contain thousands of incomplete and/or inaccurately annotated genes. Novel and critical genotype-to-phenotype linkages may be revealed through the resolution of and mechanistic understanding of transcriptional regulation of complex repetitive genes and functional tandem arrays (Eichler 2019; Miga and Eichler 2023). Filling these gaps and leveling the genomic playing field for similar advances in cats and other animal models will also require a complete genetic playbook.
Here we outline the latest advances in resolving the phylogenetic framework for feline comparative genomics, highlighting the unique promise of studying hybrid models to generate T2T genomes spanning the phylogeny in the next few years. We will discuss insights gained into genome structure and chromosome evolution through comparative genomics and recent success stories using genotype-to-phenotype mapping to clarify the basis for feline traits and species evolution. Lastly, we will explore the potential for discovering and applying genomic evidence to the management and conservation of these often threatened and endangered species. Together, we aim to show that the cat family is rapidly advancing into a valuable model system for evolutionary and biomedical research.
The cat family as a model system for comparative genomics
Forty-one currently recognized cat species are the product of eight recent and parallel radiations (Fig. 1; Johnson et al. 2006; Li et al. 2016a, 2019; Kitchener et al. 2017). Members of these lineages show both unique phenotypes and many examples of convergent evolution, including large body size (Panthera, Puma, and Acinonyx) and pelage and tail length in arboreal specialists (e.g., Neofelis sp. and Pardofelis marmorata) that misled early Felidae taxonomy. The first gene-based and mitochondrial studies that attempted to decipher the interrelationships of living species showed that the branching order within some of the eight phylogenetic lineages was robust to variation in genomic sampling, whereas the relationships within other species groups (e.g., Lynx, Panthera) and between the eight lineages have been challenging to resolve even with whole-genome data. Phylogenetic patterns are dramatically influenced by different factors, including the mode of inheritance/genomic location (i.e., mitochondria, sex chromosomes, autosomes) (Johnson et al. 2006; Trigo et al. 2008; Davis et al. 2010; Walters-Conte et al. 2014; Li et al. 2016a, 2019).
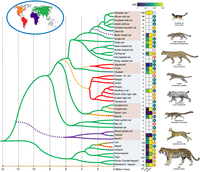
Phylogeny and evolutionary timescale of living felids. Branching relationships vary considerably across the genome, primarily as a function of gene flow/introgression and incomplete lineage sorting (Li et al. 2019). The relationships and species divergence times are derived from a consensus of regions of the genome with low recombination and of recent studies incorporating complete species sampling (Li et al. 2019; Jamieson et al. 2023; Lescroart et al. 2023; Yuan et al. 2023, 2024). The branches are color-coded based on current and inferred historical distributions from the fossil record and biogeographic reconstructions. Dashed branches indicate hypothesized dispersal events out of Europe and other regions. The columns shown to the right of the species names indicate the number of individuals with whole-genome sequence data sets available on NCBI's Sequence Read Archive (SRA) (last accessed January 11, 2024), as determined based on taxonomic classification (Kitchener et al. 2017; Lescroart et al. 2023). Heatmaps of contig (left) and scaffold (right) N50 metrics are shown from the highest-quality reference genomes from each respective species. Genomes nearing telomere-to-telomere (T2T) status are represented as assemblies with high contig and scaffold N50's (i.e., domestic cat), whereas species with low contig N50 but high scaffold N50 represent more fragmented assemblies. White boxes indicate species with no genome assembly available. IUCN conservation status logos are placed to the right of the assembly contiguity heatmaps. (NE) not evaluated, (LC) least concern, (VU) vulnerable, (EN) endangered, (NT) near threatened, (*) unpublished. Images by C. Buell.
