
A new framework for exploratory network mediator analysis in omics data
- Qingpo Cai1,6,
- Yinghao Fu2,3,6,
- Cheng Lyu1,6,
- Zihe Wang2,
- Shun Rao2,
- Jessica A. Alvarez4,
- Yun Bai3,
- Jian Kang5 and
- Tianwei Yu2
- 1Department of Biostatistics and Bioinformatics, Emory University, Atlanta, Georgia 30322, USA;
- 2Shenzhen Research Institute of Big Data, School of Data Science, the Chinese University of Hong Kong, Shenzhen (CUHK-Shenzhen), Guangdong 518172, P.R. China;
- 3School of Medicine, the Chinese University of Hong Kong, Shenzhen (CUHK-Shenzhen), Guangdong 518172, P.R. China;
- 4Department of Medicine, Emory University, Atlanta, Georgia 30322, USA;
- 5Department of Biostatistics, University of Michigan, Ann Arbor, Michigan 48109, USA
-
↵6 These authors contributed equally to this work.
Abstract
Omics methods are widely used in basic biology and translational medicine research. More and more omics data are collected to explain the impact of certain risk factors on clinical outcomes. To explain the mechanism of the risk factors, a core question is how to find the genes/proteins/metabolites that mediate their effects on the clinical outcome. Mediation analysis is a modeling framework to study the relationship between risk factors and pathological outcomes, via mediator variables. However, high-dimensional omics data are far more challenging than traditional data: (1) From tens of thousands of genes, can we overcome the curse of dimensionality to reliably select a set of mediators? (2) How do we ensure that the selected mediators are functionally consistent? (3) Many biological mechanisms contain nonlinear effects. How do we include nonlinear effects in the high-dimensional mediation analysis? (4) How do we consider multiple risk factors at the same time? To meet these challenges, we propose a new exploratory mediation analysis framework, medNet, which focuses on finding mediators through predictive modeling. We propose new definitions for predictive exposure, predictive mediator, and predictive network mediator, using a statistical hypothesis testing framework to identify predictive exposures and mediators. Additionally, two heuristic search algorithms are proposed to identify network mediators, essentially subnetworks in the genome-scale biological network that mediate the effects of single or multiple exposures. We applied medNet on a breast cancer data set and a metabolomics data set combined with food intake questionnaire data. It identified functionally consistent network mediators for the exposures’ impact on the outcome, facilitating data interpretation.
Life exposures and medical interventions impact clinical outcomes through their interactions with the complex molecular network in the human body. Unraveling the network and finding key intermediaries to the exposures can help us identify the critical mechanisms of the action exerted by the exposures. Modern omics technology (e.g., RNA-seq and LC/MS metabolomics, etc.) generates unbiased molecular profiles, which allow us to find potential molecular mediators of the exposures by examining all genes/proteins/metabolites together with the exposures and clinical outcome.
However, finding mediators is a challenging problem even in traditional low-dimensional data. The statistical mediation analysis is the major modeling framework to study the relationship between the independent variable (exposure) and the dependent variable (outcome) via the inclusion of a third variable (mediator variable). Besides the direct effect between the exposure and the outcome, it is assumed that the exposure has an effect on the mediator, which in turn has an effect on the outcome. Figure 1A depicts the typical setting in mediation analysis (Baron and Kenny 1986; VanderWeele and Vansteelandt 2009).
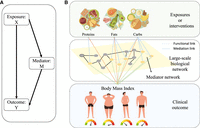
Overview of our algorithm. (A) A typical setting in mediation analysis. (B) The setup of the new method. Yellow nodes form mediator networks.
More recent advances in mediation analysis have adopted the potential outcome or counterfactual framework in causal inference (Rubin 1978; Robins and Greenland 1992; Pearl 2001) and have been widely used by researchers in the biostatistics, epidemiology, and causal inference fields (VanderWeele 2016). Built upon this framework, some approaches were developed to allow multiple mediators by including the interaction effect between the exposure and the mediator and the interaction between the mediators (VanderWeele and Vansteelandt 2009; Huang et al. 2014; Daniel et al. 2015).
In omics data analysis, our goal is to select mediators from the candidate pool of all the genes, proteins, or metabolites. The size of the candidate pool is magnitudes higher than in traditional mediation analysis settings. In the past few years, there has been a rapid development of mediation analysis methods for high-dimensional mediators. These methods can be roughly divided into three categories (Zeng et al. 2021).
The first type of method performs dimension reduction on the matrix of candidate mediators, and perform multivariate mediation analysis on a small number of latent variables. For example, through principal component analysis (PCA) or sparse principal component analysis (SPCA), the principal components of all potential mediators are extracted first (Huang and Pan 2016; Derkach et al. 2019; Zhao et al. 2020). By extracting the PCs that have no correlation with each other and using them as mediation variables, on the one hand, the dimension of the mediating variables is much lower than the dimension of the original data. On the other hand, because these principal components have no correlation, the mediation logic is relatively simple, without considering the dependencies between the various intermediary variables. But the problem is that each mediation variable is a linear combination of the original variables. Biological interpretations for the obtained mediating variables raise another question. More sophisticated methods use latent factors to integrate information about mediating variables (Derkach et al. 2019). Unlike other methods in this category, the latent factors are estimated as part of the mediation model, rather than as a step independent of the mediation analysis. However, the interpretation issue is not solved.
The second type of method performs a separate mediation analysis on each potential mediator individually and then combines the results in a false discovery rate (FDR) control process. Information about all potential mediators is used to estimate the parameters of the distribution under the null hypothesis H0. In mediation analysis, the H0 is a composite hypothesis that consists of three components: independent variable–mediator variable, mediator variable–dependent variable, and independent variable–dependent variable. This is different from regular FDR calculations. Methods such as JT-comp and JS-mixture have developed FDR theory and computational methods to accommodate this property of mediation analysis (Huang 2019; Dai et al. 2022).
