
A Bayesian framework to study tumor subclone–specific expression by combining bulk DNA and single-cell RNA sequencing data
- Yi Qiao1,6,
- Xiaomeng Huang1,6,
- Philip J. Moos2,
- Jonathan M. Ahmann3,
- Anthony D. Pomicter3,
- Michael W. Deininger3,4,
- John C. Byrd5,
- Jennifer A. Woyach5,
- Deborah M. Stephens3 and
- Gabor T. Marth1
- 1Eccles Institute of Human Genetics, University of Utah, Salt Lake City, Utah 84112, USA;
- 2Department of Pharmacology and Toxicology, University of Utah, Salt Lake City, Utah 84112, USA;
- 3Huntsman Cancer Institute, University of Utah, Salt Lake City, Utah 84112, USA;
- 4Division of Hematology and Hematologic Malignancies, University of Utah, Salt Lake City, Utah 84112, USA;
- 5The James Comprehensive Cancer Center, The Ohio State University, Columbus, Ohio 43210, USA
-
↵6 These authors contributed equally to this work.
Abstract
Genetic and gene expression heterogeneity is an essential hallmark of many tumors, allowing the cancer to evolve and to develop resistance to treatment. Currently, the most commonly used data types for studying such heterogeneity are bulk tumor/normal whole-genome or whole-exome sequencing (WGS, WES); and single-cell RNA sequencing (scRNA-seq), respectively. However, tools are currently lacking to link genomic tumor subclonality with transcriptomic heterogeneity by integrating genomic and single-cell transcriptomic data collected from the same tumor. To address this gap, we developed scBayes, a Bayesian probabilistic framework that uses tumor subclonal structure inferred from bulk DNA sequencing data to determine the subclonal identity of cells from single-cell gene expression (scRNA-seq) measurements. Grouping together cells representing the same genetically defined tumor subclones allows comparison of gene expression across different subclones, or investigation of gene expression changes within the same subclone across time (i.e., progression, treatment response, or relapse) or space (i.e., at multiple metastatic sites and organs). We used simulated data sets, in silico synthetic data sets, as well as biological data sets generated from cancer samples to extensively characterize and validate the performance of our method, as well as to show improvements over existing methods. We show the validity and utility of our approach by applying it to published data sets and recapitulating the findings, as well as arriving at novel insights into cancer subclonal expression behavior in our own data sets. We further show that our method is applicable to a wide range of single-cell sequencing technologies including single-cell DNA sequencing as well as Smart-seq and 10x Genomics scRNA-seq protocols.
Introduction
Bulk DNA/RNA sequencing is insufficient to study subclone-specific cellular behavior
Intratumoral genomic, transcriptomic, and epigenetic heterogeneity are hallmarks of cancer and driving forces of disease progression (Nowell 1976; Russnes et al. 2011; Greaves and Maley 2012; Marusyk et al. 2012; Bedard et al. 2013; Meacham and Morrison 2013; McGranahan and Swanton 2017; Dagogo-Jack and Shaw 2018; Lawson et al. 2018). We (Qiao et al. 2014) and others (Jiao et al. 2014; Miller et al. 2014; Roth et al. 2014; Deshwar et al. 2015; Vandin 2017) have developed methods for reconstructing the tumor's subclonal composition and its evolution over time and space within a patient, using somatic tumor mutation allele frequencies measured in bulk DNA sequencing data. Whereas the tumor mutations gleaned from bulk DNA sequencing can be used to define the genetic subclones and map out their evolutionary trajectory, functional (e.g., transcriptomic or epigenetic) data is necessary to investigate cellular behavior and its evolution. Because the tumor sample is typically a mix (de Ridder et al. 2005; Palmer et al. 2006; Meyerson et al. 2010) of multiple tumor subclones, normal stromal cells, and infiltrating immune cells, each with potentially divergent and a priori unknown expression behavior (Newman et al. 2015; Hao et al. 2019), measuring bulk gene expression (via RNA-seq experiments) in the tumor is insufficient to delineate the transcriptomic behavior of specific subclones.
Single-cell RNA expression profiles cannot distinguish between genetic tumor subclones
Single-cell RNA sequencing (scRNA-seq) has presented a new and promising approach to study both cell type– and tumor-specific gene expression. In these approaches, cell type– or tumor-specific expression markers are used to distinguish, for example, stromal and different types of blood cells from tumor cells. However, even in cases when unique tumor markers make it possible to identify tumor cells, gene expression data alone is insufficient to differentiate between genetically defined tumor subclones, in order to study subclone-specific expression.
The presence of subclone-defining tumor mutations in single-cell RNA sequences permits determination of subclonal cell identity
The cell-specific RNA sequences collected to characterize single-cell gene expression provide a direct avenue to link the presence or absence of the genetic mutations that define tumor subclones to the transcriptional behavior of the cell. Here we briefly summarize relevant existing methods and discuss their limitations. The VarTrix (10x Genomics at GitHub [https://github.com/10XGenomics/vartrix], accessed July 20, 2020) tool provides the ability to identify cells in which scRNA-seq data collected on the 10x Genomics platform shows the presence of a specific selected DNA mutation, allowing the examination of the transcriptional consequences of that mutation. This approach has been used successfully in a recent study (Petti et al. 2019) to compare the transcriptional behavior of distinct AML cell populations defined by key DNA mutations. However, because this method focuses on a single DNA mutation at a time, the number of cells it can highlight representing a given clone is limited. This is because a mutation may or may not be revealed in a given cell because of the inherent sparsity of scRNA-seq data (Supplemental Fig. 1). DENDRO (Zhou et al. 2020) calls genetic mutations directly from the scRNA-seq reads and uses the presence of these mutations to cluster individual cells into genetically distinct groups. However, DENDRO was only designed for data produced by full-transcript scRNA-seq technologies. Furthermore, because the method does not have the ability to distinguish between inherited variation and somatically acquired mutations in the tumor, the clusters it generates do not necessarily correspond to the genomically defined subclones in the tumor samples (see our analysis below). Finally, Cardelino (McCarthy et al. 2020) uses a Bayesian probabilistic approach to cell assignment, and presents the only comparative method to ours. But as our analyses below show, it is outperformed by scBayes in all data sets we included in this manuscript. We further note that, although not directly related to the problem this manuscript attempts to tackle, an alternative avenue exists to relate cellular genotype and phenotype via separately sequencing single-cell DNA and RNA followed by data integration (clonealign) (Campbell et al. 2019). Finally, a method (PhyloIEx [Jun et al. 2023]) has been developed very recently to use genomic and scRNA-seq data to jointly reconstruct tumor subclone structure and carry out cell-to-subclone assignment.