Detailed phylogenomic analyses using whole-genome SNP and sequence data have clarified that rampant ancient gene flow is likely the primary cause underpinning the previous challenges in resolving the cat family tree (Fig. 1). Traces of past introgression have been found in genomes of the roaring cats of the genus Panthera (Li et al. 2016a, 2019; Figueiró et al. 2017; Mochales-Riaño et al. 2023; Sun et al. 2023); the Neotropical genus Leopardus (Trigo et al. 2008, 2013; Li et al. 2019; Trindade et al. 2021; Lescroart et al. 2023); the Palearctic and Neartic genus Lynx (Li et al. 2019; Harris et al. 2022); the Asian leopard cat radiation, genus Prionailurus (Li et al. 2016a, 2019); and the domestic cat lineage, genus Felis (Li et al. 2016a; Yu et al. 2021; Yuan et al. 2024). These findings match the emerging consensus in evolutionary biology that bouts of interspecific gene flow commonly occur following speciation despite the establishment of reproductive barriers that maintain species-level distinctiveness (Mallet et al. 2016; Edelman and Mallet 2021). The selective persistence of introgressed alleles has evolved from the adaptive benefit of the novel, divergent genomic variation as felids move across incredibly large landscapes. These have included genes associated with immunity, thermoregulation, and sensory perception pathways (Figueiró et al. 2017; Harris et al. 2022; Myers et al. 2022; Howard-McCombe et al. 2023).
Curiously, three other cat lineages, each containing a trio of extant species, show little evidence of gene flow. The puma lineage contains two large cats (the mountain lion and cheetah) and the diminutive, weasel-like jaguarundi. The caracal lineage evolved in Africa and includes three extant species: the long-legged, savannah-adapted serval, the dry-habitat dwelling caracal, and the tropically adapted African golden cat of West Africa. Finally, the Asian golden cat lineage contains the arboreal marbled cat, the phenotypically variable Asian golden cat, and the moderately sized bay cat endemic to the island of Borneo. Each of these species is ecologically distinct and is separated by relatively deep genetic splits (2 million years) (Li et al. 2016a; Patel et al. 2016). The combination of these factors, and the probability that some of these specialist species likely evolved in allopatry, likely explain their pronounced reproductive isolation compared with the other five cat lineages.
The extent to which hybridization can confound the accurate inference of species relationships has gained the attention of many systematists during the past decade in studies across the tree of life (Pennisi 2016; Wallis et al. 2017; Edelman et al. 2019; Nelson et al. 2021; Yang et al. 2021; Thawornwattana et al. 2022; Wanke and Wicke 2023). An important parameter that influences the local distorting effects of gene flow, as well as random genetic processes like incomplete lineage sorting, is the local rate of recombination and its interaction with natural selection (Schumer et al. 2018). Studies of feline pedigrees (Menotti-Raymond et al. 1999, 2003; Li et al. 2016b) and direct measurement of meiotic crossover rates (Segura et al. 2013) revealed that cats have the highest reported levels of recombination in mammals, with more variation in overall rate within the family than between ordinal and superordinal clades of mammals.
Tracking phylogenetic variation with different genomic features across the cat family tree has provided a strong benchmark on how to partition genome-wide phylogenetic variation to guide phylogenomic inference across the mammalian tree of life (Murphy et al. 2021; Harris et al. 2022). In particular, recombination rate profiles combined with species tree inference estimated in windows across chromosomes have identified regions of the genome prone to gene flow (e.g., telomeric regions), fitting the previous observations of a correlation between recombination rate and frequency of introgressed haplotypes owing to interaction with natural selection (Romiguier et al. 2013; Schumer et al. 2018). Notably, cat genome comparisons have uncovered a striking genetic architecture on large swaths of the X Chromosome in which gene flow is repelled in large (tens of megabases), homologous, gene-rich recombination cold spots shared across the cat family and even with other orders of mammals (Li et al. 2016a,b, 2019; Figueiró et al. 2017, Bredemeyer et al. 2023). A recent study analyzing structural variation from near-gapless single-haplotype genomes from multiple cat species has shown that these recombination cold spots are not produced by large inversions but rather many, smaller (<2-Mb) dispersed and gene-rich inversions (Bredemeyer et al. 2023). Indeed the 45- to 50-Mb recombination cold spot may hold a genomic record within mammalian genomes, harboring more than 500 genes, including components of the female X Chromosome inactivation center and ampliconic gene arrays with testis-biased gene expression (Brashear et al. 2021). The combination of these sex-biased gene regulatory networks may comprise the components of supergene >65 million years old. Future functional and population genomic studies are needed to further understand the impact of X Chromosome structural variation on reproductive isolation and trait evolution across the mammalian tree of life.