The third type of method directly puts the independent variable, candidate potential mediators, and the dependent variable together for mediation modeling. Due to the very high dimension of the potential mediators, such methods target sparse models through regularized regression or Bayesian methods. Take high-dimensional mediation analysis (HIMA) (Zhang et al. 2016) as an example; the method first screens the potential mediators to moderately reduce the dimension of the problem, and then uses minimax concave penalty (MCP) regularization to achieve a sparse mediation model. The model was further extended to survival mediation analysis, which can decompose direct and indirect effects in the Cox proportional hazard framework (Luo et al. 2020). Bayesian models use proper priors, such as spike-and-slab prior, to achieve sparse models (Song et al. 2020, 2021). A more recent example is mediateR (Huang et al. 2023), which is a comprehensive mediation analysis framework that models high-dimensional mediators by using a regularized outcome model with ridge penalties. It can handle various types of response models, such as continuous, binary, and survival, and measure the mediation effects on appropriate scales for each type of response model.
Given the recent developments, the existing mediation analysis framework still faces tremendous difficulty in handling omics data. Firstly, some key assumptions in the traditional frameworks may not be reasonable in the large biological system, such as the sequential ignorability assumption, as the biological system is known for the pervasive existence of feedback loops, and unmeasured factors may well exist in such a complex system. Secondly, biological units are known to be organized in a modular structure. We expect mediators to be concentrated in certain pathways. The current framework cannot incorporate existing knowledge about gene/protein/metabolite relations into the inference. Thirdly, the type of relations modeled under the traditional framework is mostly linear or monotone, which cannot handle complex biological mechanisms such as nonlinear and dynamic relations. These limitations are difficult to address under the current framework.
When considering omics data, biological units function in a highly coordinated manner. Mediators to an exposure are likely to be functionally interconnected. Thus, our interest is not only to identify individual mediators from the vast pool of candidate mediators, but also to identify groups of mediators that are connected to the existing signal transduction, protein interaction, or metabolic network. The selected mediators should collectively show a coherent functional implication (Fig. 1B). We name such small subnetworks formed by the mediators as “mediator networks.”
We propose a new framework for predictive mediation analysis that can handle tens of thousands of candidate mediators, model nonlinear relations, and incorporate existing knowledge. The goal of our method is to sensitively detect potential mediators and mediator networks, which can be validated by further experiments. Instead of using the traditional mediation model which is very rigid, we focus on the prediction accuracy estimated by cross-validation as a criterion for potential mediator selection. A mediator should be significantly associated with the exposure, linearly or nonlinearly. It should also be able to improve prediction accuracy when incorporated into the model that includes the exposure. Given the limited sample size and vast search space, there is a potential issue of selecting false mediators. To address this issue, we further develop an algorithm to use the existing knowledge to help guide the selection of mediator networks that are biologically plausible.
We conducted simulation studies to illustrate the performance of the proposed statistical testing framework. We applied the proposed predictive mediation analysis framework on the molecular taxonomy of breast cancer international consortium (METABRIC) data, and the metabolomics and food intake questionnaire data set from the Emory–Georgia Tech Predictive Health Initiative Cohort. The resulting mediators and mediator networks were biologically plausible, indicating the method was effective in finding mediators and mediator networks effectively.
Results
We implemented the method as an R (R Core Team 2022) package named “medNet” (see Software availability). Within our package, we have incorporated three classical machine learning methods: logistic regression (LR), support vector machine (SVM), and random forest (RF). With the requirement of analyzing high-dimensional omics data, our package also supports parallel computing, which significantly enhances our computational efficiency. In this study, for all experiments conducted using medNet, the fold change parameter is uniformly set to 5.
Simulation study
We conducted a comprehensive set of simulation studies to evaluate the performance of our proposed algorithm in identifying predictive exposures and mediators. In these simulations, we used the Barabasi–Albert (BA) model to generate the initial candidate mediator network. The BA model is particularly well-suited for capturing the degree distribution of biological graphs. For our experiments, we configured the power law exponent to be 0.5. To simulate exposure and mediator values, we used a multivariate Gaussian distribution, the covariance structure of which depends on the structure of the candidate mediator network. We assumed the presence of three exposures and 500 candidate mediators, with only one of these exposures being truly predictive of the outcome.
We considered three distinct scenarios to establish relationships among the mediators. In scenarios (1) and (2), we strategically
selected a few initial mediators from the network with moderate degree, and then expanded cliques by incorporating a subset
of their neighboring vertices. In scenario (1), when generating mediator values using a multivariate Gaussian distribution,
we set the mean vector to 0 and the covariance matrix as 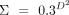 , where D represents the shortest distance between all pairwise vertices in the graph.
, where D represents the shortest distance between all pairwise vertices in the graph.
In scenario (2), while maintaining a mean vector of 0, we used the identity matrix I as the covariance matrix Σ. The true mediators were selected the same way as in scenario (1).
Unlike the preceding scenarios, scenario (3) involved hierarchical clustering on the pairwise shortest distance matrix of all mediators generated from the BA model. A specific cluster was chosen to represent the true clique of mediators. The covariance structure of the mediators was generated the same way as in scenario (1).
Exposure values were also generated from a multivariate Gaussian distribution with a mean vector of 0, and we set the identity
matrix I as the covariance matrix. One exposure was chosen as the true predictive exposure. Then we established correlations between
the true exposure and mediators, by adjusting the selected true mediator values using the corresponding exposure: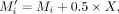 where X is the value of the true exposure. Then we used a LR model to generate the outcome Y. Y is generated as follows:
where X is the value of the true exposure. Then we used a LR model to generate the outcome Y. Y is generated as follows: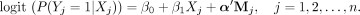
Here, n represents the sample size, for which we used values of 200, 300, and 400 in our simulation studies. We sampled coefficient values β = (β0, β1)T and α = (α1, α2, …, αm)T from a uniform distribution within the range of (1,2), and set part of them negative randomly. Mj denotes the value vector of true mediators, with a length of m. In this configuration, the chosen effect size setup aligns reasonably with real-world data sets, as illustrated in Supplemental Figure S1.