Results
We developed scBayes, a Bayesian-statistical approach to study subclone-specific phenotypes
Utilization of whole-genome or whole-exome tumor/normal DNA sequencing data ensures high-confidence reconstruction of the genetic tumor subclones, together with comprehensive identification of the somatic mutations that define each subclone. Our method then makes use of these subclone-defining mutations as a “scaffold” to assign individual cells from scRNA-seq data to specific tumor subclones (Fig. 1). Importantly, when a set of high-confidence somatic mutations are used jointly to assess cell assignment, two cells need not share any single tumor mutation with each other in the scRNA-seq data to be assigned to the same subclone. After assigning individual cells to genetic subclones, subclone-specific expression profiles can be generated and compared (Fig. 1A).
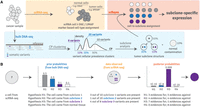
Overview of the scBayes algorithm. (A) The scBayes algorithm combines single-cell RNA sequencing-based transcriptomic analysis (shaded in yellow) with bulk DNA sequencing-based genetic subclone analysis (shaded in blue) to derive subclone-specific expression profiles (shaded in red) via assigning each cell a tumor subclone identity. Cells representing normal cell contamination are also assigned. CP stands for cellular prevalence. (B) Simplified overview of our probabilistic model. For a given cell, scBayes evaluates the Bayesian posterior probabilities that the cell represents each of the genetic subclones while taking evolution into consideration (e.g., for H3, both variants of subclone 1 and subclone 3 are considered positive evidences because subclone 3 is the descendent of subclone 1). The cell is assigned to the subclone that maximizes the posterior probability, and meets a minimum probability threshold. See Methods for a complete description of our statistics model.
The scBayes algorithm
scBayes considers the alternative hypotheses that a given cell represents any one of the tumor subclones reconstructed from the bulk DNA sequencing data, or normal (noncancerous) tissue; and it evaluates the Bayesian posterior probability of each such hypothesis (Fig. 1B). Tumor subclones are defined by sets of somatic mutations; and the “subclone” with no somatic mutations corresponds to the normal tissue. The scRNA-seq reads for a given cell provide positive evidence (i.e., show the mutant allele); negative evidence (i.e., show the germline allele); or no evidence at all (i.e., no read coverage) at the site of a particular somatic mutation. For every cell and each subclone, our algorithm calculates the data likelihood, that is, the conditional probability of observing the specific combination of positive and negative evidence at each somatic mutation site, given that the cell represents the subclone under consideration (see Methods). Under the assumption that data is missing completely at random (MCAR), we omit the sites in a cell that have no RNA sequencing coverage for the assignment of this cell. We use the subclone fraction inferred from the bulk DNA sequencing data during subclone analysis as the prior probability that the cell represents that subclone; and we assign the estimated normal cell fraction as the prior probability that the cell represents “contaminating” normal tissue. After applying Equation 1 (see Methods) to combine the priors and data likelihoods, a Bayesian posterior probability is calculated for each hypothesis, and the cell is assigned to the subclone with the highest posterior probability.
The scBayes analysis workflow
scBayes takes as input several pieces of information. The first is the subclone structure and somatic mutations defining each subclone. It is important to note that mutation clusters and subclones are two separate entities. Users will need to use either our (Qiao et al. 2014) or other methods (Vandin 2017) to perform subclone structure reconstruction from the mutation clusters. To compile the resulting subclone structure information for scBayes, users will create a YAML config file defining the subclones, the mutation clusters each subclone contains, and the paths to VCF files that describe the mutations in each cluster. With the same somatic mutations, users will use the scGenotype utility in scBayes to assess whether sequence coverage is present at these mutation sites in each scRNA-seq cell, and whether mutant alleles were found. Finally, the scAssign utility takes the genotype file from scGenotype, together with the YAML config file, and performs cell assignment. The output is a tab delimited file in which each row describes the assignment details (e.g., assigned subclone, assignment quality) for each cell, which is identified using the cell barcode.
Validation using simulation
We first attempted to validate and characterize the performance of our cell assignment algorithm using simulation. We examined the effect of the following variables: single-cell sequencing genotyping true positive rate (likelihood of observing the variant allele in cells that have it); single-cell sequencing genotyping false positive rate (likelihood of observing the variant allele in cells that do not have it); subclone structure; subclone reconstruction error (percent of somatic mutations assigned to the wrong subclone); and informed prior versus flat prior. When we vary one of these variables, all other variables are fixed to a reasonable default value (see Methods) to isolate their effects. Each simulation configuration is repeated 100 times to establish mean and variance in results. We further compared scBayes to Cardelino on these simulated data sets (Supplemental Fig. 2). In conclusion, scBayes performs well with respect to data quality variations, showing >90% medium correct assignment rate at up to 40% scRNA-seq false positive rate (Supplemental Fig. 2A), down to 50% of true positive rate (Supplemental Fig. 2B) and up to 30% subclone reconstruction error rate (Supplemental Fig. 2E,F). With arbitrarily reduced data qualities, scBayes showed a slight advantage using informed prior versus uniform prior (Supplemental Fig. 2C). An exhaustive evaluation using all six possible subclone structures containing three subclones showed that the performance of scBayes is virtually agnostic to subclone evolution patterns (Supplemental Fig. 2D).
Validation using a published pseudobulk data set reconstructed from single-cell DNA-seq data
To further validate the cell assignment performance of our method, we took advantage of a published, single-cell DNA sequencing data set with reconstructed pseudobulk clonal structure (Laks et al. 2019). This data set consists of three different samples: TOV2295(R) - SA921, OV2295(R2) - SA922, and OV2295 - SA1090. With each sample, the original paper sequenced single-cells, identified genomic alterations (including point mutations which we use in this validation experiment), and reconstructed subclone structure based on shared alterations. In total, hundreds of cells were sequenced, and from which nine subclones (labeled as A through I) were identified across three samples (Supplemental Fig. 3, top half of each panel). Treating this as the ground truth, we performed scBayes cell-to-subclone assignment using the published subclone structure and subclone-defining variants. We found that scBayes correctly assigns 75.4% (SA921), 98.6% (SA922), and 84.5% (SA1090) of cells. We further compared our performance to that of Cardelino, which correctly assigned 70% (SA921), 98.6% (SA922), and 77.9% (SA1090) of cells (Supplemental Fig. 3).
Validation using a synthetic data set with cells of known origin
We in addition performed validation for our cell assignment algorithm using a synthetic data set in which the correct origins of cells are already known. We acquired bulk DNA sequencing and single-cell RNA sequencing (10x Genomics Chromium 5′ capture protocol) data from three chronic lymphocytic leukemia patients (Supplemental Fig. 4A). The bulk DNA sequencing data are from isolated B cells and B cells (as germline control), which allow us to identify somatic mutations present in the B cells for each patient. Using these mutations, we constructed a synthetic subclone structure in which each subclone corresponds to one patient, and thus contains the patient's unique B cell mutations. Cells from scRNA-seq data of these patients are then assigned to this synthetic subclone, followed by evaluation of whether the cells from a patient are assigned to the subclone representing the same patient (Supplemental Fig. 4B). Note that we used somatic mutations alone to minimize the impact of inherited genetic differences across patients. We achieved 100%, 98.6%, and 95.5% accuracy for patient 1, 2, and 3, respectively, in assigning B cells to the correct cancer subclones (Supplemental Figs. 4C, 5). These results provide validation that scBayes can assign cells with experimentally determined ground truth and real-life data quality with very high accuracy. In addition, we evaluated the performance of Cardelino on this data set using recommended parameters (Supplemental Fig. 6), and note that Cardelino failed to assign the majority of cells.