A path toward T2T genomes for all cats
The domestic cat genome has been among the most well-developed mammalian genome assemblies since the dawn of comparative sequencing following the completion of the HGP. Beginning with the first gene map and mapped feline phenotype (O'Brien and Nash 1982; O'Brien et al. 1986), the improvement of the domestic cat's genome sequence has been a long-term effort by the feline genomic community (Murphy et al. 2007; Davis et al. 2009; Montague et al. 2014; Li et al. 2016b; Buckley et al. 2020; Bredemeyer et al. 2021a, 2023). The motivation is to bring precision medicine to domestic cats in veterinary clinics and to advance conservation genomic initiatives in the 40+ other cat species. The earliest cat gene mapping studies revealed surprising syntenic conservation with the human genome compared with most other mammals (O'Brien and Nash 1982; Lyons et al. 1997; Murphy et al. 1999, 2000, 2007; Davis et al. 2009). Genetic linkage maps based on interspecific hybrid pedigrees were integrated with the gene-dense maps for disease and trait gene mapping (Menotti-Raymond et al. 1999, 2003, 2009) and together helped anchor early generations of feline genome assemblies into chromosome-level scaffolds (Pontius et al. 2007; Montague et al. 2014; Li et al. 2016b; Buckley et al. 2020). Because a majority of felids are chromosomally homosequential (Nash and O'Brien 1982; Wurster-Hill and Centerwall 1982), this enabled the alignment of short sequence reads, assembly contigs, and scaffolds from each new wild cat genome to the domestic cat reference genome while retaining much of the expected genomic context (Cho et al. 2013; Montague et al. 2014; Dobrynin et al. 2015; Abascal et al. 2016; Kim et al. 2016; Figueiró et al. 2017). As a result, Felidae was one of the first vertebrate families in which whole-genome shotgun sequencing or draft genome assemblies were completed for virtually all species.
Evidence for rampant hybridization between the ancestors of living cat species inferred from multispecies alignments should perhaps not be surprising given the prevalence of hybridization between distantly related felid species documented in captivity (Fig. 2; Gray 1972; Davis et al. 2015). These findings are bolstered by growing genetic evidence for hybrid zones between some natural populations of cat species (Schwartz et al. 2004; Homyack et al. 2008; Trigo et al. 2008). Remarkably, the evolutionary divergence between the parent species of the more extreme cat hybrids is equivalent to the genomic differentiation between humans and the different great ape species (Bredemeyer et al. 2023). Much of this reproductive success is attributable to the remarkable karyotypic conservation across the cat family, in which nearly all species possess a diploid karyotype of 2n = 38 (Nash and O'Brien 1982; Wurster-Hill and Centerwall 1982; Davis et al. 2009). Subtle differences exist in the morphology and centromere position of some chromosomes that define specific cat lineages/species, although the analysis of highly contiguous genome assemblies indicates these apparent changes are likely owing to centromere repositioning events and not rearrangements (Bredemeyer et al. 2021a, 2023). The one notable exception is a single-chromosome fusion shared by all cats of the Neotropical genus Leopardus (2n = 36). As a result, viable hybrid offspring of crosses between the domestic cat and Leopardus are restricted to the F1 and rarely early backcross generations.