In both scenarios (1) and (2) of our simulation data set, we observed an average of approximately 22 true mediators, whereas in scenario (3), this average was around 20. To concisely summarize our simulation outcomes, we present the average values of three key performance metrics: the true positive rate (TPR), the false positive rate (FPR), and the FDR. The TPR represents the proportion of actual positives correctly identified as positives, whereas the FPR corresponds to the ratio of true negatives mistakenly identified as positives, and the FDR is the ratio of the number of false positive discoveries to the total number of positive discoveries. We provide TPR, FPR for exposures and all three metrics for mediators. The mean values of these metrics are displayed in Figure 2A, whereas the distribution for each is shown in Figure 2B. For exposure, there was a small chance of false positive when the sample size was small. For mediators, as the total number of candidate mediators is large and the graph structure conferred some correlation between true and false mediators, a certain level of false positive was present. With the increase in sample size, the TPR increased. However, without adjusting the hyperparameter H, false positives may remain stable or even marginally increase. Overall, the FDR decreased when sample size increased. Optimizing H can effectively manage the FPR, bringing it within acceptable limits. Given the large number of candidate mediators (500) and a small set of true mediators (around 20), an FDR near 0.3 is typical in such high-dimensional contexts and is considered manageable for further functional analysis in real-world data analyses. Notably, LR demonstrated the lowest FPR and FDR, in accordance with anticipated outcomes due to linear interrelations among exposures, mediators, and responses in the simulation setting. SVM and RF achieved higher TPR, at the cost of higher FPR (still ∼10%), causing the FDR to rise to ∼70%, which indicates they were overly sensitive in the linear setting. In the Discussion section, we further discuss the comparison of medNet with leading multivariate mediator identification methods MedFix (Zhang 2022) and bama (Yimer et al. 2022), and explore their capability under nonlinear settings.
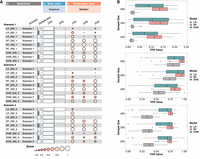
Performance of medNet under different simulation and model settings. This study assesses medNet's functionality across logistic regression (LR), support vector machine (SVM), and random forest (RF) models, in three distinct simulation scenarios. Consistent T (0.6) and H (18) settings were maintained, and each experiment was repeated 20 times. (A) The averaged true positive rate (TPR) and false positive rate (FPR) values for exposures, along with the scaled averages of TPR, FPR, and false discovery rate (FDR) for mediators, categorized by model_sample size_scenario and (B) the distributions of TPR, FPR, and FDR values for mediators.
In summary, the simulation studies demonstrate the robustness and good selection accuracy of our proposed method, which can effectively identify mediators across diverse scenarios, even with a limited number of samples. To further validate the efficacy of our approach, we apply the proposed procedure to analyze two real-world data sets.
Real data application: molecular taxonomy of breast cancer international consortium data
The data set contains clinical traits, gene expression, and single nucleotide polymorphism (SNP) genotypes derived from breast tumors collected from participants of the METABRIC trial. A total of 1363 breast cancer patients between the ages of 21 and 96 were included, with 31 clinical traits and gene expression levels for 489 genes. In our analysis, the outcome variable used was the 5-yr survival status, with all gene expression levels used as high-dimensional mediators and the Nottingham prognostic index (NPI) as the exposure variable.
NPI is calculated using specific clinical and pathological features of the tumor: NPI = (0.2 × S) + N + G, where S is the size of the index lesion in centimeters, N is the node status, and G is the grade of the tumor. NPI is widely used in prognostication and can be combined with other predictive factors to select patients for systemic adjuvant treatments. Our aim is to identify markers or pathways that underlie the molecular mechanism of NPI's predictive power, and potentially enhance the prognostic ability of NPI. We obtained the functional relation network between the genes from the High-quality INTeractomes (HINT) database (Das and Yu 2012). The candidate network consisted of 378 genes connected by 1313 edges. We then used medNet to identify combinations of gene features that can potentially mediate the relation between NPI and the survival rate. We used a LR model for this analysis with H set to 18 and T set to 0.6.
After identifying the potential mediators, a functional analysis was conducted to gain insight into the underlying biological processes. The GOstats (Falcon and Gentleman 2006) package in R was used to test the enrichment of Gene Ontology (GO) biological processes. We considered biological processes with P-values <0.01 and adjusted P-values <0.05 as significant GO terms. The top 10 pathways are shown in Table 1 for further analysis.
Top 10 GO biological processes in the subgraph identified by medNet on the METABRIC data set
Our analysis shows that biological processes mediating NPI and breast cancer progression are focused around themes of increased cell proliferation, invasive behavior, and metastasis, which is in line with prior research. Notably, we identified nuclear transport as a key pathway involved in breast cancer development and transformation. The dysregulation of nuclear–cytoplasmic transport of oncogenes and tumor suppressors is a hallmark of cancer cells, and modulation of nuclear–cytoplasmic transport activity has been shown to inhibit tumorigenesis (Kau et al. 2004).
Metastasis is a major contributor to treatment failure and is responsible for the majority of breast cancer-related deaths. Moreover, cellular metabolism is a vital player in breast cancer progression and metastasis. Numerous genes have been implicated in the regulation of cellular metabolism, and several pathways have been identified as critical players in this process. Specifically, pathways such as “Regulation of generation of precursor metabolites and energy,” “Negative regulation of catabolic process,” and “Regulation of macromolecule metabolic process” are all related to the regulation of cellular metabolism and have shown potential implications in breast cancer. Among the genes involved in the regulation of metabolism in breast cancer, STAT3 and AKT1 have been extensively studied and found to be enriched in the aforementioned pathways. In fact, there is substantial evidence for the association between STAT3 and AKT1 (Park et al. 2005), with major STAT3 response elements located within exon1 and intron1 regions of the AKT1 gene, indicating that AKT1 is a direct target gene of STAT3. Therefore, gaining a better understanding of the regulation of cellular metabolism in breast cancer, particularly the role of genes such as STAT3 and AKT1, may lead to the development of more effective treatments and personalized strategies for breast cancer patients.