Application of scBayes in a published data set from a refractory breast cancer patient
We used scBayes to reanalyze a published data set (Brady et al. 2017) in which we previously described subclonal tumor evolution in a breast cancer patient across multiple rounds of chemotherapy, response, and relapse. Here we focus on the bulk whole-genome DNA sequencing data collected on the Illumina platform, and single-cell RNA sequencing data collected using the Fluidigm / Smart-seq platform, at two critical time points in the patient's disease progression: before and after doxorubicin treatment. According to our analysis based solely on bulk DNA sequencing data, doxorubicin treatment eliminated all (SC2, SC3, and SC4) predoxorubicin subclones (Fig. 2A). The sole postdoxorubicin subclone (SC5), a mutated version of the predoxorubicin subclone (SC3), became dominant and resistant to doxorubicin, leading to the patient's further progression and death.
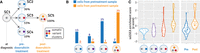
Single-cell assignment and subclone-specific EMT signature enrichment analysis in a longitudinal breast cancer data set. (A) Tumor subclone evolution reconstructed from bulk whole-genome DNA sequencing data across doxorubicin treatment of a refractory breast cancer patient from our previous study (Brady et al. 2017). (B) Cell-to-subclone assignment from scRNA-seq data collected from the tumor samples before and after doxorubicin treatment. Blue bars and numbers represent the number of cells from the pretreatment sample assigned to each subclone; orange bars and numbers represent the number of cells from the posttreatment sample assigned to each subclone. (C) Z-score of ssGSEA enrichment scores for epithelial to mesenchymal transition (EMT) signature: grouped by subclone (left), and by time point (right).
First, as part of our reanalysis, we used scBayes to assign pre- and postdoxorubicin cells from the scRNA-seq data to the genetically defined pre- and postdoxorubicin subclones. We found that the majority of the predoxorubicin cells were assigned to predoxorubicin subclones (SC2, SC3, SC4) and postdoxorubicin cells to the postdoxorubicin subclone (SC5), indirectly yet further validating our cell-to-subclone assignment algorithm (Fig. 2B; Supplemental Fig. 7). Furthermore, this analysis provided additional insight that we could not glean from the bulk DNA analysis. Specifically, three cells from the postdoxorubicin sample were assigned to predoxorubicin subclones SC3 and SC4 that, according to the bulk DNA analysis, became extinct as a result of the treatment. This suggests that a small fraction of the cells representing these clones, in fact, could have survived past the treatment. Conversely, we found two predoxorubicin cells that were assigned to the postdoxorubicin subclone (SC5), potentially indicating that the mutant subclone only found in the bulk DNA analysis after the treatment was already present before the treatment at extremely low fractions. Although more comprehensive sampling and investigation are needed to arrive at definitive biological conclusions, these results show how the integration of the scRNA-seq data and cell assignments afford us new perspectives in examining the evolution of the tumor subclones through this critical treatment regimen for the patient.
Second, we compared the scRNA-seq data collected pre- and postdoxorubicin treatment (Fig. 2C, right), and measured their epithelial to mesenchymal transition (EMT) signature levels using ssGSEA (Fig. 2C; Mootha et al. 2003; Subramanian et al. 2005). Overall, and consistent with previous described findings (Brady et al. 2017), the cells after the treatment showed elevated EMT signature as compared to before the treatment. Using the subclonal cell assignments performed with scBayes, we analyzed the subclone-specific expression signatures, and found that the predoxorubicin subclone (SC3) showed intermediate EMT enrichment that fell between the predoxorubicin subclones (SC2, SC4) eliminated by the treatment and the postdoxorubicin subclone (SC5) which derived from it. Here, cell-to-subclone assignment enabled the elucidation of the gradual transformation of the subclonal EMT phenotype.
Third, we asked whether it was possible to arrive at the same conclusion with DENDRO and Cardelino, two relevant existing computational methods. Because DENDRO is only applicable to full-transcript scRNA-seq data, our breast cancer data set is ideal for this comparison. To quantify cell assignment quality, we introduce the number of expected alleles (NEA) and the number of allelic collisions (NAC) which we define as the total number of somatic mutations, for which we observed a mutant allele in a cell, that is present (and not present, respectively) in the genetic subclone the cell is assigned to; and allelic collision rate (ACR) defined as the number of allele collisions divided by the total number of observed mutation alleles across all assigned cells. We acknowledge that these metrics do have limitations, especially in the presence of bulk subclone reconstruction errors. However, we do believe that it provides a fair comparison across methods when applied to the same data set. Running on our scRNA-seq data set, DENDRO grouped the cells into four clusters (D1, …, D4). However, as illustrated in Supplemental Figure 8, there was no clear correspondence between the four DENDRO clusters and the five genetically defined subclones SC1, …, SC5, which were extensively validated in the original study (Brady et al. 2017). Each DENDRO cluster contained cells that show sequencing evidence for somatic mutations that span multiple subclone-exclusive mutation clusters (Supplemental Fig. 8A, top) as opposed to the consistent grouping between cells and subclones as shown in scBayes results (Supplemental Fig. 8B, top). Indeed, even when we assigned DENDRO clusters to the genetic subclones in a way that minimizes NAC and ACR, this best assignment shows higher ACR compared to scBayes (Supplemental Fig. 8A, bottom). Cardelino cell assignments (Supplemental Fig. 8C) also show a higher ACR similar to DENDRO.
Application of the method in a novel data set: longitudinal samples from a chronic lymphocytic leukemia (CLL) patient
We then applied our method to a data set we collected from a chronic lymphocytic leukemia (CLL) patient receiving ibrutinib treatment, a Bruton tyrosine kinase (BTK) inhibitor. From bulk DNA sequencing on B cells using T cells as normal control, we identified a dominant B cell clone (SC1) at the pretreatment time point (T1), see Figure 3. Over the course of the treatment, another clone (SC2) emerged at year 1 (T2) and expanded at year 2 (T3), whereas SC1 shrunk (Fig. 3A). We also performed scRNA-seq on unsorted PBMC samples at all three time points, which allowed us to identify different blood cell types (Fig. 3B), and to pinpoint the malignant B cells. We then performed subclone assignment on the B cell population, which showed that the majority of the cells at T1 represented SC1 (Fig. 3C; Supplemental Fig. 9). At T2 and T3, the fraction of cells assigned to SC1 decreased, and the fraction of cells assigned to the emerging and expanding subclone SC2 increased. It is worth noting that the total B cell count has decreased over time, in agreement with the clinical remission of the disease observed in this patient as a result of treatment. In addition, we evaluated the performance of Cardelino on this data set (Supplemental Fig. 10). We note that Cardelino failed to assign the majority of cells.