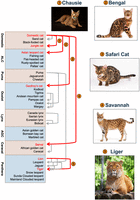
Cat hybrids have played an important role throughout the history of feline genomics. We have displayed many of the better-documented hybrids with gray and red arrows. Red arrows indicate some of the most common hybrid cat breeds that have been used in the generation of single-haplotype genome assemblies (Bredemeyer et al. 2021a, 2023; A.J. Harris, L.A. Lyons, T. Raudsepp, et al., unpubl.), as well as several genotype-to-phenotype mapping studies (Kaelin et al. 2024). (ALC) Asian leopard cat, (AGC) Asian golden cat. Chausie © tania_wild/Adobe Stock; Bengal cat © karandaev/Adobe Stock; Safari cat © karpichenko/Adobe Stock; Savannah © Eric Isselée/Adobe Stock; liger © Aleksey Ipatov/Adobe Stock.
Interspecific hybrids are of special note, given their prominence in multiple foundational aspects of feline genetics and genomics. Many cat species are popular exhibitions in zoos worldwide, and the cohabitation of similar yet distinct species led to many unexpected examples of interspecific hybridization spanning the cat phylogeny (Fig. 2; Gray 1972). For example, F1 crosses between the tiger and lion produce divergent growth patterns depending on the direction of the cross (see Gray 1972). Lion sire × tiger dam crosses produce ligers that are characterized by overgrowth phenotypes. In contrast, the reciprocal F1 cross produces tigons, which are roughly the same size as the parent species. These aberrant F1 growth phenotypes are often attributed to species-specific imprinting effects that arise from different mating strategies in lions (social) and tigers (solitary), although the precise genetic mechanism is unknown.
In the 1970s and 1980s, domestic cat breeders began experimenting with interspecific crosses with the Asian leopard cat (Prionailurus bengalensis) to introduce more exotic spotting patterns of wild felids into domestic cat lines. These first foundational crosses ultimately led to the creation of the Bengal domestic cat breed, which is among the most popular cat breeds worldwide (Roberts 2014). These same crosses were used for early biomedical research at the National Institutes of Health on feline leukemia virus (Benveniste and Todaro 1975) and later the first cat genetic linkage maps (Menotti-Raymond et al. 1999, 2003). Another early exotic cross between the domestic cat and a species from the Neotropical lineage of spotted cats, the Geoffroy's cat (Leopardus geoffroyi), gave rise to the exotic cat breed dubbed the Safari cat. Safari cats were a biomedical research model for hematopoietic stem cell research (Abkowitz et al. 1990, 1995, 1998). Two other popular domestic cat breeds derive from interspecific crosses: the Chausie (domestic cat × jungle cat, Felis chaus) and the Savannah (domestic cat × serval, Leptailurus serval). Others are developing, including the Marguerite, a cross between the domestic cat and the sand cat (Felis margarita) (Fig. 2).
Perhaps most importantly, the large prevalence of divergent hybrids derived from interspecific crosses in captivity provides one of the most powerful resources for generating T2T genome assemblies for many cat species and multiple individuals of domestic cats. Trio binning was developed to phase and independently assemble divergent parental haplotypes from F1 hybrids (Koren et al. 2018) and has been applied to multiple animal hybrids (Bredemeyer et al. 2021a; Oppenheimer et al. 2021; Delorean et al. 2023; Jevit et al. 2023). This approach uses a combination of parental short-read Illumina and long-read Pacific Biosciences (PacBio) sequences from the F1 offspring to efficiently phase >99.5% of the long reads (and often 99.999% of Hi-Fi reads) before assembly, thus removing phasing issues for diploid genome assembly. When implemented in the Verkko T2T assembler software for diploid genomes, trio-based approaches remain one of the most efficient techniques to achieve T2T genome assemblies (Li and Durbin 2024). Trio-binning was used to phase and assemble six ultracontiguous single-haplotype genome assemblies from three divergent F1 interspecific cat hybrids from PacBio continuous long reads (CLRs) (Bredemeyer et al. 2021a, 2023). These resources allowed gene discovery within and interspecific comparisons between large and complex regions that were fragmented or lacking in both short-read and long-read diploid-derived genome assemblies from the same species. These difficult-to-assemble regions are increasingly understood as playing important roles in disease resistance and susceptibility, as well as lineage-specific differences that may manifest as reproductive isolation (Bredemeyer et al. 2023; Miga and Eichler 2023; Brannan et al. 2024). In one notable example, the gene FELDI, which encodes the major cat allergen (Bergeron and Luquiau 1994), had been missing from all previous cat chromosome assemblies. Cat allergies affect 10% of the human population (Trifonova et al. 2023). Only in the newest single-haplotype genome assemblies (Bredemeyer et al. 2021a) were FELDI and its flanking regions properly assembled, opening the door toward producing allergen-free domestic cats (Brackett et al. 2022). Ongoing progress coupling these cat F1 hybrid resources with the latest PacBio HiFi+ Oxford Nanopore Technologies ultralong reads will deliver T2T quality genomes for several cat species within the coming year.