The subnetwork consisting of AKT1, RASSF1, MYC, TGFBR2, CASP6, BCHE, TGFBR1, SMAD2, and ZFYVE9 plays a crucial role in regulating various aspects of tumor cell behavior, including survival, mobility, epithelial–mesenchymal transition (EMT), and metastasis. Specifically, the TGFB/SMAD and PI3K/AKT pathways are central in regulating these processes and show extensive cross talk and bidirectional regulation (Zhang et al. 2013). Additionally, it is worth noting that TGFB stimulates the phosphorylation of STAT3, with STAT3 being involved in the expression of TGFB1 gene. Also, AKT is believed to have a critical role in the progression and maintenance of cancer stem cells or cancer stem-like cells, as suggested in a previous study (Zhong et al. 2019). Moreover, it is important to highlight that MYC transcription is negatively regulated by SMAD2/3, and the down-regulation of MYC is a crucial event for achieving growth inhibition induced by TGFB, which is frequently impaired in cancer cells (Chen et al. 2002).
Our analysis suggests that the genes AKT1, BARD1, CDK, and others are involved in nuclear transport and that their dysregulation may significantly contribute to the development and progression of breast cancer. Studies have demonstrated that AKT1 regulates the subcellular localization of certain proteins involved in nuclear transport, and its activation has been linked to breast cancer cell proliferation and survival (Ju et al. 2007). Furthermore, AKT1 has been shown to promote the migration of mammary epithelial tumor cells across an endothelial cell barrier, thereby increasing their persistence and directionality. Besides, extensive evidence highlights the role of BARD1 as an oncogene in breast cancer patients, with potential uses as a prognostic/diagnostic biomarker and as a therapeutic target for cancer susceptibility testing and treatment (Hawsawi et al. 2022). These features include high histologic grade, large tumor size, lymph node metastases, and negative progesterone receptor status (Kim et al. 2008), all of which are correlated with the NPI. Collectively, our findings provide novel insights into the molecular mechanisms underlying breast cancer and identify potential targets for diagnostic, prognostic, and therapeutic interventions.
As shown in the network in Figure 3, ATR and CHEK1 are interconnected, and their network has been validated in the research (Abdel-Fatah et al. 2015); ATR–CHEK1 pathway is critical for genomic stability, and the deregulation of the ATR–CHEK1 network may influence breast cancer pathogenesis. High ATR protein and high cytoplasmic CHEK1 are associated with aggressive phenotype and poor prognosis. Another gene of interest, FANCD2, has also been well-studied in regard to cancer susceptibility and initiation. Tissue microarray analysis showed that up-regulation of FANCD2 is positively associated with tumor size and adverse prognosis in breast cancer (Feng and Jin 2019). FANCD2 is a pivotal player in the FA/BRCA repair pathway and is important for maintaining genome stability in response to various forms of DNA damage. The intra-S phase ATR–CHEK1 checkpoint promotes FANCD2 monoubiquitination and the assembly of subnuclear foci in response to DNA damage (Andreassen et al. 2004).
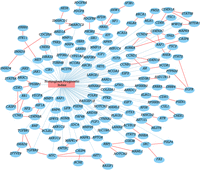
Predictive mediators identified by medNet in the METABRIC data set. Blue edges are statistical association edges and red edges are functional links.
Given the sample size is reasonably big, we studied the impact of sample size on the performance on the real data set. We conducted a random sampling procedure, where different subsets of the complete data set were selected based on specified sample size values. We then identified the number of intersecting terms and genes between results from the subsets and the full data set. As expected, the performance deteriorates with the reduction of sample size (Supplemental Fig. S2).
The Emory/Georgia Tech predictive health data set: metabolomics and food questionnaire data
This data set includes untargeted metabolomics and food questionnaire data, along with variables like age, gender, weight, and height. It encompassed 179 subjects, each with 86 nutrition variables from the questionnaire, and 856 metabolic features mapped to the KEGG human metabolic network. Our goal was to identify metabolic features and pathways mediating nutrient intake's impact on body mass index (BMI).
To analyze the data, we considered nutrition variables as the exposure, metabolomics variables as the mediator, and binary overweight status based on BMI as the outcome (BMI ≥ 25.0). We used the KEGG metabolic pathway map (Kanehisa and Goto 2000) as the candidate network. The corresponding candidate graph constructed for our analysis comprised 918 edges. Within this graph, 728 metabolic features were connected. Then for each nutrition variable, we applied the algorithm to find combinations of metabolic features that potentially mediate the effect of the nutrition variable on overweight status.
After finding the metabolic features that served as mediators for a nutrition variable, to facilitate interpretation, we further conducted a hypergeometric test to assess the overrepresentation of metabolic pathways among these selected metabolic features, based on the annotations of metabolic feature to pathways using metapone (Tian et al. 2022). The metabolic pathways that exhibited significant overrepresentation (with P-value <0.01) among the selected metabolic features were considered to be the mediator pathways, as illustrated in Figure 4.
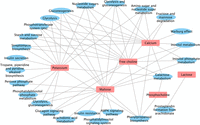
Identified mediator pathways (with P-value <0.01) for individual exposures. Significant associations were found for potassium, free choline, calcium, maltose, phosphocholine, and lactose.
