
Navigating the landscape of epitranscriptomics and host immunity
- 1Bioinformatics Interdepartmental Program, University of California, Los Angeles, California 90095, USA;
- 2Department of Integrative Biology and Physiology, University of California, Los Angeles, California 90095, USA;
- 3Molecular Biology Interdepartmental Program, University of California, Los Angeles, California 90095, USA;
- 4Molecular Biology Institute, University of California, Los Angeles, California 90095, USA
Abstract
RNA modifications, also termed epitranscriptomic marks, encompass chemical alterations to individual nucleotides, including processes such as methylation and editing. These marks contribute to a wide range of biological processes, many of which are related to host immune system defense. The functions of immune-related RNA modifications can be categorized into three main groups: regulation of immunogenic RNAs, control of genes involved in innate immune response, and facilitation of adaptive immunity. Here, we provide an overview of recent research findings that elucidate the contributions of RNA modifications to each of these processes. We also discuss relevant methods for genome-wide identification of RNA modifications and their immunogenic substrates. Finally, we highlight recent advances in cancer immunotherapies that aim to reduce cancer cell viability by targeting the enzymes responsible for RNA modifications. Our presentation of these dynamic research avenues sets the stage for future investigations in this field.
Throughout their life cycle, RNAs undergo numerous processing events. Among these activities are RNA modifications, which describe changes to the chemical composition of individual nucleotides. Over 150 types of RNA modifications, also referred to as epitranscriptomic events or marks, have been characterized to date (Boccaletto et al. 2022). RNA modifications can regulate a broad range of biological processes, including those mounted by the host immune system. Here, we broadly classify the effects of immune-related epitranscriptomic events into the following categories: regulation of immunogenic RNAs, regulation of genes involved in innate immune response, and facilitation of adaptive immunity. Given extensive evidence for their immune relevance, we focus on reviewing the contributions of the following internal (non-cap) host RNA modifications: N6-methyladenosine (m6A), 5-methylcytosine (m5C), adenosine deamination to inosine (A-to-I editing), cytosine deamination to uracil (C-to-U editing), and pseudouridine (Ψ) (Fig. 1). We also summarize salient methods for genome-wide characterization of RNA modifications and their immunogenic substrates. This is followed by a discussion of how manipulation of RNA modifications has been leveraged in cancer immunotherapies. An exhaustive account of all studies related to epitranscriptomics or immunity is outside the scope of this review; however, our goal is to provide a landscape view of research being conducted at the intersection of these exciting fields.
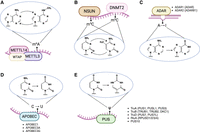
RNA modifications and their corresponding writer/editing enzymes: (A) N6-methyladenosine (m6A) deposited by a methyltransferase complex that includes the METTL3–METTL14 heterodimer, (B) 5-methylcytosine (m5C) deposited by the DNA methyltransferase homolog DNMT2 and methyltransferases from the NSUN family, (C) deamination of adenosine to inosine by ADAR enzymes, (D) deamination of cytosine to uracil by APOBEC proteins, and (E) conversion of uridine to pseudouridine (Ψ) by pseudouridine synthase enzymes. When applicable, the generic writer/editing enzyme name is depicted alongside the list of specific protein names. Pseudouridine synthases are grouped by family. Created with BioRender (https://www.biorender.com).
The cast of immune-related RNA modifications
Most known RNA modifications involve the alteration of residues that simply “decorate” the nucleoside (e.g., the addition of methyl groups). This category of epitranscriptomic marks includes m6A and m5C, which refer to the methylation of the sixth nitrogen atom of adenine and the fifth carbon atom of cytosine, respectively (Fig. 1A,B). m6A modifications are deposited by a methyltransferase complex that includes a heterodimer (Liu et al. 2014) composed of the writer proteins METTL3 (Bokar et al. 1997) and METTL14. The METTL3–METTL14 complex is tethered to target sites by the WT1 associated protein (WTAP) (Ping et al. 2014). Although all three proteins are required for m6A methyltransferase activity, METTL3 is the primary nuclear methyltransferase that adds m6A during transcription (Wang et al. 2016). Additional accessory proteins are also involved with methyltransferase complex activity, including VIRMA (Yue et al. 2018), RBM15-RBM15B (Patil et al. 2016), and ZC3H13 (Wen et al. 2018). On the other hand, m5C is catalyzed by the DNA methyltransferase homolog DNMT2 (also known as TRDMT1) and the NOP2/Sun (NSUN) methyltransferase family (Reid et al. 1999; Goll et al. 2006). Although RNA m5C methyltransferases were first studied in the context of tRNAs and rRNAs, they were later shown to target mRNAs and other noncoding RNAs (Squires et al. 2012).
A handful of RNA modifications—often referred to as RNA editing—can change the identity of the RNA nucleoside itself (i.e., a conversion from the four basic nucleosides: adenosine, cytosine, guanosine, or uridine). Two prominent types are A-to-I and C-to-U RNA editing (Fig. 1C,D), which describe deamination events performed by members of the ADAR (Bass et al. 1997) and APOBEC (Wedekind et al. 2003) protein families, respectively. The ADAR family consists of ADAR (also known as ADAR1, encoded by the ADAR gene), ADARB1 (also known as ADAR2, encoded by the ADARB1 gene), and the catalytically inactive ADARB2 (also known as ADAR3, encoded by the ADARB2 gene) (Cho et al. 2003). ADAR consists of two major isoforms: the constitutively expressed isoform ADAR-p110 and the interferon-induced isoform ADAR-p150 (Patterson and Samuel 1995). Although the shorter p110 isoform is predominantly nuclear, the longer p150 isoform can be found in both the nucleus and cytoplasm (Poulsen et al. 2001). Nonetheless, both isoforms have exhibited the ability to shuttle between these two compartments (Strehblow et al. 2002; Fritz et al. 2009). Although A-to-I editing enzymes target RNAs, some members of the APOBEC family exclusively deaminate DNA. However, APOBEC1 (Teng et al. 1993), APOBEC3A (A3A) (Sharma et al. 2015), and APOBEC3G (A3G) (Sharma et al. 2016) have been shown to perform RNA editing. C-to-U editing is far less prevalent than A-to-I editing in humans; although millions of A-to-I editing sites have been identified in the human genome (Bazak et al. 2014; Mansi et al. 2021), most studies have not identified more than a few hundred C-to-U editing sites (Rosenberg et al. 2011; Sharma et al. 2015; Sharma et al. 2019; Alqassim et al. 2021).