Genotype-to-phenotype mapping
The increasingly complete genomic resources for the domestic cat genome have transformed the genetic analysis of diseases and traits in domestic cats and have similarly begun to reap benefits for health-related studies in captive and natural populations of wild relatives in the family Felidae (Oh et al. 2017; Carroll et al. 2024). Genome-wide association studies (GWAS) in feline models have been very effective in identifying candidate loci that can be further interrogated for pathogenic mutations. These include diseases that are major health concerns in the feline population, including progressive retinal atrophy (Alhaddad et al. 2014), hypertrophic cardiomyopathy (Meurs et al. 2021), and many others (Buckley and Lyons 2020). The majority of these mapped traits and diseases are monogenic. However, numerous complex traits such as feline asthma, hyperthyroidism, and diabetes mellitus are polygenic and will likely require substantially larger cohorts to identify causal loci (Hernandez et al. 2022). Nonetheless, the diagnostic advances thus far are a testament to the organization of the cat genetic community under the umbrella of the 99 Lives Project (Aberdein et al. 2017; Oh et al. 2017). With variants called from genome alignments of hundreds of breed and random-bred domestic cats to the latest reference genomes (Buckley et al. 2020; Bredemeyer et al. 2023), discoveries of new causal variants are occurring at a regular, if not accelerated, pace (Lyons et al. 2021; Kopke et al. 2022; Lyraki et al. 2022; Occelli et al. 2022; Bilgen et al. 2023; Habacher et al. 2023; McElroy et al. 2023; Katz et al. 2024). We refer readers to recent reviews that comprehensively discuss the advances in domestic cat disease models and genetic testing (Buckley and Lyons 2020; Lyons and Buckley 2020; Lyons 2021).
A unique and attractive aspect of comparative genomic studies in the Felidae family is the remarkable amount of pelage color, pattern, and other trait variation found within and across the eight species lineages (Eizirik and Trindade 2021). This rich phenotypic resource provides multiple, often convergently evolved, phenotypes to interrogate with traditional genetic mapping and phylogenetic-based genotype-to-phenotype (PhyloG2P) (Smith et al. 2020) approaches to identify loci that are common in nature but not typically available for interrogation in mouse models. The past decade has witnessed a surge in this area, with work by Barsh and colleagues leading the identification of causal mutations in several genes, chromosomally mapped in earlier studies (Lyons et al. 2006; Eizirik et al. 2010), that regulate the timing and distribution of pigment cells (Kaelin and Barsh 2013). These include a master regulator gene, TAQPEP, that regulates tabby markings in domestic cats (i.e., spots vs. stripes) and cheetahs (Kaelin et al. 2012). For example, loss-of-function mutations in TAQPEP transform the regular, narrow stripes in a mackerel tabby cat into large whorls in a blotched cat (Kaelin et al. 2021). Kaelin and colleagues also identified another key gene, DKK4, that epistatically regulates TAQPEP and generates the dominant, uniform ticked coat pattern of the Abyssinian cat breed, observed in many wild felids (e.g., mountain lions). Using PhyloG2P approaches, Yuan et al. (2023) identified intriguing parallel amino acid substitutions in the clouded leopard and marbled cat, two arboreal specialists that inhabit similar Southeast Asian rainforests and have remarkably convergent traits: a long relative tail length, supination of the ankle joints for head-first descents, and marbled/clouded pelage patterns. Yuan and colleagues identified two parallel amino acid substitutions in two candidate genes (MYSM1 and GOLGB1) with known roles in mammalian pigmentation formation that are otherwise highly conserved across the cat family. Future PhyloG2P studies screening for relevant noncoding variants within improved T2T-quality genomes may reveal the genetic basis for these and other remarkable examples of ecomorphological convergence across the cat family.