Well-established pathways, including insulin secretion (Seino et al. 2011), insulin resistance (Kahn et al. 2006), and amino sugar and nucleotide sugar metabolism (Mir et al. 2022), were discovered as mediator pathways. In addition, our analysis found the links between the nutrition variables to obesity are also mediated by carbohydrate metabolism and lipid metabolism, which has been demonstrated in numerous studies. Excessive food intake leads to insulin resistance and leptin resistance, preventing cells from taking up glucose and the brain from receiving leptin signals to stop eating. This results in a persistent feeling of hunger, leading to a vicious cycle of overeating, increased fat gain, and elevated blood sugar levels.
Many pathways that were found by our analysis were related to diet, hormones, and obesity. Examples include prostaglandin formation from arachidonate (Sonnweber et al. 2018), galactose metabolism (Mhd Omar et al. 2021), gluconeogenesis (Gastaldelli et al. 2000), glycolysis (Sharma et al. 2020), AMPK signaling pathway (Rojas et al. 2011), glucagon signaling pathway (Del Prato et al. 2022), and so on. Our analysis also revealed that potassium plays a crucial role in obesity mediated through insulin secretion. Foods such as pork, rockfish, cod, tuna, milk, yogurt, soybeans, lima beans, kidney beans, and winter squash are rich in potassium, and their ingestion influences the plasma concentration of potassium (Udensi and Tchounwou 2017). Keeping potassium levels balanced is essential for proper insulin secretion, as imbalances in potassium concentrations can disrupt the normal functioning of ATP-sensitive potassium channels and affect insulin release. Low serum potassium concentrations decrease insulin secretion, leading to glucose intolerance, and hypokalemia induced by diuretics increases the risk of diabetes (Heianza et al. 2011).
Another microelement calcium also has been found to play a significant role in anti-obesity studies. Recent research suggests that calcium regulation is linked to glycolysis, the process by which glucose is converted into pyruvate and subsequently oxidized to generate ATP, the primary energy source for cells (Dejos et al. 2020). Whereas glycolysis is essential for normal cellular metabolism, there is some evidence to suggest that altered glycolytic metabolism may contribute to the development of obesity (Wu et al. 2005; Sharma et al. 2020). Studies of the cancerous Warburg effect, typically characterized by increased glucose uptake and glycolysis, have also shown that glycolytic metabolism may play a role in obesity and related diseases. Obesity is often associated with chronic low-grade inflammation, which can promote insulin resistance and impact cellular metabolism. Recent findings have demonstrated that the Warburg effect is related to a variety of inflammatory diseases (Palsson-McDermott and O'neill 2013), suggesting that its metabolic impact may be connected to obesity as well.
There are many mediators on our list that link with obesity closely and can be verified by existing evidence. 15-Hydroxyeicosatetraenoic acid (15-HETE, C05966, arachidonic acid metabolism pathway) is an important mediator that is found to be related to maltose. 15-HETE is known to promote angiogenesis, a process that is considered a major precursor to obesity and a key pathological characteristic of diabetic microvascular complications. In adipose tissue, 15-HETE up-regulates vascular endothelial growth factor and promotes angiogenesis, which can also regulate vascular remodeling and brain ischemia (Ma et al. 2011; Chen et al. 2017). However, tissue levels of 15-HETE are significantly reduced in the adipose tissue of people on high-fat diets (HFDs) (Wang et al. 2017); during the progression of obesity, inadequate angiogenic remodeling in adipose tissue can drive fibrosis and persistent hypoxia, leading to unhealthy adipose tissue expansion.
Another example is the mediator nicotinate (C00253, tropane, piperidine, and pyridine alkaloid biosynthesis pathway), which is closely linked with potassium. Nicotinate is a form of niacin, also known as vitamin B3, which is found in grains, the main source of carbohydrates in the human diet. Nicotinate has been found to be a potent stimulator of appetite and niacin deficiency may lead to appetite loss. Nicotinate impairs hepatic glucose assimilation by increasing both glycogenolysis and gluconeogenesis through some action. This effect is not related to antilipolysis (Miettinen et al. 1969), but induces insulin resistance of obesity.
Some of the most common food components that are related to obesity did not show a significant relation with BMI on their own. To explore the relationship between such common exposures and BMI through metabolites, we conducted an additional experiment using the data set, eliminating the threshold on exposure variable to initiate the computation. Given these nutrition exposures are correlated with each other, we examined the combined effects of exposures and their corresponding mediators using Algorithm 2. If the combination of a specific exposure and its corresponding mediators resulted in a greater impact compared to the effect of the exposure alone, we included this subnetwork in the predictive mediator network. This approach allowed us to identify significant associations between exposures and metabolites, providing valuable insights into the potential effects of common exposures and their interconnections. The results of this analysis are visually presented in Figure 5.
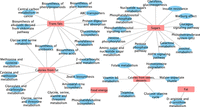
Potential mediator pathways associated with six common obesity-inducing food components: Trans fats, Sugars, Fat, Calories from fat, Food energy, and Calories from sweets and desserts.
Our findings align closely with current research. High caloric and energy intake is an established risk factor for obesity (Gulati and Misra 2014; Mlodzik-Czyzewska et al. 2020), which we found acts through various amino acid metabolism pathways, including cysteine and methionine metabolism, glycine, serine and threonine metabolism, 2-oxocarboxylic acid metabolism, methionine and cysteine metabolism, and so on. Studies have confirmed that changes in bacterial metaproteome, particularly in pathways related to amino acid metabolism, are most notable after a HFD. We observed that l-leucine (C00123) is highly correlated with the exposure of high calories from fat, as well as the other mediators. l-leucine is an essential amino acid classified as a branched-chain amino acid (BCAA) and is found abundantly in foods such as ground beef, peanuts, almonds, and salmon. Research indicates that BCAAs, particularly l-leucine, play a critical role in maintaining glucose balance by improving glucose recycling through the glucose–alanine cycle and regulating muscle protein synthesis via the insulin signaling pathway (Layman 2003). Restricting or removing l-leucine from the diet may disrupt the balance of different amino acids, resulting in reduced body fat levels and improved glucose tolerance, thereby protecting against obesity (Zhou et al. 2021).