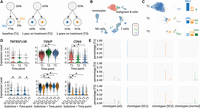
Subclone-specific differential expression and clonotype analysis in a data set collected from a CLL patient undergoing ibrutinib treatment. (A) Genetic subclone structure over three time points from bulk DNA-seq analysis. (B) UMAP of cell clusters of scRNA-seq data collected at the three time points, colored by time point and labeled by cell types. (C) Cell assignment results of B cells, colored by subclone identity assigned via scBayes: blue, orange, green, and gray represents SC1, SC2, normal, and unassigned, respectively. (D) Sample and subclone-specific expression profiles of genes TNFRSF13B, TXNIP, and CD69, highlighting different patterns of expression change across the three time points. (*) P < 0.005 and FDR < 0.05. (E) Overall and subclone-specific V(D)J clonotype diversity. Each bar represents a unique clonotype; the height of a bar corresponds to the percentage of that clonotype within the B cell (left most column) or specific B cell subclone (right three columns) population.
Our ability to associate cells with subclones, and to perform subclone-specific differential expression analysis allows us to deconvolute the effects of cell population dynamics versus treatment induced expression change (Fig. 3D), as illustrated by gene expression changes in three CLL-relevant genes. The first gene is TNFRSF13B, which encodes the TNF receptor superfamily member 13B, also known as TAC1. TNFRSF13B is a receptor for the B cell–activating factor of tumor necrosis factor family (BAFF) signaling, which can induce the activation of the canonical NF-kappaB pathway and promote CLL cell survival (Endo et al. 2007). Although the overall expression level of TNFRSF13B decreased over time in the tumor, subclone-specific expression analysis revealed that it was not the direct effect of treatment altering the expression in the cells. Rather, TNFRSF13B expression level was different between the subclones: high in SC1, and low in SC2, and remained constant within each subclone over time; and the overall decrease of TNFRSF13B expression over time was the result of subclone SC1 shrinking, and SC2 expanding. The second gene is thioredoxin interacting protein (TXNIP), which has been reported as a tumor suppressor that inhibits glucose uptake, tumor cell proliferation, and cell cycle progression (Jeon et al. 2005; Kwon et al. 2010; Park et al. 2018). Overall TXNIP expression increased from T1 to T2. Subclone-specific analysis shows that TXNIP expressions were similar between the two subclones at each time point, and similarly increased from T1 to T2, likely because of the effect of treatment. Finally, we examined CD69, an early activation marker in CLL (Montraveta et al. 2016), a gene that has been shown to be down-regulated by ibrutinib treatment (Herman et al. 2014). We see a corresponding decrease in CD69 expression in our patient. According to our analysis, the overall decrease is the result of a subclone-specific expression decrease in SC1 alone (CD69 expression remained unchanged in SC2 between the consecutive time points). These results highlight the level of resolution obtainable only via subclone-specific gene expression analysis made possible by cell-to-subclone assignment.
We performed genome-wide differential expression analysis between cells assigned to SC1 and SC2 and found 44 up-regulated and 22 down-regulated genes in SC1 relative to SC2 (Supplemental Fig. 11A). We further compared the expression of cells of SC1 and SC2 to B cells that were assigned as genetically normal cells. We found 94 up-regulated and 56 down-regulated genes in SC1 relative to normal (Supplemental Fig. 11B); and 23 up-regulated and 39 down-regulated genes in SC2 relative to normal (Supplemental Fig. 11C). SC2 expressed less of these differential genes compared to SC1. Most relevant to CLL malignancy, we found that genes MIR155 (oncogene and associated with aggressive CLL disease [Cui et al. 2014]), ID3 (prosurvival of CLL cells [Weiler et al. 2015]), RAC2 (involved in B cell receptor signaling [Arana et al. 2008]), and FCER2 (involved in B cell activation and proliferation) were overexpressed in SC1 relative to normal; whereas two B cell markers CD22 and MS4A1 were underexpressed in SC1 relative to normal (Supplemental Fig. 11D). The expression levels of these genes in SC2 cells were between SC1 and normal (Supplemental Fig. 11D), suggesting that SC2 is phenotypically less aggressive than SC1.
The overexpression of the B cell activation markers in subclone SC1 described above led us to the hypothesis that this was the dominant malignant CLL subclone in the patient; whereas SC2 represents a less aggressive CLL cell population. To test this hypothesis, we investigated the single-cell V(D)J profiles, or clonotypes (Fig. 3E), collected for each cell as part of the scRNA-seq experiment. We found that one clonotype was dominant (93.16% of all B cells, see Fig. 3E, column 1) in this patient at pretreatment (T1), likely the result of the monoclonal expansion of a leukemic population. This observation is also consistent with published studies (Campbell et al. 2008; Boyd et al. 2009). The fraction of this clonotype declined over the course of the treatment (T2, T3), and the total set of clonotypes became more diverse. Because the single-cell clonotype data share the same cell barcodes as the scRNA-seq results, we are able to derive subclone-specific clonotype profiles using the scBayes cell assignment results (Fig. 3E, columns 2–4). We found that the dominant clonotype in the patient at T1 was also the dominant clonotype among cells of SC1 (Fig. 3E, column 2). Cells assigned to SC2 and the normal subclone showed more diverse clonotypes (Fig. 3E, columns 3–4) compared to those assigned to SC1. Taken together, these observations are consistent with the patient's response to ibrutinib treatment, that is, that the BTK inhibitor successfully targeted the fast-proliferating malignant subclone SC1, shrinking it during the course of treatment to become only a minor subclone within the B cell population. These results highlight the importance of subclone-level understanding of tumor cell behavior and treatment response, enabled by cell-to-subclone assignment.
Discussion
We have developed a method that effectively reconstructs subclonal phenotypes via the assignment of cells from scRNA-seq to genetically defined tumor subclones. Our method accounts for data sparsity and sequencing errors occurring in scRNA-seq data via a Bayesian statistical framework (Fig. 1). While assigning cells from scRNA-seq experiments (both full-transcript and end-capture protocols) is the primary goal of our method, we showed that it is generally applicable to a wide range of single-cell sequencing technologies that are capable of assessing the presence of somatic mutations. We provided proof of concept for applying scBayes to single-cell DNA sequencing data, as well as 10x Genomics scATAC sequencing data, while acknowledging that applications in these areas likely require further evaluation and method characterization. We validated the correctness of our algorithm using simulated data sets (Supplemental Fig. 2), synthetic pseudobulk data set from single-cell DNA sequencing (Supplemental Fig. 3), a synthetic data set using real 10x sequencing data in which we know the true cell identities (Supplemental Fig. 4), as well as using a previously published breast cancer data set in which both the subclone structure and the subclonal phenotypes are orthogonally or experimentally validated (Fig. 2). We note that our method does not explicitly model certain aspects of data that could potentially affect the cell assignment process, such as dead cells, empty droplets, or doublets, as some other tools do (Roth et al. 2016). Although these are important aspects to consider, there have also been a number of dedicated tools developed for such purposes (Xi and Li 2021). Instead of incorporating these functionalities directly into scBayes, we would refer analysts to using these more suitable tools to generate a cell barcode list for cells considered to be high quality. scBayes can then use this barcode list to assign only high quality cells. Subclone reconstruction from bulk DNA sequencing data often yields competing, alternative subclone structures. Because the assignment quality of scBayes can be considered a measurement of the concordance between the bulk DNA and scRNA-seq data, we propose that the sum of assignment qualities across all cells be used to identify the correct subclone structure. To show the utility of this approach, we manually changed the subclone structure in Figure 3 to two incorrect subclone structures, and found the total assignment quality to be consistently lower for the incorrect structures (Supplemental Fig. 12).