The many millions of years of evolutionary distance separating the parents of the hybrid cat breeds have made them instrumental in furthering studies of mammalian hybrid incompatibilities in cat breed morphology and speciation. Kaelin et al. (2024) analyzed the hybrid ancestry of 904 Bengal cats to test the hypothesis that leopard cat alleles governing more exotic pelage traits originally desired during the breed's formation were selected for during breed formation. They observed a twofold depletion of leopard cat ancestry below the expected percentage based on the breeding paradigm. This included the absence of leopard cat ancestry across the large 50-Mb recombination cold spot on the X Chromosome, confirming this region's role in hybrid incompatibilities and speciation (Li et al. 2019; Bredemeyer et al. 2023). Surprisingly, they found that most traits associated with Bengal-specific coloration traits were owing to strong selection and near fixation of domestic cat alleles, as well as no evidence for strong selection for leopard cat alleles. In particular, Kaelin et al. (2024) mapped a domestic cat-derived FGFR2 color variant that confers the popular “glitter” phenotype in Bengals. In contrast, two other desirable coat color traits in Bengals, “charcoal” (a partial melanism manifesting as a “mask and cape” appearance) (Gershony et al. 2014) and pheomelanin intensity (i.e., the range of yellowish to rufous hair colors), were found to be explained by recent introgression of leopard cat haplotypes spanning the ASIP and CORIN genes, respectively. In both genes, leopard cat allelic expression is reduced in a domestic cat background, consistent with a hybrid incompatibility between parental genotypes from the two species.
Haldane's rule is one of two “rules of speciation” (Coyne and Orr 1989) and is the taxonomically widespread manifestation of hybrid incompatibility, in which the reproductive fitness of the heterogametic sex (XY males, ZW females) is most heavily impacted in crosses between two species. In mammalian interspecific hybrids, male sterility is characterized by consistent biomarkers: meiotic arrest at the pachytene stage of spermatogenesis and chromosome-wide overexpression of the X Chromosome owing to failure of meiotic silencing of chromatin (Gray 1972; Good et al. 2010; Campbell et al. 2013; Larson et al. 2017, 2018). In the four interspecies cat hybrids that have been examined, hybrid males suffer from spermatogenic failure and X Chromosome-wide up-regulation relative to the autosomes (Davis et al. 2015; Allen et al. 2020; Bredemeyer et al. 2021b). A GWAS performed in the Chausie breed of hybrid cats identified a rapidly evolving, functional X-linked macrosatellite repeat locus (DXZ4) as a major effect hybrid sterility gene, the first X-linked gene affecting hybrid sterility identified in mammals (Bredemeyer et al. 2021b). Subsequent comparative genomic analyses across the cat family tree revealed this repeat array is one of the most rapidly evolving regions of felid genomes and fits the model for a hybrid sterility gene (Bredemeyer et al. 2023). Future T2T genome-enabled evolutionary studies of ancestry and hybrid incompatibilities in other hybrid cat breeds will be informative toward understanding the role of DXZ4 and other rapidly evolving genomic elements in mammalian speciation and adaptation.