Traditionally, it was believed that excessive caloric intake in the form of a HFD was the primary risk factor for obesity. Consuming excessive dietary fat can disrupt crucial metabolic pathways, including the function of nitric oxide synthesis (NOS), which affects the production of nitric oxide (NO). This disruption can have implications for blood flow, glucose uptake, and lipid metabolism (Togo et al. 2018). When NO levels are reduced, these processes can be disrupted, potentially promoting obesity. An essential substrate for NOS is l-arginine (C00062), which was significant in our predictive mediator network. Extensive research supports the benefits of l-arginine, often recommended for individuals dealing with obesity, to aid in fat loss and promote muscle mass formation (McKnight et al. 2010). Additionally, a HFD can disturb the balance of amino acid metabolism over time, resulting in reduced protein synthesis and increased protein breakdown in muscle tissue.
In recent years, there has been a shift in perspective, with more attention being given to the effects of sugars. It has been discovered that sugars can contribute to obesity from various angles (MacDonald 2016). Excessive sugar intake can lead to insulin resistance, which was confirmed in our results (Fig. 5). The overconsumption of sugars, which contributes to excessive energy intake, is associated with increased fat accumulation in the liver and muscles, and it impairs insulin signaling specifically in the liver (MacDonald 2016).
Calories from sweets and desserts are the main driver of weight gain and can have significant effects on various metabolic processes (Romieu et al. 2017). Pyridoxal phosphate (PLP, C00018, thiamin metabolism, glucose–alanine cycle, malate–aspartate shuttle, homocysteine degradation, and folate metabolism) plays a crucial role in the relationship between obesity and calories from sweets and desserts. PLP is the biologically active form of vitamin B6 and serves as a vital cofactor for key enzymes involved in kynurenine (Kyn) catabolism. Several studies have provided substantial evidence that PLP facilitates Kyn catabolism, leading to the inhibition of the AHR/STAT3/IL6 signaling pathway and enhanced insulin sensitivity (Huang et al. 2022). Additionally, PLP acts as a vital mediator in the homocysteine degradation pathway, which involves breaking down homocysteine, an amino acid derived from methionine metabolism. It is essential for maintaining normal homocysteine levels in the body, as elevated levels have been associated with various health issues, including obesity (Preninka et al. 2022). Furthermore, excessive consumption of sweets, desserts, and unhealthy fats, particularly those high in refined sugars, often lacks essential nutrients like vitamins B6, thiamin, and folate. These nutrient imbalances can disrupt energy metabolism and contribute to weight gain and obesity (Aasheim et al. 2008; Mlodzik-Czyzewska et al. 2020).
Discussion
In this work, we developed a new method for exploratory mediator analysis, and its associated R package medNet, which provides a robust mediation analysis framework focusing on predictive modeling. We proposed new definitions for predictive exposure, predictive mediator, and predictive network mediator. An estimation procedure was proposed to identify predictive exposure and predictive mediator. Following the definition of predictive mediator, the proposed method builds a network that combines the mediation dependence between the mediator and exposure and the functional relations between mediators. We also proposed two greedy algorithms that can incorporate various machine learning algorithms to find a subnetwork of exposures and mediators.
Our proposed “predictive mediation analysis” is not equivalent to the causal mediation analysis, but it can be used as a screening method for detecting the potential causal mediation relations among exposure X, mediator M, and outcome Y. Basically, suppose X has predictive mediation effects on Y through M. This does not necessarily imply that X also has the causal mediation effects on Y through M. On the other hand, if X does not have predictive mediation effects on Y through M, then it is not likely that X has the casual mediation effects on Y through M. For example, in the METABRIC data, we acknowledge that NPI may be the outcome of some genes, but it may also affect the expression of other genes. The biological process can be complicated and thus we do not claim the identified mediation effects have the causal interpretations. Instead, we define the concept of the “predictive mediation effects” where we believe the predictive power of NPI plus M is higher than that of only using NPI. That indicates M can facilitate improving the predictive power of NPI. The selected mediator network for NPI can be candidate causal mediators for future confirmatory studies.
The candidate network is a pivotal element in our methodology; its integration limits the search space and leads to more meaningful and biologically relevant results. Although the quality of the candidate network can influence medNet's performance, predictive mediators that are by themselves strongly related to the exposure and outcome can be detected irrespective of the network's quality. On the other hand, a low-quality network may result in less functionally relevant mediators being detected, as demonstrated in Supplemental Analysis S1, Supplemental Figures S6–S8, and Supplemental Tables S1 and S2. The justification of medNet regarding predictive exposures and mediators hinges on the predictive capacity of the incorporated variables. Even when considering a potential false mediator as a candidate, it will be discarded during the evaluation process if it lacks predictive power. This stringent evaluation criterion ensures that only variables with genuine predictive contributions are retained, enhancing the reliability and accuracy of the medNet methodology, and controlling the false positives even in the presence of a low-quality candidate network.
In the selection of hyperparameter settings of medNet, we need to consider the balance between sensitivity and specificity. The hyperparameter T is pivotal in mitigating false positives among the exposures. A lower threshold for T allows a greater number of exposures to enter the predictive network, but at the risk of increasing the false positives among the exposures and mediators (Supplemental Fig. S3). Both computational considerations and prior knowledge regarding the exposures may be taken into account. The hyperparameter H plays a crucial role in determining the inclusion of mediators. A higher value of H means more stringent control over false positives among the potential predictive mediators (Supplemental Fig. S4).