In low mutation coverage situations, we see the utility of our method in automating what can be an ad hoc, tedious, and error prone process if performed manually. This is especially beneficial when a large number of cells are considered, many of which can be ambiguous (cells having positive evidence for mutually exclusive clones). To manually account for all observations can be unattractive (even five variants can give a total of 243 different per-cell presence/absence combinations because each variant has three potential states: no coverage, reference only coverage, and mutation-confirming coverage). Our method provides an automated, documented, and repeatable process to handle this complexity programmatically. In extremely low mutation coverage and cell assignment situations, assigning scRNA clusters instead is a viable option. We did not build this feature directly into our tool because it makes an implicit assumption that cellular genotype and phenotype are correlated. This may or may not be valid, as cell plasticity can drive differential expressions in spite of similar genetic background; and genetically divergent subclones can have convergent expression behavior. If this is an assumption the analyst is willing to make, scBayes results can be used to perform cluster-to-subclone assignment (e.g., assigning a consensus subclone identity to each cluster).
Our method has the capacity to generate biological insights and novel hypotheses. The analysis results we presented on the breast cancer pre- versus postdoxorubicin treatment data set and the CLL longitudinal data set highlights the value of applying our method to delineate and study the evolution of subclonal phenotypes. Our analysis on the breast cancer data set (Fig. 2) not only recapitulated the finding that the elevated EMT signature was associated with the doxorubicin resistant clone, we in addition (1) discovered that its predecessor subclone in pretreatment time point was already on the trajectory of EMT enrichment; and (2) generated novel hypotheses regarding the clonal dynamics that were not investigated in the original study such as the resistant clone might have already been present at small fractions at pretreatment. Our analysis on the CLL longitudinal data set showed several novel perspectives that would otherwise have been missed without subclone-specific phenotypes (Fig. 3), including (1) the interpretation of the newly emerging subclone as less aggressive instead of being treatment resistant; (2) the elucidation that similar differential expression patterns over time at bulk level can be the result of altered subclonal expressions, altered subclone population composition, or both; and (3) the integration of B cell V(D)J clonotype data at subclonal level, which further strengthened our hypothesis that SC2 was less aggressive than SC1.
Our algorithm outperforms existing relevant methods. We identified two relevant existing methods: DENDRO and Cardelino. We compared scBayes to DENDRO and Cardelino using the full-length transcript breast cancer data set (Supplemental Fig. 8); and we additionally compared scBayes to Cardelino using the simulated data sets, the published synthetic pseudobulk data set, and our own 10x Genomics UTR capture and sequencing data set (Supplemental Figs. 6, 8, 10. DENDRO only works with full-transcript data and therefore cannot be included in the latter comparisons). We ran DENDRO and Cardelino according to their published instructions including input data processing. Consistently, we showed that scBayes is capable of assigning a much higher fraction of cells, and produces assignment results that incur the least conflicts with the DNA-seq-based subclone structure. Furthermore, although correctness is the most important criterion, scBayes also runs much faster. Using simulation on false positive rate as an example, scBayes finished analyzing 500 simulated data points (100 repeats at 5%, 10%, 20%, 30%, and 40% false positive rate each, Supplemental Fig. 2A) in 53 sec, while Cardelino took 5.5 h to finish on the same input.
Finally, our method is applicable to practical cancer genomic studies with typical data sets. Simulation results indicated that our method is highly accurate with respect to a wide range of single-cell RNA sequencing true / false positive rates and subclone reconstruction error rates, and agnostic to subclone structure patterns. The breast cancer and CLL data sets we analyzed in our manuscript represent high and low tumor somatic mutation burden, respectively (Bailey et al. 2018). We further reviewed recent articles studying cancer heterogeneity, and summarized the number of subclones and somatic mutations per subclone reported by these studies (Supplemental Table 1). We found that the data sets included and analyzed in our manuscript are representative of the level of genetic information obtainable from a typical tumor sample. Furthermore, our approach is plausibly applicable for other sequence-based single-cell-omic data types, for example to study tumor- and subclone-specific chromatin accessibility using cell-to-subclone assignment in single-cell ATAC-seq (scATAC-seq) data sets (see Supplemental Fig. 13 for proof-of-concept analysis with a chronic myelomonocytic leukemia data set).
Methods
scBayes approach
scBayes works in two stages. Stage 1, scGenotype, is the single-cell genotyping stage, during which the sequencing evidence (reads) are tallied at each somatic variant position for each cell barcode. This results in a matrix where each row corresponds to a somatic variant, and each column a cell. The content of the matrix has the format DP:RO:AO where DP stands for sequencing depth (total read count overlapping the variant position); RO stands for number of reference observations (reads having the reference allele at the variant position); and AO stands for number of alternate observations (reads having the variant allele at the variant position). This matrix is used for the next stage. The pseudocode for scGenotype is as follows:
somatic_variants = load_variants(somatic_variants_vcf);
genotype_matrix = dictionary of dictionary
for_each v in somatic_variants do
pileup = pileup_single_cell_alignments_at(v.position);
for_each read in pileup do
genotype_matrix[read.cell_barcode][v].DP += 1;
if read.allele == REF_ALLELE do
genotype_matrix[read.cell_barcode][v].RO += 1;
else if read.allele == v.allele do
genotype_matrix[read.cell_barcode][v].AO += 1;
end-if
end-for
end-for
print(genotype_matrix)
Stage 2, scAssign, is the cell-to-subclone assignment stage, during which the posterior probability for each cell to have
come from each subclone is calculated using the Bayesian posterior probability equation (Eq. 1):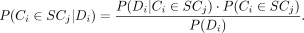 (1)
(1)
The term P(Ci ∈ SCj) is the prior probability for cell i to have come from subclone j. We use the cellular prevalence of subclone j from the bulk DNA sequencing-based subclone analysis if it is reasonable to assume that the single-cell subclone composition
represents that which was in the bulk DNA sequencing sample. This is often a good assumption if the bulk DNA sequencing library
was made from the same biological sample as the single-cell sequencing library. Alternatively, a flat prior 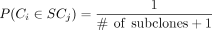 can be used (the +1 in the denominator accounts for the additional, no-variant subclone representing the normal tissue).
can be used (the +1 in the denominator accounts for the additional, no-variant subclone representing the normal tissue).