Pseudouridine (Ψ) is an isomer of uridine that is formed via the activity of pseudouridine synthases (Fig. 1E). Currently, 13 pseudouridine synthases have been identified in eukaryotes (Borchardt et al. 2020). These enzymes are classified into six families: RsuA, RluA, TruA, TruB, TruD, and PUS10 (Hamma and Ferré-D'Amaré 2006; Borchardt et al. 2020), although RsuA-type synthases have only been found in bacteria (Rintala-Dempsey and Kothe 2017). Previous efforts have revealed various characteristics of different pseudouridine synthases. For example, PUS1, PUS7, and RPUSD4 have been shown to regulate splicing and 3′ end processing (Martinez et al. 2022), whereas TRUB1 has been shown to confer higher pseudouridylation levels relative to other synthases (Dai et al. 2023; Zhang et al. 2023). Notably, pseudouridine has been shown to increase mRNA stability based on reporter assays (Karikó et al. 2008). However, reports of the effects of pseudouridine within in vivo systems have drawn mixed conclusions, including increasing (Schwartz et al. 2014; Dai et al. 2023), decreasing (Nakamoto et al. 2017), and having no effect on mRNA stability (Zhang et al. 2023).
Genome-wide identification of RNA modifications
A-to-I and C-to-U RNA editing events can be identified in RNA-seq data based on mismatches to the reference genome, although careful consideration is required to exclude false positives that stem from SNPs or sequencing errors (Bahn et al. 2012; Ramaswami et al. 2012; Lee et al. 2013; Porath et al. 2014), especially within coding regions (Gabay et al. 2022). Because inosines resulting from A-to-I editing are converted to guanosines by reverse transcription, A-to-G mismatches can be indicative of A-to-I editing. Using general linear models, the GIREMI and L-GIREMI methods leverage lack of allelic linkage as a feature to distinguish editing sites from SNPs (Zhang and Xiao 2015; Liu et al. 2023b). Although a pair of SNPs in the same read is expected to exhibit allelic linkage owing to originating from the same haplotype, an editing site and a SNP are more likely to exhibit variable allelic linkage. This is because in most cases (i.e., in the absence of allele-specific editing), RNA editing occurs post-transcriptionally to either allele in a stochastic manner. Aside from these computational methods, specialized experimental protocols have also been developed for inosine detection. One of these methods is inosine chemical erasing (ICE)-seq, which performs ICE using cyanoethylation combined with reverse transcription to identify inosines based on a decrease in G signals in a sequence chromatogram of cDNA (Suzuki et al. 2015). Another method is EndoVIPER-seq, which uses immunoprecipitation (IP) with an inosine-binding enzyme endonuclease V (EndoV) to enrich for edited transcripts before sequencing (Knutson and Heemstra 2020). Despite these alternatives, most studies opt for RNA-seq when performing editing analysis owing to its broader accessibility.
On the other hand, RNA modifications that do not entail changes to the actual nucleotides cannot be detected solely based on RNA–DNA mismatches in standard RNA-seq data. m6A modifications are commonly identified through IP-based techniques. These include MeRIP-seq (Meyer et al. 2012) and m6A-seq (Dominissini et al. 2012), both of which can be used to identify modified sites at a relatively low resolution of ∼100–200 nucleotides (nt) using IP-based techniques. Single-nucleotide resolution methods that use cross-linking-based technologies such as m6A-CLIP (Ke et al. 2015), miCLIP (Linder et al. 2015), and PA-m6A-seq (Chen et al. 2015) were developed a few years later. In addition, m6A-LAIC-seq was developed to enable isoform-level characterization of methylated transcripts (Molinie et al. 2016). These methods were followed by single-nucleotide resolution antibody-free approaches, including m6A-REF-seq (Zhang et al. 2019) and MAZTER-seq (Garcia-Campos et al. 2019), both of which enable m6A detection using the MazF endoribonuclease. Other antibody-free methods include DART-seq (in which m6A residues are found adjacent to induced C-to-U mismatches) (Meyer 2019), m6A-SEAL-seq (which involves FTO-assisted m6A selective chemical labeling) (Wang et al. 2020c), and m6A-label-seq (in which m6A sites are metabolically modified with N6-allyladenosine) (Shu et al. 2020). More recent methods enable m6A quantification at single-base resolution. Although m6A-SAC-seq (Hu et al. 2022) and eTAM-seq (Xiao et al. 2023) use regression-based methods to predict m6A levels, GLORI (Liu et al. 2023a; Shen et al. 2024) enables absolute m6A quantification.
Although early m6A detection methods featured IP-based techniques, m5C modifications were first identified using bisulfite sequencing (Schaefer et al. 2009; Squires et al. 2012), in which cytosine (but not m5C) is chemically converted to uracil. Later in the early 2010s, RNA immunoprecipitation (RIP) approaches followed by deep sequencing—such as m5C-miCLIP (Hussain et al. 2013b) and 5-Aza-IP (Khoddami and Cairns 2013)—were introduced for m5C identification. These methods were followed by TAWO-seq (Yuan et al. 2019), which enables bisulfite-free and base-resolution identification of m5C based on peroxotungstate oxidation. Recently, metabolic propargyl labeling and sequencing (MePMe-seq) has been shown to facilitate the simultaneous identification of m6A and m5C sites at single-nucleotide resolution based on distinct termination profiles (Hartstock et al. 2023). Although bisulfite-based approaches are still regarded by some researchers as the gold standard detection method (Hussain et al. 2013a; Ma et al. 2022), the high variability in the number of m5C sites detected across these studies highlights the importance of developing stringent computational pipelines for removing false positives (Huang et al. 2019).