Simulation studies showed that the estimation procedure can identify predictive exposure and predictive mediators with good accuracy. However, given the majority of the large number of candidates are not predictive mediators, a certain level of false positives remain, which requires follow-up confirmatory studies to identify true mediators. We further compared the performance between medNet and two leading multivariate mediator analysis methods, MedFix and bama (Supplemental Fig. S5). The comparison uses LR models, the same as those used in the “Simulation study” found in the Results section, to evaluate each method's ability to discern mediators. As bama's default parameter setting detected very few mediators, we tuned bama such that the FDR level is similar to the other two methods. While achieving similar FDR, MedFix tended to detect much fewer mediators than the other two. medNet was less sensitive compared to bama when the sample size was low. However, when the sample size was higher, medNet was more sensitive than bama, detecting more true positives while maintaining a similar FDR level.
Our medNet method includes three distinct modeling approaches: LR, SVM, and RF. LR tends to emphasize the near-linear associations among exposures, mediators, and outcome. In contrast, SVM and RF are adept at capturing the nonlinear relations that may exist between these variables. These latter models may be more useful in high-dimensional settings where complex interrelations exist. The choice between these models should be guided by the nature of the underlying biological relationship. When the associations are presumed linear, LR offers simplicity, robustness, and interpretability. However, for more intricate biological systems where nonlinear interactions are suspected, SVM and RF may provide the necessary computational muscle to uncover these relationships. We conducted a simulation study where all relations were nonlinear (Supplemental Analysis S2), while medNet using RF and SVM achieved good results (Supplemental Fig. S9). The associated simulation data are available in Supplemental Simulation Data. Each model's performance should be evaluated in the context of the specific biological question at hand, ensuring the selection of the most appropriate tool for mediator discovery.
Overall, we introduced a new predictive mediator framework for omics data, which takes into account prior knowledge of functional associations between candidate mediators, and allows nonlinear associations. Applications on two real data sets showed that our method can detect subnetworks that not only mediate the relation between exposure and outcome, but also have meaningful biological interpretations. We believe the new framework and its associated methods can be a valuable addition to the computational tools that aim to unravel complex mechanisms of exposures and treatments.
Methods
Suppose there are m exposure variables 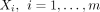 and v candidate mediator variables
and v candidate mediator variables 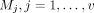 , and a binary outcome variable Y. We also assume the knowledge of a network (graph) G = (V, E), where the vertices V correspond to the candidate mediator variables, and E denotes the collection of functional relations between the variables. The graph G is optional.
, and a binary outcome variable Y. We also assume the knowledge of a network (graph) G = (V, E), where the vertices V correspond to the candidate mediator variables, and E denotes the collection of functional relations between the variables. The graph G is optional.
Predictive mediation analysis framework
We propose several new definitions for building a predictive mediation analysis framework in this section. We base our selection criteria on the area under the curve (AUC) of the receiver operating characteristic (ROC) curve (Bamber 1975; de Carvalho et al. 2013), which has a strong connection with the popular nonparametric Mann–Whitney U test (Bamber 1975; Faraggi and Reiser 2002).
We use AUC(Xi) and AUC(Xi, Mj) to denote the AUC for classifier that only uses feature Xi and uses features Xi, Mj, respectively. In the definitions proposed for predictive mediation analysis, we also need to state whether the evaluation metric AUC has predictive power or not. We consider this as a subjective statement to say whether a certain AUC value can be considered as predictive or not. Thus, we set up a predefined threshold value T as user input here, for example, T = 0.6. An AUC value is considered as predictive if it is larger than the threshold value T.
Predictive exposure
An exposure variable Xi is a predictive exposure about the outcome Y if AUC(Xi) > T. This definition can be used as a screening procedure to find predictive exposures.
Predictive mediator
The predictive mediator is defined for a given exposure. Mj is a predictive mediator of exposure Xi if the following conditions hold:
-
Xi, Mj are significantly associated,
-
AUC(Xi, Mj) > T,
-
AUC(Xi, Mj) > AUC(Xi),
-
AUC(Xi, Mj) > AUC(Mj).
Note here Xi does not need to be a predictive exposure. The first condition requires that Xi and Mj are significantly statistically associated. The other three conditions require that the AUC of the combination of exposure and mediator should be larger than the AUC of exposure or mediator alone.
Candidate network
Our goal is to find the combination of candidate mediators and single or multiple exposure variables while incorporating the dependence/functional structure that confers high predictive performance and good interpretability. There are two kinds of relations here. One is the dependence between the exposures and the candidate mediators, which is established using the definition of predictive mediator. The other is the functional relation between mediators, which is given by the network that denotes the functional links between mediators. We define that a mediation link between an exposure Xi and a mediator Mj exists if Mj is a predictive mediator of Xi. Using this definition for mediation links, we can build a network between all exposures and their predictive mediators. Then we merge it with the given functional network between mediators to form a combined network G. Given network G, we can adopt machine learning algorithms to find subnetworks combining exposures and mediators that are not only highly predictive of the outcome, but also are easy to interpret from a biological perspective.
Predictive network mediator for single exposure
Given the network G, we can now define the predictive network mediator for a single exposure. This definition is motivated by the fact that sometimes
researchers might have an exposure of particular interest and they would like to see which mediators combined with this exposure
are highly predictive about the outcome. Given an exposure X and a set of mediator nodes 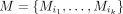 from the combined network G, M is predictive network mediator of exposure X if the following three conditions hold:
from the combined network G, M is predictive network mediator of exposure X if the following three conditions hold:
-
At least one mediator in M is a predictive mediator of X.
-
Removal of any one mediator from M will decrease AUC(X, M).
-
Adding another candidate mediator to M will not increase AUC(X, M).
The first condition requires that exposure X is connected to at least one mediator in M. The other two conditions require that the predictive performance of exposure X and mediator set M is optimal.
Estimation procedure and algorithm
In this section, we propose estimation procedures and algorithms to find predictive exposure, predictive mediators, and predictive network mediators.