The term P(Di|Ci ∈ SCj) is the data likelihood, and measures how likely it is to have observed the genotype information Di from single-cell sequencing if cell i had come from subclone j. We denote 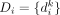 in which
in which 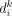 is a binary value indicating whether somatic variant k is present in the sequencing data from cell i; and
is a binary value indicating whether somatic variant k is present in the sequencing data from cell i; and 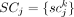 in which
in which 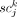 is a binary value indicating whether subclone j contains variant k. We further assume that the ability for single-cell sequencing to assess the presence of any somatic variant in a cell is
independent from the presence of other somatic variants. We can then calculate the data likelihood as follows:
is a binary value indicating whether subclone j contains variant k. We further assume that the ability for single-cell sequencing to assess the presence of any somatic variant in a cell is
independent from the presence of other somatic variants. We can then calculate the data likelihood as follows: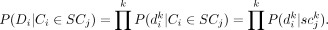 (2)
(2)
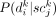 in Equation 2 (henceforth denoted as Pd) has four possible cases. In the case of
in Equation 2 (henceforth denoted as Pd) has four possible cases. In the case of 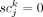 , which indicates that subclone j does not contain somatic variant k, Pd(0|0) represents the likelihood of the single-cell sequencing data not showing a presence of the variant allele, which
intuitively is very high (scBayes uses 0.99 by default); and Pd(1|0) represents the likelihood of the single-cell sequencing
data showing a presence of the variant allele, which intuitively should be very low as only sequencing error and false negative
in somatic variant calling can result in this situation (scBayes uses 0.01 by default). In the case of
, which indicates that subclone j does not contain somatic variant k, Pd(0|0) represents the likelihood of the single-cell sequencing data not showing a presence of the variant allele, which
intuitively is very high (scBayes uses 0.99 by default); and Pd(1|0) represents the likelihood of the single-cell sequencing
data showing a presence of the variant allele, which intuitively should be very low as only sequencing error and false negative
in somatic variant calling can result in this situation (scBayes uses 0.01 by default). In the case of 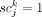 , which indicates that subclone j does contain somatic variant k, Pd(0|1) represents the likelihood of the single-cell sequencing data not showing a presence of the variant allele. This
is known as “allele-dropout” and has a fairly high likelihood to occur in single-cell sequencing data. Pd(1|1) represents
the likelihood of the single-cell sequencing data showing a presence of the variant allele. This is expected to occur at a
high probability. We estimated Pd(0|1) and Pd(1|1) for Fluidigm (92 cells), 10x Genomics scRNA-seq (4572 cells), and 10x Genomics
scATAC-seq (2524 cells) platforms using inherited variants (therefore all cells are expected to have them). See below for
the detailed method and Supplemental Figure 14 for the results.
, which indicates that subclone j does contain somatic variant k, Pd(0|1) represents the likelihood of the single-cell sequencing data not showing a presence of the variant allele. This
is known as “allele-dropout” and has a fairly high likelihood to occur in single-cell sequencing data. Pd(1|1) represents
the likelihood of the single-cell sequencing data showing a presence of the variant allele. This is expected to occur at a
high probability. We estimated Pd(0|1) and Pd(1|1) for Fluidigm (92 cells), 10x Genomics scRNA-seq (4572 cells), and 10x Genomics
scATAC-seq (2524 cells) platforms using inherited variants (therefore all cells are expected to have them). See below for
the detailed method and Supplemental Figure 14 for the results.
P(Di) is the normalization factor, which can be calculated by 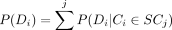 .
.
It is worth noting that Equation 2 highlights the benefit of scBayes, that is to allow a large number of somatic variants to participate in the task of assigning cells subclone identities. The pseudocode for scAssignment is as follows:
subclones, Pd = load_config(); // Pd stands for data likelihoods
genotypes = load_genotypes(scGenotype_result);
for_each c in genotypes.cells do
normalization = 0;
for_each sc in subclones do
d = 0;
data_likelihood = 1;
for_each v in c.genotypes do
if v.DP == 0 do continue; // no sequencing depth at v position; skipping
if v.AO > 0 do d = 1; // if 1 or more reads contain variant
data_likelihood *= Pd(d | 1 if sc.has(v) else 0);
end-for
c.joint_prob[sc] = data_likelihood * sc.fraction; // joint_prob = data likelihood * prior
normalization += c.joint_prob[sc];
end-for
max_prob = 0;
argmax_sc = null;
for_each sc in subclones do
c.posterior[sc] = c.joint_prob[sc] / normalization;
if c.posterior[sc] > max_posterior do
max_posterior = c.posterior[sc];
argmax_sc = sc;
end-if
end-for
print(“Cell “, c, “ is assigned to subclone”, sc, “ with confidence”, max_posterior);
end-for
Estimation of data likelihoods
We used inherited variants with consistently high allele frequencies in all samples (>0.3) to measure Pd(0|1) and Pd(1|1) because all cells are expected to have these variants in their genomes. We deliberately give no further distinction to heterozygous/homozygous status, intronic/exonic status, or potential allele preferences by cell types as the same confounding factors affect somatic mutations too. For each of these variants, we counted the number of cells, denoted as Nc, that have sequencing coverage at the variant site. We then further counted the number of cells, denoted as Np, that had positive evidence for the presence of the variant (1 or more sequencing reads having the variant allele). A ratio r = Np / Nc is calculated. Repeating this procedure over all considered variants yields a distribution that models Pd(1|1), which we term as “pick-up rate”. Averaging the pick-up rates over all considered variants gives the estimated Pd(1|1), which is used as the default data likelihood in scBayes. We then calculate Pd(0|1) as 1 – Pd(1|1). We estimated the data likelihoods separately for Fluidigm, 10x Genomics scRNA-seq, and 10x Genomics scATAC-seq technologies (Supplemental Fig. 13). If users have estimated their own data likelihood values for different platforms, with different data sets, or with different parameterizations, they can override the default when performing cell assignment.
scBayes cell assignment algorithm validation using simulated data sets
To test the performance of scBayes and competing methods with respect to various properties of the input data, we created a simulation framework with which we can simulate somatic mutations, subclone structures, true cell assignments, and single-cell genotypes. The following properties can be altered for each simulation:
-
Variant coverage rate: the likelihood of observing ≥ 1 scRNA-seq read at a specific somatic mutation site in a cell. A default value of 5% is used if not specified otherwise.
-
scRNA-seq coverage distribution parameter P: from our data sets, we found that a geometric distribution models the sequence coverage (at sites with at least 1 read) well in scRNA-seq data. Through distribution fitting, we estimated that a P = 0.318 parameter best fit the actual observation. This is the default value if not specified otherwise.