Several transcriptome-wide pseudouridine-detection methods have been published since the mid-2010s (Schwartz et al. 2014; Carlile et al. 2015; Li et al. 2015). These methods rely upon a N-cyclohexyl-N′-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate (CMC) reaction with pseudouridines, which results in a stop signature during reverse transcription (Sun et al. 2023). More recently, bisulfite-based approaches adapted from improved m5C detection protocols (Khoddami et al. 2019) have been developed. These detection methods, which include BID-seq (Dai et al. 2023) and PRAISE (Zhang et al. 2023), are capable of quantitatively estimating the modification level of each site based on nucleotide deletion signatures.
In addition to these antibody and chemical-based detection methods, nanopore direct sequencing of full-length native RNA molecules is increasingly receiving attention as a modality for modification detection. Nanopore direct RNA sequencing features a helicase motor that facilitates the movement of an individual RNA strand through a protein nanopore, generating a monovalent ionic current signature that is used by base-calling algorithms to predict the corresponding nucleotide sequence (Garalde et al. 2018; Jain et al. 2022). Strategies for identifying RNA modifications in this type of data either involve the detection of modification-induced base-calling errors or involve prediction of modified sites through analysis of the raw signal itself (Furlan et al. 2021; Leger et al. 2021). Many detection algorithms use training data consisting of sequences with the modification of interest alongside sequences lacking such modifications. An example includes data sets in which a writer enzyme has been knocked down. Most of these methods have been applied to identify m6A. Signal intensity-based methods for m6A include Nanocompore (Leger et al. 2021), xPore (Pratanwanich et al. 2021), Nanom6A (Gao et al. 2021b), MINES (Lorenz et al. 2020), and m6Anet (Hendra et al. 2022). On the other hand, base-calling methods include EpiNano (Liu et al. 2019b), DiffErr (Parker et al. 2020), DRUMMER (Price et al. 2020), and ELIGOS (Jenjaroenpun et al. 2021). Recently, NanoSPA (Huang et al. 2024) was developed to simultaneously profile both m6A and pseudouridine in the transcriptome by leveraging m6A-SAC-seq (Hu et al. 2022) and the group's previously published model, NanoPsu (Huang et al. 2021), respectively. NanoPsu and other methods (Begik et al. 2021; Tavakoli et al. 2023) are base-calling error-based approaches, although methods trained on features extracted from nanopore sequencing data raw signal have also been developed (Hassan et al. 2022). Nanopore-based methods for A-to-I editing site detection also exist, including DeepEdit (Chen et al. 2023) and Dinopore (Nguyen et al. 2022). Currently, m5C sites has not been explored transcriptome-wide to the same extent as other modifications, although they have been detected in synthetic oligonucleotides using Nanocompore (Leger et al. 2021). Additionally, CHEUI was developed to perform simultaneous prediction of m6A and m5C using nanopore direct RNA sequencing data (Acera Mateos et al. 2022). With the slew of methods being developed to uncover RNA modifications in nanopore data, we anticipate the imminent growth of this list of tools.
RNA modifications and innate immunity
Modulation of transcript immunogenicity
Pattern recognition receptors (PRRs) initiate antiviral responses upon recognition of pathogen-associated molecular patterns (PAMPs). Nucleic acids, including RNAs, are among the PAMPs that can activate these pathways. The presence or absence of RNA modifications can determine whether this sensing, and subsequent antiviral activation, is initiated. In other words, epitranscriptomic marks can modulate the immunogenicity of their host transcripts. Previous investigations have shown that double-stranded RNAs (dsRNAs), circular RNAs (circRNAs), and long noncoding RNAs (lncRNAs) are subject to such regulation.
Perhaps the most well-known class of endogenous immunogenic RNAs is dsRNAs (Chen and Hur 2022), particularly those that are subject to A-to-I editing (Fig. 2A). The immunogenicity of dsRNAs arises from their structural similarity to dsRNAs, which are often produced upon viral infection (Weber et al. 2006). Indeed, mounting evidence suggests that a major function of ADAR editing is to modulate immune response (Heraud-Farlow et al. 2017; Eisenberg and Levanon 2018). Following observations that double knockout with Mavs (Mannion et al. 2014) rescues the embryonic lethality of Adar mutant mice (Hartner et al. 2004, 2009; Wang et al. 2004), the absence of ADAR editing activity (Liddicoat et al. 2015; Pestal et al. 2015) was shown to activate the upstream cytosolic dsRNA sensor IFIH1 (also known as MDA5), subsequently triggering interferon response. ADAR editing can destabilize immunogenic dsRNAs by introducing mismatches into perfectly paired duplex regions (Liddicoat et al. 2015), thus preventing MDA5 sensing. Specifically, the IFN inducible (Patterson and Samuel 1995) p150-isoform of ADAR is responsible for regulating MDA5-mediated recognition of dsRNAs (Pestal et al. 2015). ADAR-p150's status as both an interferon stimulated gene (ISG) and an ISG regulator through its modulation of immunogenic dsRNAs highlights the importance of its expression in antiviral response (Ward et al. 2011). Recently, several groups have shown that the Zα domain that is specific to the p150 isoform also plays an important role in inflammatory signaling. Specifically, ADAR-p150 inhibits Z-RNA Alu–Alu duplexes that would otherwise activate the left-handed Z-nucleic acid sensor ZBP1, which elicits caspase-8-dependent apoptosis and MLKL-mediated necroptosis (de Reuver et al. 2022; Hubbard et al. 2022; Jiao et al. 2022). Other than allowing cells to avoid activation of MDA5- and ZBP1-dependent inflammatory pathways, ADAR has also been shown to prevent translational shutdown induced by another dsRNA sensor: EIF2AK2 (also known as Protein Kinase R [PKR]) (Chung et al. 2018). Upon dsRNA activation, PKR globally represses cellular translation by phosphorylating eukaryotic initiation factor 2 (EIF2A) (Dar et al. 2005). Notably, a more recent investigation reported that ADAR-p150 binding—rather than editing activity—protects cells from hyperactivation of PKR (Hu et al. 2023), although the investigators suggest that editing may still play a subtle role.