Finally, a study by Myers et al. (2022) revealed that ancient hybridization may have introduced wild cat alleles that later manifested as breed-specific disease susceptibility into the ancestors of domestic cat populations. In a genetic hunt for genes conferring susceptibility to ringworm that is prevalent in the Persian cat breed, Myers and colleagues identified a single major effect haplotype encompassing an S100 gene family cluster that encodes proteins with known roles in antifungal immunity. This haplotype, notably the region encompassing S100A9 that encodes a subunit of the antimicrobial protein calprotectin, was surprisingly divergent (13 nonsynonymous variants) between the case and control haplotypes, more typical of between-genus felid divergences. This prompted an investigation of the ancestry of the locus, which revealed that members of some species of the domestic cat lineage possessed two divergent haplotypes, one of which arose early in felid history as a separate branching event that is only present in the domestic cat, the Asiatic wildcat (Felis ornata) and the sand cat (F. margarita). The investigators posit that these two divergent alleles have been maintained by balancing selection and that the variant conferring disease susceptibility in Persian cats likely arose in arid climates occupied by the sand cat and the ancestors of the domestic cat, where it may have conferred resistance to other local pathogens. This study mirrors similar observations in human populations in which immunity loci are often enriched with ancient, admixture-derived Neanderthal ancestry (Kerner et al. 2021; Corcoran et al. 2023; Urnikyte et al. 2023).
A surge in wild cat genome assemblies inform conservation efforts
The iconic story of the cheetah's genetic legacy described in Tears of the Cheetah (O'Brien 2003) remains a paradigmatic example of the effects of loss of genomic diversity on species health and conservation. First reported nearly 40 years ago (O'Brien et al. 1985), the striking lessons of genetic monomorphism in this species set off a race and arguably sparked an entire discipline to develop genomic resources that could be applied to other endangered and threatened cat species (Menotti-Raymond and O'Brien 1995a,b). Eighteen felid species are currently listed as vulnerable or endangered by the IUCN, and six others are near threatened (Fig. 1). This effort to document and improve our understanding of genomic diversity in all living species of cats would lead to the revelation of similar effects of population declines and genetic inbreeding on health-related traits in many cat species, including lions, clouded leopards, cheetahs, and Florida panthers (Wildt et al. 1986, 1987; Roelke et al. 1993; O'Brien 2003; Johnson et al. 2010; Dobrynin et al. 2015).
Today, a burgeoning number of chromosome-level assemblies from different felid species populate the NCBI Assembly database. As of this writing, long-read genome assemblies are available or under development for nearly all of the “big cats” (Fig. 1), and a disproportionate amount of genomic sequencing has been focused on these species. For several threatened and endangered felid species, these genome assemblies have enabled population genomic sequencing studies that reveal critical insights into the genetic consequences of rapid demographic change and inbreeding load in small populations. Multiple population studies from large felids have shown a marked loss of heterozygosity and elevated proportions of runs of homozygosity (Dobrynin et al. 2015; Kim et al. 2016; Saremi et al. 2019; de Manuel et al. 2020; Armstrong et al. 2021; Paijmans et al. 2021; Ochoa et al. 2022; Prost et al. 2022; Shukla et al. 2022; Yuan et al. 2023), as well as evidence for purging of deleterious variation in small, isolated populations, including Arabian leopards (Mochales-Riaño et al. 2023), Iberian lynx (Kleinman-Ruiz et al. 2022), and Indian tigers (Khan et al. 2021).
Small felids, by comparison, are more poorly studied and lag behind the large cats. Similarly, the status, quality, and number of whole-genome sequences are comparatively small relative to those of the big cats (Fig. 1). Nonetheless, recent genome-enabled studies of black-footed cats identified high levels of inbreeding and elevated risks associated with amyloidosis in captive and natural populations (Yuan et al. 2024) and supported the management of captive breeding programs (Oh et al. 2017). In contrast, the critically endangered Scottish wildcat (a subpopulation of the European wildcat) has suffered massive genetic swamping owing to recent introgression by domestic cats (Howard-McCombe et al. 2023). In most of these case studies, recent anthropogenic activities have altered and fragmented the habitats of small and declining populations, leading to genetic isolation. Collectively, these population genomic studies identify the critical importance of collecting and interpreting genomic data for proper implementation and management of in situ and ex situ conservation programs.