Estimation for predictive exposure
Predictive exposure is defined as an exposure variable Xi that satisfies AUC(Xi) > T. This AUC is for evaluating predictive performance and should be evaluated on test data. In this paper, we adopt cross-validation to estimate the AUC value. We propose an estimation procedure to identify predictive exposure using repeated cross-validation. We use the mean of all the repeated cross-validation AUC as the final estimation for AUC. See below for details of the estimation procedure for predictive exposure:
-
Given a repeated number of R, for r = 1, …, R, calculate
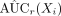 .
.
-
Check
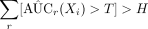 .
.
We can declare an exposure as a predictive exposure about the outcome if 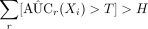 , where the threshold value is also a user input value. A higher value for H results in a low TPR and FPR and a lower value for H results in a high TPR and FPR. Estimation for AUC(Xi) is
, where the threshold value is also a user input value. A higher value for H results in a low TPR and FPR and a lower value for H results in a high TPR and FPR. Estimation for AUC(Xi) is 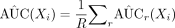 .
.
Estimation for predictive mediator
Assume a given exposure Xi and a given mediator Mj. First, we need to check whether Xi and Mj are significantly associated. There are several association metrics that are commonly used in statistics, that is, Pearson's correlation, Kendall rank correlation, and Brownian distance correlation (Székely and Rizzo 2009). Among all those choices, Brownian distance correlation can measure complex dependence between two random vectors with arbitrary dimensions. Thus, we adopt the Brownian distance correlation in the following content to measure the association between exposure and mediator. A statistical hypothesis testing procedure is provided in Székely and Rizzo (2013) and Székely et al. (2007) to test the significance of the Brownian distance correlation.
Given Xi and Mj are significantly correlated, we propose an estimation procedure for predictive mediator similar to the estimation procedure given in predictive exposure. See below for details:
-
Given a repeated number of R, for r = 1, …, R, calculate
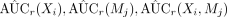 .
.
-
Check
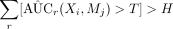 .
.
-
Check
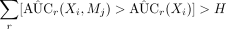 .
.
-
Check
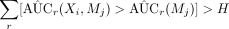 .
.
If all three inequalities hold, Mj is a predictive mediator of Xi.
Greedy algorithms for predictive network mediator
In this section, we first propose a greedy algorithm (Algorithm 1) for finding a predictive network mediator for a single exposure of interest. To address the need to study multiple exposures simultaneously, we propose another greedy algorithm (Algorithm 2). Algorithm 2 can be used as a screening procedure to find subnetwork of exposure and mediators from network G that are highly predictive about the outcome.
Greedy algorithm to select mediators for a single exposure
1: Input: exposure Xi, network G
2: Step 1: select 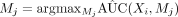 , where Mj is unvisited mediator neighbor nodes of Xi
, where Mj is unvisited mediator neighbor nodes of Xi
3: If Mj exists
4: mark Mj as visited and denote C = {Xi, Mj}
5: Else
6: Stop. Procedure is finished
7: Step 2: find all unvisited mediator neighbor nodes Ne for C
8: For each Nei ∈ Ne
9: calculate 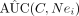
10: adopt estimation procedure for AUC(C, Nei) > AUC(C)
11: If there exists Nei that survives the estimation procedure
12: Set C = {C, Nei} where 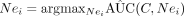 for Nei ∈ Ne.
for Nei ∈ Ne.
13: Mark Nei as visited. Go to Step 2
14: Else
15: Store result C. Go to Step 1
Greedy algorithm for mediator selection for multiple exposures
1: Input: network G
2: Step 1: select unvisited pair 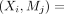
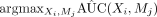 among all unvisited mediators
among all unvisited mediators
3: If Mj exists
4: mark Mj as visited and denote C = {Xi, Mj}
5: Else
6: Stop. Procedure is finished
7: Step 2: find all unvisited neighbor nodes Ne for C
8: For each Nei ∈ Ne
9: calculate 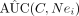
10: adopt estimation procedure for AUC(C, Nei) > AUC(C)
11: If there exists Nei that survives the estimation procedure
12: Set C = {C, Nei} where 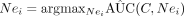 for
Nei ∈ Ne.
for
Nei ∈ Ne.
13: Mark Nei as visited if Nei is a mediator. Go to Step 2
14: Else
15: Store result C. Go to Step 1
Software availability
The medNet software package, along with its source code, has been made available for download on GitHub (https://github.com/EddieFua/medNet). Furthermore, scripts used in conducting the primary analyses are accessible in this repository. The medNet source code and scripts for reproducing supplemental analyses are also available as Supplemental Code.
Data access
The METABRIC data set provides phenotype data and gene expression data, which are available for download from cBioPortal (https://www.cbioportal.org/study/summary?id=brca_metabric). For researchers interested in obtaining the raw data from the metabolomics and food intake questionnaire data set, it is recommended that they make a request with the Emory–Georgia Tech Predictive Health Institute (https://med.emory.edu/departments/medicine/research/centers-institutes/predictive-health/index.html). The preprocessed data of these two data sets can be downloaded from GitHub (https://github.com/EddieFua/medNet).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was partially supported by National Institutes of Health grants (R01DA048993, R01MH105561, R01GM124061), National Science Foundation grant IIS2123777, Guangdong Talent Program (2021CX02Y145), Guangdong Provincial Key Laboratory of Big Data Computing, and Shenzhen Key Laboratory of Cross-Modal Cognitive Computing (ZDSYS20230626091302006).
Author contributions: J.K., T.Y., and Q.C. conceived the concept. Q.C., C.L., and Y.F. programmed the method. Y.F., C.L., Z.W., S.R., and Y.B. conducted simulation studies and real data analyses. Y.F., C.L., J.A.A., and Y.B. conducted the interpretation of real data results. Q.C., Y.F., Y.B., J.K., and T.Y. wrote the draft manuscript. All authors participated in corrections and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278684.123.
-
Freely available online through the Genome Research Open Access option.
- Received November 2, 2023.
- Accepted April 11, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