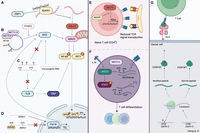
Effects of RNA modifications on innate and adaptive immunity (see text for references). RNA modifications impart various effects on innate immune pathways. (A) A-to-I editing of dsRNA regions by ADAR prevents IFIH1 (also known as MDA5) sensing and subsequent activation of the type I IFN signaling pathway. ADAR also binds Z-RNA, thus inhibiting ZBP1 activation. (B) m6A modifications in circRNAs can function as markers of “self,” preventing RIGI sensing. (C) The presence of pseudouridine and m5C can repress TLR signaling. Pseudouridine has also been shown to inhibit 5-triphosphate mediated RIGI sensing. (D) Depletion of m5C modifications results in increased transcription of Pol III transcripts, including RPPH1. RPPH1 itself facilitates transcription of 7SL RNAs, which are RIGI ligands. RNA modifications play roles in adaptive immunity: (E) Deletion of ADAR results in excessive ISG expression, thus impairing T cell receptor (TCR) signaling. (F) Loss of m6A leads to destabilization of Tcf7 transcripts, consequently impairing naïve T cell differentiation into T follicular helper (Tfh) cells. On the other hand, the absence of m6A stabilizes JAK-STAT signaling inhibitors (SOCS family), leading to subsequent inhibition of T cell homeostatic proliferation and differentiation. (G) An A-to-I editing site in cyclin I (CCNI R75G) leads to the production of an altered peptide (CCNI-ED). When presented by tumor cells, this peptide could be recognized by tumor-infiltrating lymphocytes. Created with BioRender (https://www.biorender.com).
Like A-to-I editing, the presence of m6A may also prevent the formation of endogenous dsRNAs. Compared with control cells, METTL3-depleted murine fetal liver cells were found to exhibit increased staining with the dsRNA-specific antibody J2 (Gao et al. 2020). Given that the oligoadenylate synthetase (Oas) family of dsRNA sensors and ISGs exhibited up-regulated expression upon METTL3-depletion, the investigators suggest that these dsRNAs are likely to mediate innate immune response upon m6A depletion.
circRNAs are generated via a back-splicing circularization mechanism in which a downstream 5′ donor site of an exon is joined to an upstream 3′ acceptor site (Chen 2016). Multiple layers of coordination between circRNA production and innate immunity have been established (Chen et al. 2017; Li et al. 2017b; Liu et al. 2019a). Building upon these investigations, m6A modifications in endogenous circRNAs have been found to function as a marker of “self.” These marks allow the immune system to distinguish them from exogenous circRNAs, which activate immune signaling via RIGI (Chen et al. 2019). Indeed, the presence of m6A recruits the YTHDF2 reader protein, which the investigators suggest may inhibit the RIGI conformational transformations necessary for downstream immune gene signaling (Fig. 2B).
As implied by their name, lncRNAs are long (>200-nt) transcripts that are not translated into functional proteins (Statello et al. 2021). Nonetheless, they can contribute to various important biological processes. Indeed, J2 pulldown of putatively immunogenic dsRNA transcripts in METTL3-depleted murine cells included two lncRNAs: Malat1 and Rian (Gao et al. 2020). Notably, Malat1 was previously found to contain destabilizing m6A residues within its predicted hairpin structures (Liu et al. 2013), although future studies are needed to determine whether a causal relationship exists.
In addition to A-to-I editing and m6A, other types of RNA modifications may also regulate innate immunity. Nearly two decades ago, incorporation of modified nucleosides—including pseudouridine (Ψ), m5C, and m5A—into otherwise immunostimulatory RNA transcripts was reported to abrogate TLR signaling (Fig. 2C; Karikó et al. 2005). Pseudouridines were also found to prevent 5-triphosphate-mediated RIGI activation (Hornung et al. 2006), inhibit dsRNA recognition (Karikó et al. 2011), and enhance translation by diminishing activation of PKR (Anderson et al. 2010). Indeed, these findings proved instrumental in the development of the COVID-19 mRNA vaccines, in which the incorporation of N1-methyl-pseudouridines reduced RNA immunogenicity and enhanced protein production (Karikó et al. 2008; Polack et al. 2020). Thus, although A-to-I editing and m6A mediate transcript immunogenicity by inducing structural changes, these findings suggest that the existence of pseudouridine itself dampens immunostimulatory potential.
RNA modifications may also modulate the immunogenicity of endogenous transcripts in a less direct manner. Depletion of the m5C writer NSUN2 in human A549 cells resulted in up-regulation of various Pol III transcribed ncRNAs, including 7SL RNAs (Zhang et al. 2022). Notably, 7SL is known to activate the RIGI sensor (Nabet et al. 2017). Although 7SL itself was not a target of NSUN2-mediated m5C methylation, a m5C site was found within ribonuclease P RNA component H1 (RPPH1), which is known to facilitate Pol III transcription of various noncoding RNAs (Guerrier-Takada et al. 1983; Kleinert et al. 1988; Reiner et al. 2006). The investigators propose that abrogation of m5C methylation increases RPPH1 levels, which subsequently increases transcription of 7SL RNA. In other words, although m5C modifications may not directly target the RNAs that are sensed by RIGI, their absence does lead to increased expression of an upstream regulator of these immunostimulatory transcripts (Fig. 2D).
Strategies for genome-wide identification of immunogenic substrates
RNA modifications can regulate the abundance or structure of transcripts that have the propensity to trigger cytosolic sensors. However, the landscape of these immunogenic transcripts—including how they differ across different biological systems and contexts—remains to be fully elucidated. Although certain circRNAs and ncRNAs have exhibited immunogenic potential, the most widely recognized and potent substrates (owing to their abundance and resemblance to foreign nucleic acids) are dsRNAs. For this reason, along with the fact that strategies for identifying circRNAs (Jeck and Sharpless 2014; Szabo and Salzman 2016; Li et al. 2018; Kristensen et al. 2019) and ncRNAs (Duan et al. 2021; Mattick et al. 2023) have been previously reviewed, here we focus on methods that can facilitate the identification of immunogenic dsRNAs. We categorize the discussion of these methods into two sections: first, strategies for the identification of dsRNAs in general, and second, strategies for zeroing in on the immunogenic substrates.