Conclusions and future prospects
Now entering its fifth decade, the Feline Genome Project has blossomed into a driving force for the discovery and application of genetic information to advance basic biology, veterinary medicine, species management and conservation, and translational biomedical research. In the current era of genomic technological innovation, it is easy to imagine a day soon when all cat species will have at least one highly contiguous reference genome assembly, with a representative of each of the eight clades at or close to the T2T standard. The ability to make genotype-to-phenotype connections and medical advances with the cat model is only limited by appropriate animal cohorts and the completeness of genome assemblies. It is worth noting that most genomic studies on domestic and wild cat species have been based on highly fragmented diploid genome assemblies with much missing adaptive genetic variation. Complex genomic regions harboring genes influencing feline immunity (e.g., multimegabase major histocompatibility and immunoglobulin chain receptor loci), reproduction (e.g., sex chromosomes), and more remain collapsed or poorly represented. What impact do the elevated recombination rates in felids have in generating diversity that is not captured by genomic summaries of runs of homozygosity? How much functionally adaptive variation is present in these and other unresolved genomic regions? With T2T genomes around the corner for domestic and wild cat species, we anticipate exciting discoveries when the complete set of genetic variation is interrogated in genome-wide screens for disease and signatures of natural selection.
Of great importance to future feline genomic advances is improving the quality of the gene and regulatory annotations in genome assemblies so that researchers can confidently associate genetic variants with a disease or trait phenotype. Most causal mutations identified in GWAS, particularly those underlying complex traits, reside in noncoding regions adjacent to multiple genes (Pickrell 2014). In many cases, it is difficult to know which genes are influenced by noncoding intergenic SNPs or to know whether structural variation (e.g., inversions, deletions, translocations, etc.) within noncoding sequences will impact gene function. Prioritizing genetic variants for functional and translational applications based on evolutionary constraint has become a powerful approach to this end (Lindblad-Toh et al. 2011; Christmas et al. 2023; Dindot et al. 2023). It is then critical to continue to develop feline genomic resources that define regulatory regions against the backdrop of evolutionary constraint defined by mammalian history. As feline pangenomes develop with the acquisition of many new gapless genomes, integrating these with resources like the Zoonomia Consortia's 240-species genome alignment (Zoonomia Consortium 2020) will provide new opportunities to reveal novel regions of evolutionary constraint and innovation and facilitate PhyloG2P approaches across the cat family.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Greg Barsh, Victor David, Carlos Driscoll, Evan E. Eichler, Eduardo Eizirik, Jose Godoy, Kristofer Helgen, LaDeana Hillier, Chris Kaelin, Greger Larson, Kerstin Lindblad-Toh, Shujin Luo, Leslie Lyons, Marilyn Menotti-Raymond, Bill Nash, Steve O'Brien, Terje Raudsepp, Alfred Roca, Melody Roelke, Mike Tewes, and Wes Warren for discussions and collaborations on these topics over many years. Special thanks go to current and former members of the Murphy laboratory for their inspiration for and support of feline genomic studies, especially Brian Davis, Jan Janečka, Alison Pearks, Gang Li, Wesley Brashear, Kevin Bredemeyer, Alex Myers, and Nicole Foley. We also thank three anonymous reviewers for constructive comments on an earlier version of this manuscript. W.J.M. acknowledges recent research support on these topics from the Morris Animal Foundation (D16FE-011, D19FE-004), the U.S. National Science Foundation (DEB-1753760, DEB-2150664), and the Winn Feline/EveryCAT Health Foundation (MT16-015, W19-010). A.J.H. was supported in part by a National Institutes of Health training grant (T32 GM135115).
Footnotes
-
Article published online before print. Article and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278546.123.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