-
scRNA-seq false positive rate: This is the likelihood that scRNA-seq would erroneously show the presence of mutant alleles at a somatic mutation site when the cell does not have the mutation, or Pd(1|0). A default value of 5% is used if not specified otherwise.
-
scRNA-seq true positive rate: This is the likelihood that scRNA-seq would correctly show the presence of mutant alleles at a somatic mutation site when the cell does indeed have the mutation, or Pd(1|1). If a value is specified, it is used as a fixed rate through simulation. Otherwise, we use a single-cell allele frequency distribution estimated from our data sets, which breaks down roughly as the following:
-
30% chance of observing AF = 0 (or a false negative)
-
61% chance of observing AF = 1 (e.g., if five reads overlap the mutation site, all five reads would show the mutant allele)
-
9% chance of observing an AF that fits a normal distribution with mean of 0.5 and standard deviation of 0.2
-
-
Subclone reconstruction error rate: The likelihood of attributing somatic mutations to the wrong subclone during subclone reconstruction.
-
# of subclones to simulate
-
Whether to fix the subclone structure during simulation
Although we attempted our best to estimate a sensible set of default values from a real data set, we do not claim that our simulation framework produces realistic sequencing data as one would expect from an actual sequencing experiment. Rather, this simulation framework aims to provide data with known truth, and a single variable (e.g., scRNA-seq true positive rate) to isolate its effect on algorithmic performance while keeping other variables fixed. In some cases, such as in the simulation of informed versus flat prior, we had to deliberately reduce the data quality significantly in order to render the effects from the simulated variable apparent. Nonetheless, this framework allowed us to examine various data quality aspects in isolation, and provided a way to compare our methods to Cardelino with respect to these aspects.
To carry out the actual performance evaluation as presented in Supplemental Figure 2, we simulated 100 sets of data per each experiment category, and performed scBayes and Cardelino cell assignment. The source code of our simulation framework, as well as the scripts used to carry out the different simulations, are available at GitLab (https://gitlab.com/yiq/scbayes-simulator).
scBayes cell assignment algorithm validation using scDNA sequencing-based pseudobulk data set
To further validate the performance of scBayes, and compare it to Cardelino, using data sets with known ground truth, we took advantage of a published, single-cell DNA sequencing data set (Laks et al. 2019) in which a subclone structure was reconstructed directly from the genotypes of hundreds of individual cell gathered across three samples from the same patient. This provides a direct ground truth we can use for method validation. The original publication provides data including the mutations believed to be present in each subclone, as well as the cells that are assigned to each subclone. We performed minimal data reformatting to extract the information scBayes and Cardelino to perform cell-to-subclone assignment. Specifically, we extracted the variants for each subclone, and reformatted into the VCF format for scBayes. We in addition filtered out variants that do not appear to fit the hierarchical subclone structure (e.g., mutations that seem to independently occur on two unrelated branches). We further created a mutation / subclone matrix according to Cardelino's input format specification using the same set of variants. The minimal cell assignment quality for scBayes is changed to 3 (Phred score, or 50.12%) to be in line with the default value (50% posterior probability) used by Cardelino. The processed data files and scripts used to perform cell assignment are available at GitLab (https://gitlab.com/yiq/scbayes-datasets).
scBayes cell assignment algorithm validation using a synthetic subclone structure from a CLL data set
We isolated B cells and T cells from three CLL patients at pretreatment time point using the EasySep Magnet particles (B cell isolation: EasySep Human B Cell Enrichment Kit II Without CD43 Depletion; T cell isolation: EasySep Release Human CD3 Positive Selection Kit). We collected whole-exome sequencing data from B cells and T cells separately. Sequencing reads were aligned using BWA-MEM (Li 2013) (v0.7.17) to GRCh37 using default parameters. We chose GRCh37 because at the time the analysis was performed, 10x Genomics only provided VDJ reference builds based on GRCh37. We note that realigning the sequencing reads to GRCh38 (a more recent assembly) will not significantly affect the conclusions of the study, as the somatic mutations we used for cell assignment were recapitulated when we realigned to GRCh38, and resulted in highly concordant single-cell coverage using the GRCh38-VDJ scRNA-seq reference that became available later. We called somatic variants in B cells using T cells as the normal control with FreeBayes (Garrison and Marth 2012) (v1.2.0). We filtered for single nucleotide variants, and identified somatic variants, using variant toolkit (https://github.com/atks/vt) with the following criteria:
-
Sequencing coverage in both T cell and B cell is greater than 30×
-
Variant allele frequency in T cell is lower than 0.1 and alternative allele count is lower than 5
We made a synthetic subclone structure where each subclone contains the somatic variants of one patient. We also collected 10x Genomics single-cell RNA sequencing data (5′ expression) from the peripheral blood cells of these patients. We used the 10x Genomics cellranger pipeline (v3.0.2) to preprocess the data. Then we used the R package Seurat (Butler et al. 2018; Stuart et al. 2019) (v3.0) to remove low quality cells and to normalize the expression. B cells were identified by B cell markers, for example, MS4A1. Then we performed scBayes assignment on B cells from each of the patients to the synthetic subclones, and compared the results to the cells’ original patient identities that served as the ground truth.
Cell assignment and subclone-specific comparative phenotype analysis on the longitudinal breast cancer data set
Somatic mutation data and scRNA-seq gene expression data of this data set are available on the European Genome-phenome Archive (EGA; https://ega-archive.org/) under accession EGAS00001002436. We identified subclone-defining variants according to the published subclone structure and subclone frequencies. Cell assignment with scBayes was performed using the subclone structure described in the original publication (while giving subclones not found at a time point a very small, 0.01 prior likelihood to enable cross-sample assignment, e.g., allowing posttreatment cells to be assigned to pretreatment subclones). Cells with assignment quality lower than 5 are considered UNASSIGNED, and excluded from subclone-specific expression analysis. Single-cell RNA sequencing data was processed as described in the original publication. We performed ssGSEA (Barbie et al. 2009) analysis using the C2 curated signatures from MSigDB (including 5637 signatures) on each cell. We ran an online module ssGSEAProjection (Subramanian et al. 2005) (version 9.1.1) of GenePattern (Reich et al. 2006). ssGSEA enrichment score was calculated for each cell and used for subclone-specific phenotype analysis. We grouped cells based on their subclone identity and compared ssGEA score among cells in different subclones using two-tailed Student's t-test.
Calculation of the expected alleles ratio and allelic collision rate to evaluate the quality of cell-to-subclone assignment
To quantitatively evaluate different DENDRO clusters to genetic subclones assignments, as well as to compare DENDRO and Cardelino
with scBayes, we devised two scores for unexpected alleles: number of allelic collisions (NAC) and allelic collision rate (ACR). Because each genetic subclone is defined by one or more somatic mutation clusters (e.g., C1–C5 in Fig. 3), when a group of cells are assigned to a specific genetic subclone, we can tabulate the number of mutations observed in
these cells that are either “expected” or “in collision” with the assignment. A mutation is “expected” when the genetic subclone
these cells are assigned to is supposed to contain this mutation. As an example, if a group of cells are assigned to the subclone
“SC3” in Figure 3, which contains mutation clusters C1 and C3, any mutations of C1 or C3 observed in these cells are counted as “expected”
evidence; and any mutations of C2, C4, or C5 observed in these cells are counted as “in collision”. We define the total number
of “expected” mutations observed in all cells for a given assignment as NEA, the total number of “in collision” mutations
observed in all cells for a given assignment as NAC, and compute ACR as the ratio between NAC and total observed somatic mutations
across all cells (NEA + NAC). Although these metrics do not capture all aspects of assignment qualities, have no interpretable
meanings in their absolute values, and are susceptible to other data quality issues, for example, subclone reconstruction
errors, they offer an unbiased measurement of relative assignment goodness across methods when the same underlying data sets
are used. Intuitively, if the cell assignments are correct, and sequencing and mutation calling perfect, NAC and ACR should
both be 0. A more consistent cells-to-genetic-subclones assignment would result in lower NAC and ACR values. Formally, we
define 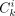 as the number of consistent mutations observed in cell cluster i and mutation cluster k;
as the number of consistent mutations observed in cell cluster i and mutation cluster k; 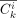 as the number of inconsistent mutations observed in cell cluster i and mutation cluster k. We define NEA, NAC, and ACR as follows:
as the number of inconsistent mutations observed in cell cluster i and mutation cluster k. We define NEA, NAC, and ACR as follows: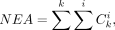
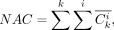
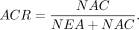
Cell assignment and subclone-specific comparative expression analysis on the CLL BTKi treatment data set
We collected whole-exome sequencing data from a CLL patient (one of the patients described above, patient 3), who received Brunton tyrosine kinase inhibitor, at three time points: pretreatment, 1 yr, and 2 yr after starting the treatment. Sequencing reads were aligned using BWA-MEM (Li 2013) (v0.7.17) to GRCh37 using default parameters. We called somatic variants in B cells using T cells as the normal control with FreeBayes (Garrison and Marth 2012) (v1.2.0). We used SubcloneSeeker (Qiao et al. 2014) to resolve the subclone structure at the three time points. We also collected 10x Genomics single-cell RNA sequencing data (5′ expression coupled with V(D)J profiling) from the same patient using the peripheral blood samples at the same three time points. We used the 10x Genomics cellranger pipeline (v3.0.2) to preprocess the data. Then we used the R package Seurat (Butler et al. 2018; Stuart et al. 2019) (v3.0) to remove low quality cells and to normalize the expression. We used canonical cell markers to identify different cell types: CD14 for monocytes, CD3D, CD3E for T cells, MS4A1 for B cells, GNLY and NKG7 for NK cells. We used scBayes to assign each B cell to a subclone. We grouped cells with the same subclone identity and compared gene expression among different subclones at different time points using likelihood ratio test (negbinom; assuming an underlying negative binomial distribution) implemented in Seurat. Significantly expressed genes were defined as adjusted P value (based on Bonferroni correction using all features in the data set) < 0.05 when different B cell subclones were compared (SC1 vs. normal, SC2 vs. normal, or SC1 vs. SC2).
Cell assignment analysis on the CMML scATAC-seq data set
We collected WGS data on the mononuclear cells sample and skin sample from a CMML patient. Sequencing reads were aligned using BWA-MEM (Li 2013) (v0.7.17) to GRCh38 using default parameters. We called somatic variants with FreeBayes (Garrison and Marth 2012) (v1.2.0). We also collected 10x Genomics single-cell ATAC sequencing data from the same patient using the peripheral blood sample. We used the 10x Genomics cellranger-atac pipeline (v1.1.0) to preprocess the data. Subclone identity was assigned to each cell by using scBayes.
Data sets
Somatic mutations from three chronic lymphocytic leukemia used for the synthetic data set are available as Supplemental Dataset 1. Somatic mutations from the CLL longitudinal patient samples are available as Supplemental Dataset 2. Somatic mutations identified from the chronic myelomonocytic leukemia patient are available as Supplemental Dataset 3. Genotypes from scRNA-seq data of all presented data sets are available as Supplemental Dataset 4.
Software availability
We compiled all necessary data sets, sample scripts to compile and run scBayes, and R (R Core Team 2021) scripts to generate visualizations from scBayes results into a single archive with extensive documentation and a snapshot of the scBayes code base as before submission. This archive is available as Supplemental Code 1 as well as at GitLab (https://gitlab.com/yiq/scbayes-datasets). This archive further contains the scDNA-seq-based pseudobulk synthetic data set. The scBayes software is open source, and available as Supplemental Code 2 and at GitLab (https://gitlab.com/yiq/scbayes). The simulation framework is open source, and available as Supplemental Code 3 and at GitLab (https://gitlab.com/yiq/scbayes-simulator). Custom code for analyses beyond cell assignment, such as subclone-specific expression profile and clonotype analysis, is available as Supplemental Code 4 and at GitHub (https://github.com/xiaomengh/scBayes_analysis).
Data access
The single-cell expression data for the functional analysis of the CLL data set generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) and under accession number GSE186150.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
Y.Q., X.H. and G.T.M. were supported by National Institutes of Health (NIH) Grant No. U24CA209999 to G.T.M. This work was partly supported by the Utah Genome Project (award to G.T.M. and M.W.D.). Work performed in the laboratory of M.W.D. (includes J.M.A. and A.D.P.) was supported by a Translational Grant from The V Foundation for Cancer Research. Research reported in this publication used the High-Throughput Genomics and Bioinformatic Analysis Shared Resource at Huntsman Cancer Institute at the University of Utah and was supported by the National Cancer Institute of the NIH under Award No. P30CA042014. The support and resources from the Center for High Performance Computing at the University of Utah are gratefully acknowledged. The computational resources used were partially funded by the NIH Shared Instrumentation Grant No. 1S10OD021644-01A1. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions: Y.Q., X.H., and G.T.M. conceived and designed the project. Y.Q. developed and implemented the scBayes algorithm. Y.Q. and X.H. performed cell assignment analysis. X.H. performed differential expression analysis and clonotype analysis. J.C.B., J.A.W., and D.M.S. coordinated the patient recruitment, sample collection, and benchwork for the data collection on the CLL data set; P.J.M. performed the single-cell sequencing benchwork on the CLL data set; J.M.A., A.D.P., and M.W.D. coordinated the patient recruitment, sample collection, and benchwork for the data collection on the CMML data set. Y.Q., X.H., and G.T.M. wrote the manuscript. All authors read and edited the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278234.123.
-
Freely available online through the Genome Research Open Access option.
- Received June 29, 2023.
- Accepted November 22, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