Genome-wide identification of dsRNAs
Several experimental protocols can be used to enable global identification of dsRNAs. Pulldown-based methods with the dsRNA-specific antibody J2, including J2 anti-dsRNA IP (Lybecker et al. 2014; Blango and Bass 2016), dsRIP-seq (Gao et al. 2020, 2021a), and J2 fCLIP-seq (Kim et al. 2018), can be used to enable high-throughput sequencing of dsRNAs. Assuming that an IP-grade antibody is available, CLIP-based approaches for dsRNA-binding proteins can be leveraged as well. Another set of techniques involves use of chemicals that preferentially digest (Kertesz et al. 2010; Underwood et al. 2010) or modify (Lucks et al. 2011; Homan et al. 2014; Kielpinski and Vinther 2014; Loughrey et al. 2014; Cheng et al. 2015; Watters et al. 2016) unpaired nucleotides, subsequently generating individual sequence position scores that correspond to their propensity of being base-paired.
Various dsRNA identification strategies can be applied to conventional RNA-seq data sets as well. Because ADAR enzymes require a double-stranded substrate for editing (Eisenberg and Levanon 2018), the presence of A-to-I editing within a transcript indicates that it has the potential to form a dsRNA structure. Indeed, one method to identify dsRNA candidates on a genome-wide scale is to identify A-to-I editing enriched regions across the genome, followed by examination of their RNA secondary structures using computational folding algorithms (Whipple et al. 2015; Blango and Bass 2016; Reich and Bass 2019). A fundamental shortcoming of this strategy is that it relies upon known A-to-I editing sites. Thus, dsRNAs that are depleted of editing sites are likely to be missed. To fill this gap, machine-learning approaches capable of performing editing-independent dsRNA prediction with long-read RNA-seq can serve as a complementary detection method. Indeed, one recently developed method is dsRID (Yamamoto et al. 2023), which predicts dsRNA regions based on features related to region skipping in long-read sequencing data. This approach was motivated by previous observations that highly structured regions may induce skipping in RNA-seq reads as a result of intramolecular template switching during reverse transcription (Cocquet et al. 2006; Houseley and Tollervey 2010; Tardaguila et al. 2018; Liu et al. 2023b). Aided by machine-learning strategies and RNA structure-folding methods, dsRID captures long dsRNA structures in an editing-independent and sample-specific manner.
Genome-wide searches for retrotransposable elements (RTEs) can also facilitate dsRNA identification. RTEs, which include endogenous retroviruses (ERVs), long interspersed elements (LINEs), and short interspersed nuclear elements (SINEs) (Chen and Hur 2022), are DNA sequences that had or still have the ability to insert into new genomic locations via an RNA intermediate. More than 99% of A-to-I editing in humans is found within a type of RTE known as Alu elements (Kim et al. 2004; Levanon et al. 2004). Proximal inverted repeat Alu elements (IRAlus) have the propensity to fold intramolecularly owing to their sequence similarity, giving rise to dsRNA structures. Because Alu elements are primate specific, the search for inverted Alus does not apply to species beyond primates. Rather, a generalized search for inverted duplicated sequences (IDSs) is possible, as was performed to characterize dsRNA structures across dozens of species (Barak et al. 2020). Although IDS regions likely form intramolecular dsRNA structures, dsRNAs can also result from intermolecular base-pairing in which two separate molecules anneal together to form a structure. Indeed, RTEs such as ERVs and LINE1 elements can form intermolecular duplexes (Yang and Kazazian 2006; Chiappinelli et al. 2015) and have been shown to activate dsRNA-triggered antiviral signaling pathways (Kassiotis and Stoye 2016; Zhao et al. 2018). Several high-throughput sequencing methods for identifying RTEs and quantifying their abundance have been developed (Jin et al. 2015; Lerat et al. 2017; Jeong et al. 2018; Valdebenito-Maturana and Riadi 2018) and can be leveraged to identify additional dsRNA candidates that are regulated by RNA modifications. Because not all RTEs may necessarily form dsRNAs, RNA secondary structure prediction algorithms can be used to ascertain their dsRNA-forming propensity (Zuker 2003; Markham and Zuker 2008; Reuter and Mathews 2010; Lorenz et al. 2011).
Honing in on immunogenic substrates
By virtue of their design, several of the experimental strategies discussed in the previous section not only facilitate dsRNA identification but also lend evidence for their immunogenic potential. CLIP-based approaches can use dsRNA cytosolic sensors, such as MDA5, RIGI, and PKR, to identify RNAs bound to activators of antiviral signaling. Although CLIP experiments of PKR (Kim et al. 2018) and RIGI (Jiang et al. 2018) have been performed, attempts to perform MDA5 CLIP have encountered numerous technical challenges (Herzner et al. 2021). As an alternative approach, Ahmad et al. (2018) used an RNase protection assay coupled with RNA-seq to identify endogenous ligands for MDA5. These experiments revealed that hybrid-forming IRAlus were primary immunostimulatory targets of MDA5.
Immunogenic substrates can also be identified using approaches other than identifying targets of dsRNA sensors. Solomon et al. (2017) coupled ADAR knockdown with a high-throughput structure probing method known as PARS-seq. Contrary to the prevailing notion that the main role of ADAR editing is to destabilize duplexes, this study found that ADAR silencing led to a lower global double-stranded-to-single-stranded RNA ratio. Although this finding suggests that editing stabilizes RNA duplexes in most cases, the subset of transcripts that exhibited the opposite trend (i.e., those that are likely destabilized by editing) was significantly enriched for potential MDA5 ligands. Indeed, many of these transcripts harbored IRAlus that fold into long, nearly perfectly paired dsRNAs (Kato et al. 2008). These results present a pool of immunogenic dsRNA candidates that may be an intriguing starting point for future studies.
Several groups have identified candidate immunogenic dsRNAs based on regions that are specifically edited by ADAR p150 (rather than p110) and found that they constituted a limited portion of all edited regions (Kim et al. 2021; Sun et al. 2022). Although these results suggest that only a small subset of dsRNAs are immunogenic, an alternative—and not necessarily mutually exclusive—model is that all dsRNA targets are subject to weak editing by ADAR. In other words, immune homeostasis can be maintained as long as the overall dsRNA abundance level is under control (Levanon et al. 2024). To better understand the veracity of these models, additional investigations into the landscape of immunogenic dsRNAs and how ADAR exerts its protective effects are even more highly warranted.
Regardless of how immunogenic dsRNA candidates are identified, one straightforward assessment of their immunogenicity is the correlation of their expression with a measure of interferon response, such as the expression of ISGs. For example, the output of IFN signaling has been previously summarized using an ISG score: a metric formulated based on the expression profile of 38 signature genes (Liu et al. 2019c). Alternatively, genetics-based approaches can be used to identify supporting evidence for dsRNA immunogenicity. Colocalization analysis between genetic variants associated with A-to-I editing (edQTLs) and risk loci for autoimmune and immune-mediated diseases facilitated the identification of dsRNAs composed of IRAlus, as well as cis-natural antisense transcripts (cis-NATs) (Li et al. 2022). Cis-NATs are composed of sense RNA annealed to natural antisense transcripts (NATs) that are transcribed from their opposite strand, thus forming long, perfectly complementary dsRNAs (Faghihi and Wahlestedt 2009). Reduced editing of these dsRNAs was associated with inflammatory disease risk variants, thus supporting their potential immunogenic status (Li et al. 2022).
Although most studies to date have been limited to exploring ADAR substrates, future work—implementing approaches discussed above—can further expand the known atlas of immunogenic dsRNAs and characterize their roles in different cell types and cellular conditions. These methods may also be adapted to facilitate the identification of immunogenic circRNAs and ncRNAs, which, to our knowledge, has not previously been subject to review. Importantly, cytoplasmic localization is a key factor in determining substrate immunogenicity. Thus, ascertaining cellular localization ought to be a critical consideration in any investigation on immunogenic targets.
Regulation of genes involved in immune system defenses
In the above sections, we discussed how RNA modifications regulate the immunostimulatory potential of their target transcripts. Another class of immune-relevant RNA modifications is those that regulate the life cycle of genes with known immune system relevance, such as ISGs or their upstream signaling molecules.
As revealed by transcriptome-wide investigations, the presence of RNA modifications can impact transcript stability (Wang et al. 2014; Brümmer et al. 2017), translational efficiency (Wang et al. 2015; Choi et al. 2016; Mao et al. 2019), cellular localization (Roundtree et al. 2017), splicing (Hsiao et al. 2018; Kapoor et al. 2020; Tang et al. 2020), and RNA secondary structure (Liu et al. 2015; Solomon et al. 2017). For m6A and m5C, these regulatory effects are mediated by reader proteins that either selectively recognize or preferentially bind to these modifications. On the other hand, the effects of A-to-I and C-to-U editing often occur by virtue of the resultant nucleotide changes. Thus, global studies on the regulatory impact of RNA modifications often use knockdown of reader proteins and/or the editing enzymes themselves (i.e., ADARs, APOBECs, and methyltransferases) before analyses of the resultant transcriptome. Characterization of modified transcripts that are bound to the reader/editing protein of interest via IP-based approaches (Wang et al. 2014, 2015; Bahn et al. 2015; Roundtree et al. 2017; Yang et al. 2017) may also yield insights about the function of RNA modifications.
Immunostimulatory treatments have been found to modulate the abundance of proteins involved in the deposition of RNA modifications (Peng et al. 2006; Rubio et al. 2018; Winkler et al. 2019). These findings suggest that RNA modifications play an important role in immune response. Indeed, multiple studies have revealed that genes involved in immune system defenses are regulated by RNA modifications. One mechanism of regulation is the modulation of mRNA turnover rate. m6A modifications in particular mediate mRNA decay by recruiting the destabilizing m6A reader protein YTHDF2 (Wang et al. 2014). In an IFN-I-independent pathway of host/viral resistance, m6A modifications were found to destabilize OGDH, which encodes metabolic intermediates that boost viral replication (Liu et al. 2019d). Multiple genes encoding type I interferon–related genes are subject to m6A-mediated stability regulation as well, including the IFNB1 transcript (Rubio et al. 2018; Winkler et al. 2019) and Mavs (Qin et al. 2021). Other modifications can also regulate the stability of type I IFN signaling pathway components. For example, depletion of the m5C-writer NSUN2 led to increased stability of IRF3, resulting in amplification of the type I interferon response (Wang et al. 2023). Additionally, a pan-cancer analysis of A-to-I editing in epithelial and mesenchymal tumors presented evidence that RNA editing regulates the abundance of immune-related genes, including the transcript encoding PKR (Chan et al. 2020). Here, the investigators propose that A-to-I editing modulates the binding affinity of the RNA-binding protein (RBP) ILF3, which is known to stabilize its target mRNAs.
RNA modifications can also alter the translational efficiency of their target genes. Translation of the ISGs IFITM1 and MX1 (McFadden et al. 2021), as well as the serine/threonine kinase gene RIOK3 (Gokhale et al. 2020), was enhanced by m6A. RNA modifications can also indirectly modulate translational efficiency by regulating transcript localization. Indeed, demethylation of the antiviral transcripts Mavs, Traf3, and Traf6 was found to induce nuclear retention and prevent subsequent protein translation (Zheng et al. 2017). As evidenced by the improved translational efficiency of pseudouridine-containing transcripts that were synthesized in vitro, pseudouridine may also boost the translational efficiency of their host transcripts (Karikó et al. 2008). Using a nanopore native RNA sequencing method for pseudouridine prediction (NanoPsu), interferon pathway and antiviral response genes were found to exhibit the highest increase in pseudouridine modification probability (referring to either increased number of sites and/or increased modification fraction) upon IFNG or IFNB1 treatment in HeLa cells (Huang et al. 2021). The investigators suggest that pseudouridine enhances the translation of these ISG transcripts upon immunostimulatory treatment. Future studies are needed to determine other mechanisms in which pseudouridine may regulate immune-relevant transcripts.
RNA modifications and adaptive immunity
Unlike type I IFN production and signaling, which can be mounted by nearly all cells owing to their ability to produce IFNA1/IFNB1 upon PAMP recognition (McNab et al. 2015), adaptive immunity relies upon the activities of antigen-specific T cells and antibody-producing B cells (Marshall et al. 2018). Epitranscriptomic events have been found to play critical roles in the successful maturation, activation, and differentiation of these cells. Using a mouse model in which ADAR was ablated specifically in CD4+ T cells, Nakahama et al. (2018) showed that ADAR supported T cell maturation by preventing excessive MDA5-activated ISG expression (Fig. 2E). Without the editing activity of ADAR, the excessive ISG expression impaired T cell receptor (TCR) signaling. In the same vein, deletion of METTL3 from CD4+ T cells impeded their differentiation into effector cells by stabilizing m6A-modified mRNAs encoding JAK-STAT signaling inhibitors in the SOCS family (Fig. 2F; Li et al. 2017a). Inhibition of STAT signaling subsequently inhibited T cell homeostatic proliferation and differentiation. m6A modifications were also found to be important for CD4+ T cell differentiation into T follicular helper (Tfh) cells. Loss of METTL3 lead to destabilization of Tcf7 transcripts, consequently impairing production of the TCF7 (also known as TCF-1) protein, a key regulator of Tfh differentiation (Yao et al. 2021). Depletion of the m6A methyltransferase complex protein WTAP has also been used to elucidate the impact of m6A on TCR signaling and T cell survival (Ito-Kureha et al. 2022). This study revealed that thymocyte differentiation, activation-induced death of peripheral T cells, and regulatory T cell function are dependent on WTAP and m6A methyltransferase functions. For a comprehensive review of past studies on the regulatory roles of m6A in different immune cells, we refer readers to Han and Xu (2023).
T cells must discriminate between self- and non-self-antigens. This learning process is largely mediated by medullary thymic epithelial cells that can express a large fraction of body antigens. In comparison to other cell types, medullary thymic epithelial cells display significantly higher levels of RNA processing, including A-to-I and C-to-U RNA editing, thus expanding the diversity of their self-antigen repertoire (Danan-Gotthold et al. 2016). The existence of peptides derived from edited RNA transcripts was later confirmed via a specialized proteogenomic screening approach that identified five edited peptides, one of which was derived from an A-to-I edited site of cyclin I (CCNI R75G) (Fig. 2G). When presented by tumor cells, this edited peptide was shown to be recognized by and to elicit a robust T cell effector cytokine IFNG response by tumor-infiltrating lymphocytes (Zhang et al. 2018). In the same vein, Zhou et al. (2020) identified an edited neoantigen (arising from an A-to-I editing site in the OSBPL9 gene) that was complexed to MHC I based on high-resolution mass-spectrometry in an ovarian cancer sample.
m6A has also been found to regulate antigen presentation. In dendritic cells, transcripts encoding lysosomal proteases are subject to m6A modifications, which enhance their translation via YTHDF1 binding. Because lysosomal proteases can destruct internalized antigens, inhibiting their activity enhances antigen cross-presentation of wild-type dendritic cells (Han et al. 2019). Thus, YTHDF1 represents a therapeutic target that could aid in a more robust antitumor response.
Exploiting epitranscriptomics for cancer immunotherapies
As a disease characterized by the uncontrollable growth and spread of abnormal cells, cancer pathology is inextricably tied to aberrations in immune system response. The advent of immunotherapies has revolutionized the treatment of cancer, but these therapeutics are only effective in a subset of patients (Snyder et al. 2014; Gao et al. 2016; Zaretsky et al. 2016; Sambi et al. 2019). Thus, further exploration of how treatment resistance can be overcome is highly warranted.
Emerging evidence suggests that the manipulation of RNA modifications can be leveraged to reduce cancer cell viability and subsequent tumor growth. Here we focus specifically on how ADAR and METTL3 depletion can function as promising therapeutic strategies, given that inhibitors specifically targeting their activity have been developed and characterized in an immuno-oncogenic context. The relevance of both enzymes to cancer etiology has been amply demonstrated in previous studies. The expression of the A-to-I editing enzyme ADAR is up-regulated in a myriad of cancers (Fumagalli et al. 2015; Anadón et al. 2016), likely owing to amplification of the corresponding chromosomal region (Knuutila et al. 1998) and as a consequence of inflammatory signaling (Fumagalli et al. 2015). Indeed, the positive correlation between ADAR and the type I interferon response in cancer cell lines is primarily explained by the p150 isoform, consistent with its role as an ISG (Fumagalli et al. 2015). Moreover, a global analysis of A-to-I editing profiles across 17 cancer types from The Cancer Genome Atlas (TCGA) revealed that tumors exhibit distinct profiles from normal tissues, including numerous clinically relevant cross-tumor editing events (Han et al. 2015). Other examples in which increased ADAR editing have promoted cancer progression have been previously reviewed (Fritzell et al. 2018).
Several studies have shown that ADAR deletion reduces cancer cell viability, at least partially through activation of PKR (Gannon et al. 2018; Bhate et al. 2019; Ishizuka et al. 2019; Liu et al. 2019c; Kung et al. 2021). Loss of ADAR enhances sensing of immunogenic ligands, subsequently triggering levels of inflammation and IFN sensitivity sufficient to sensitize tumor cells to immunotherapy, and overcomes resistance to checkpoint blockade (Ishizuka et al. 2019). Combating therapeutic resistance through ADAR inhibition may be facilitated by drugs such as rebecsinib (17S-FD-895), an ADAR splice isoform switching inhibitor, which binds within the spliceosome core complex to induce intron retention in ADAR p150. Rebecsinib was shown to obviate editing-mediated leukemia stem cell self-renewal and has already completed preinvestigational new drug (IND) studies (PIND 153126) (Crews et al. 2023). Moreover, epigenetic therapies, such as treatment with the DNA methyltransferase inhibitor (DNMTi) 5-AZA-CdR, have been shown to enhance the antitumor effects of ADAR depletion by inducing immunogenic dsRNAs derived from Alu retroelements (Mehdipour et al. 2020). Future investigations may explore the clinical tractability offered by the combination of these two drugs and other related treatments.
In the same vein, dysregulation of m6A modifiers is commonly observed across cancer types (Deng et al. 2023) and contributes substantially to various facets of the disease, including cancer metabolism (Shen et al. 2020; Wang et al. 2020b, 2021), tumor microenvironment (Wang et al. 2019; Song et al. 2021; Yin et al. 2021), and immunotherapy response (Wang et al. 2020a). Therapeutic promise through m6A can be found in two recently developed METTL3 inhibitors: STM2457 (Yankova et al. 2021) and STM3006 (Guirguis et al. 2023). METTL3 inhibition with STM2457 in AT3 TNBC cells (triple-negative breast cancer mouse cell line) was shown to increase the formation of dsRNAs as well as MHC-I expression on the tumor cell surfaces. Enhanced T cell–mediated tumor-cell killing in the presence of STM2457 was shown to be dependent on the tumor cells’ ability to sense dsRNAs. This was demonstrated by the observation that repression of dsRNA sensor proteins abolished the increased cell surface expression of MHC-I in METTL3-inhibited cells (Guirguis et al. 2023). STC-15, a METTL3 inhibitor exhibiting comparable potency to STM3006 but featuring enhanced metabolic stability, is currently undergoing assessment in a phase I clinical trial for solid cancers (NCT05584111) (Guirguis et al. 2023).
Conclusions and future outlook
Here, we highlight investigations that have explored the multifaceted ways in which epitranscriptomic events contribute toward immune system responses (Fig. 3). Given the accumulation of findings regarding their relevance to cellular immunogenicity, we focused specifically on A-to-I/C-to-U editing, m6A/m5C, and pseudouridine modifications in this review. Moving forward, several considerations are important when executing or evaluating results from these types of studies. First, it is important to clarify whether a phenotype of interest (e.g., some measure of immune response) arises owing to RNA modification per se or rather some modification-independent feature of the editor/writer enzyme, such as RNA binding. Mutant proteins with impaired enzymatic activity (Yang et al. 2017; Tang et al. 2020) can be used to aid in this endeavor. Additionally, given that identification of RNA modifications often suffers from noise originating from sequencing errors, SNPs, and mapping biases, the implementation of appropriate filters and statistical frameworks is imperative to minimize false-positive identification of sites (Lee et al. 2013; Huang et al. 2019; McIntyre et al. 2020). Using complementary detection methods in the same biological systems can also be a helpful approach to boost sensitivity and specificity.
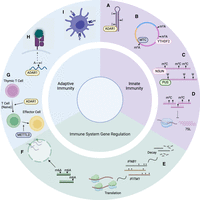
Categories of immune-related epitranscriptomic events and corresponding examples. (A) A-to-I editing by ADAR destabilizes dsRNA structures, inhibiting sensing by MDA5 and the subsequent activation of the IFN signaling pathway. (B) m6A modifications of circRNAs facilitate its recognition as “self” RNA, thus preventing RIGI sensing. (C) Pseudouridine and m5C modifications within immunostimulatory transcripts can inhibit TLR signaling. (D) m5C influences the abundance of transcripts that, in turn, up-regulate immunogenic substrates, such as 7SL. (E) RNA modifications can influence the stability and translation rate of genes associated with interferon responses. (F) m6A modification of antiviral transcripts affects nuclear export and subsequent translation. (G) ADAR promotes T cell maturation by mitigating ISG expression, and METTL3 impacts CD4+ T cell differentiation into T follicular helper cells by destabilization of Tcf7 transcripts. (H) Modifications that result in amino-acid substitutions could lead to the expression of modified peptides, which could be recognized by T cells when presented on tumor cells. (I) m6A modifications of lysosomal proteases affect antigen cross-presentation of dendritic cells. Created with BioRender (https://www.biorender.com).
An ongoing question is the exact identity of immunogenic RNAs that are targeted by RNA modifications, as well as whether they vary across different cell types. Although it is likely that PRRs target distinct groups of immunogenic RNAs based on their substrate binding preferences, it remains unknown whether they consistently bind the same groups of transcripts. Previous studies present populations of immunostimulatory RNA candidates that can be used as starting points for future investigations. These include cis-NATs (Li et al. 2022), transcripts exhibiting increased pseudouridylation upon IFN treatment (Huang et al. 2021), and genes destabilized by ADAR depletion (Solomon et al. 2017). The immunogenicity of target transcripts may also depend on biological context. Although cross-species transcriptome analysis has revealed purifying selection against the expression of long, nearly perfect dsRNAs (Barak et al. 2020), factors such as the dsRNA editing level and genetic variants may contribute to enhanced transcript immunogenicity, either by increasing sensor sensitivity (Ahmad et al. 2018) or by altering dsRNA structure. The cell type under consideration may also be an important factor. Recently, ADAR deficiency was found to result in elevated dsRNA levels in several cell types (hESCs, NPCs, and neurons), but increased type I IFN levels were observed only in neurons (Dorrity et al. 2023). Single-cell RNA-seq approaches may help further elucidate the cell type–specific effects of RNA modifications and the fate of their regulatory targets, such as whether immunogenic dsRNAs are secreted to activate specific immune cell populations.
The convergence of pathways involving different types of RNA modifications raises questions regarding their collective biological impact. This is particularly noteworthy for ADAR editing and m6A methylation. Both of these modifications occur at adenosines on the same nitrogen in a mutually exclusive manner. Once adenosine is deaminated into inosine, it can no longer be subject to m6A modification; similarly, m6A is a poor substrate for deamination (Véliz et al. 2003). Knockdown of METTL3 and METTL14 in HEK293T cells resulted in widespread increases in A-to-I RNA editing, suggesting a genome-wide inverse relationship between A-to-I editing and m6A modifications (Xiang et al. 2018). A similar trend of increased A-to-I editing levels was observed in K562 cells upon knockdown of the pseudouridine synthase 1 (PUS1) (Quinones-Valdez et al. 2019). Future investigations are required to fully elucidate the cumulative impact of epitranscriptomic events, perhaps aided by novel methods that allow for simultaneous detection of different types of modifications (Hartstock et al. 2023).
Investigations into the interplay between RNA modifications and host immunity have unearthed a trove of intriguing discoveries that show promise in human health contexts. Future implementations of experimental and computational methods will continue to illuminate the landscape of RNA modifications and their pivotal roles in shaping immune functions and phenotypes, forging new insights into the dynamic relationship between RNA epitranscriptomics and host defense mechanisms.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (R01CA262686, and R01AG078950 to X.X.). E.H. was supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE-2034835. We thank Giovanni Quinones-Valdez and Cong Liu for lending their expertise on pseudouridine and m6A-related literature.
Footnotes
-
Article published online before print. Article and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278412.123.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵








