
Genome sequencing of C. elegans balancer strains reveals previously unappreciated complex genomic rearrangements
- Tatiana Maroilley1,2,
- Stephane Flibotte3,
- Francesca Jean1,2,
- Victoria Rodrigues Alves Barbosa1,2,
- Andrew Galbraith1,2,
- Afiya Razia Chida1,2,
- Filip Cotra1,2,
- Xiao Li1,2,
- Larisa Oncea1,2,
- Mark Edgley4,
- Don Moerman4 and
- Maja Tarailo-Graovac1,2
- 1Departments of Biochemistry, Molecular Biology and Medical Genetics, Cumming School of Medicine, University of Calgary, Alberta T2N 4N1, Canada;
- 2Alberta Children's Hospital Research Institute, University of Calgary, Calgary, Alberta T2N 4N1, Canada;
- 3UBC/LSI Bioinformatics Facility, University of British Columbia, Vancouver, British Columbia V6T 1Z3, Canada;
- 4Department of Zoology, University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada
Abstract
Genetic balancers in Caenorhabditis elegans are complex variants that allow lethal or sterile mutations to be stably maintained in a heterozygous state by suppressing crossover events. Balancers constitute an invaluable tool in the C. elegans scientific community and have been widely used for decades. The first/traditional balancers were created by applying X-rays, UV, or gamma radiation on C. elegans strains, generating random genomic rearrangements. Their structures have been mostly explored with low-resolution genetic techniques (e.g., fluorescence in situ hybridization or PCR), before genomic mapping and molecular characterization through sequencing became feasible. As a result, the precise nature of most chromosomal rearrangements remains unknown, whereas, more recently, balancers have been engineered using the CRISPR-Cas9 technique for which the structure of the chromosomal rearrangement has been predesigned. Using short-read whole-genome sequencing (srWGS) and tailored bioinformatic analyses, we previously interpreted the structure of four chromosomal balancers randomly created by mutagenesis processes. Here, we have extended our analyses to five CRISPR-Cas9 balancers and 17 additional traditional balancing rearrangements. We detected and experimentally validated their breakpoints and have interpreted the balancer structures. Many of the balancers were found to be more intricate than previously described, being composed of complex genomic rearrangements (CGRs) such as chromoanagenesis-like events. Furthermore, srWGS revealed additional structural variants and CGRs not known to be part of the balancer genomes. Altogether, our study provides a comprehensive resource of complex genomic variations in C. elegans and highlights the power of srWGS to study the complexity of genomes by applying tailored analyses.
Balancer chromosomes typically contain chromosomal rearrangements that cover a large portion of genomes in model organisms (Edgley et al. 2006). They permit the maintenance of lethal or sterile mutations affecting essential genes in the heterozygous state by either suppressing recombination during meiosis, preventing the recovery of recombinant chromosomes, or leading to inviable recombinants.
Balancers in Caenorhabditis elegans implicating large structural variants (SVs) or chromosomal rearrangements stem from diverse origins. Some were created decades ago by applying physical mutagens such as X-ray, UV, and gamma radiation on C. elegans strains. They are referred to in this manuscript as “traditional balancers.” The mutagenesis processes generate random DNA breakages and chromosomal rearrangements. Rarely, rearrangements can also occur naturally (Edgley et al. 2006). Altogether, these traditional balancers cover ∼70% of the C. elegans genome. More recently, the advent of CRISPR-Cas9 technology has allowed the creation of a new set of targeted genetics balancers, balancing previously uncovered regions (Chen et al. 2018) and covering ∼85% of the genome (Iwata et al. 2016; Dejima et al. 2018), referred to as “CRISPR-Cas9 balancers” in this study.
C. elegans balancers with randomly generated chromosomal rearrangements were created before DNA sequencing was available. Their structure has mostly been explored with low-resolution labor- and resource-intensive techniques such as genetic linkage mapping, fluorescence in situ hybridization, PCR analysis (Zhao et al. 2006), SNP mapping (Kadandale et al. 2005), and oligonucleotide array comparative genomic hybridization (Maydan et al. 2007; Jones et al. 2009, 2007). As such, the exact base-pair-resolution DNA location of most balancer breakpoints is unknown. In addition, the structure of the balancers remains poorly characterized as previous approaches were unable to fully resolve complex rearrangements (Jones et al. 2007, 2009). Furthermore, we and others (Jones et al. 2007, 2009; Edgley et al. 2021; Flibotte et al. 2021; Maroilley et al. 2021) have shown that the molecular structure of genetic balancers is often more complex and larger than the descriptions made decades ago when high-resolution techniques were not available. They frequently combine several overlapping SVs and, in some instances, compounding to create chromoanagenesis-like complex rearrangements.
In C. elegans genetic studies, an incomplete characterization of balancer rearrangements prevents their reliable use in mapping experiments and can confound data analysis (Kadandale et al. 2005). Further, given that most C. elegans laboratories rely on balancers for studying essential genes, it is important to decipher the precise molecular structure of the rearrangements occurring in each C. elegans balancer.
Short-read whole-genome sequencing (srWGS) is currently the most cost-effective sequencing technology. Recently, Miller et al. (2016a,b,c) used genome sequencing to characterize genetic balancers in Drosophila melanogaster, illustrating the utility of this technology. Similarly, we recently showed that srWGS contains enough information to accurately detect breakpoints and characterize complex genome rearrangements of various types in C. elegans balancer strains (Edgley et al. 2021; Flibotte et al. 2021; Maroilley et al. 2021). In human diseases, long reads are often preferred (Gorzynski et al. 2022; Pei et al. 2022). However, recent studies have also benefitted from the power of srWGS to diagnose rare disease patients with complex genomic rearrangements (CGRs) (Ostrander et al. 2018; Minoche et al. 2021; Singer et al. 2021; Nicholas et al. 2022; Zenagui et al. 2022). However, in practice, it remains challenging to detect and characterize the whole spectrum of genomic variations with the available sequencing technologies as there are no standardized analysis pipelines. In addition, a complete characterization of the complexity of events like chromoanagenesis is especially difficult when the length of the reads precludes the overlap of the entire rearrangement. Indeed, chromoanagenesis events are characterized by sudden chromosome shattering and religation. They can be divided into three types based on their molecular properties: chromothripsis, chromoanasynthesis, and chromoplexy (Holland and Cleveland 2012; Collins et al. 2017; Pellestor et al. 2022). Chromothripsis and chromoplexy events are often balanced with little to no change in copy number and arise from nonhomologous end joining restitching after chromosome pulverization. Chromoplexy involves several chained translocations, whereas chromothripsis is mainly characterized by inversions and rearrangements in often one or occasionally a few chromosomes. However, chromoanasynthesis displays multiple copy number variations (CNVs) that emerge during replication from defective DNA repair mechanisms owing to microhomologies at breakpoints (Pellestor et al. 2022).
Here, we set out to use a srWGS approach (Maroilley et al. 2021) to systematically detect and characterize the molecular structure and genomic mapping of C. elegans genetic balancers. In addition, we report specific SVs and CGRs for each strain included in this study, constituting the first catalog of molecular characterization of complex genomic variation in C. elegans. We anticipate that this data set will provide a valuable resource for the extensive userbase of traditional genetic balancers in C. elegans genetic studies. In addition, it will give researchers a better appreciation of the complexity of genomic variations, often overlooked in genomic studies (Maroilley and Tarailo-Graovac 2019).
Results
Balancers in C. elegans genetic studies
The first balancers in C. elegans were reported as early as 1976 (Herman et al. 1976). As previously performed for four traditional balancers (Edgley et al. 2021; Flibotte et al. 2021; Maroilley et al. 2021), in this study, we report breakpoints for 17 tested and reliable traditional balancers described by Edgley et al. (2006), which are used to balance over half of the genome (Fig. 1A). The list of selected strain(s) for each balancer is detailed in the Methods and Supplemental Table S1. They were ordered from the Caenorhabditis Genetics Center (CGC). Over time, morphologically marked and lethal variants have been generated for many of these balancers. Currently, extremely stable balancers such as nT1 are carried by more than 500 different strains available from the CGC (Fig. 1B; Supplemental Table S2), suggesting that balancers are a prominent tool in C. elegans genetics studies. Since 1991, strains with each balancer have been ordered from the CGC by the C. elegans research community many times (Fig. 1C; Supplemental Table S2), illustrating the great use that has been made of the balancers over the past decades. For instance, strains with nT1 have been ordered at the CGC almost 12,000 times. With over 1500 C. elegans laboratories worldwide (as listed in CGC and WormBase as of May 8, 2022), one could estimate that every research group working with lethal or extremely deleterious mutations uses balancers.
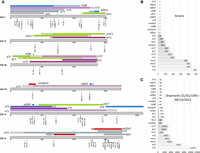
Overview of balancers. (A) Map of the C. elegans genomic regions balanced by rearrangements previously characterized at molecular level by our group (Edgley et al. 2021; Flibotte et al. 2021; Maroilley et al. 2021) or included in this study. Blue bars are for deletions. Green bars are for inversions or complex inversions. Purple and pink bars are for rearrangements involving translocations without large variation in copy number. Red bars are for rearrangements with copy number gains. Gray bars are for balancers not retrieved in this study, probably owing to breakage of balancers, or for which data are supporting the presence of a derivative. Darker colors represent the region covered by each balancer as described in previous studies. Lighter colors are the additional regions found balanced in this study. (B) Chart representing the number of strains available at the CGC carrying each balancer. (C) Chart representing the number of shipments between 1991 and 2022 of strains containing each balancer.
Using srWGS to detect known and unexpected high complexity in genomic variation
We first sequenced five balancer strains with well-defined and structurally complicated intrachromosomal rearrangements to reassess our approach with srWGS as published by Maroilley et al. (2021). The workflow is summarized in Supplemental Figure S1. These balancers were CRISPR-Cas9 engineered by applying two sequential and overlapping inversions (Dejima et al. 2018).
We successfully retrieved the breakpoints of these CRISPR-crossover suppressors previously described by Dejima et al. (2018): tmC3 V (FX30133), tmC6 II (FX30144), tmC20 I (FX30235), tmC24 X (FX30237), and tmC25 IV (FX30257). The precise genomic position of each breakpoint is described in Table 1 and Figure 2 and corroborates the data of their original study (Dejima et al. 2018). Experimental validation data (primers, PCR gels, and sequences from Sanger sequencing) are available in Supplemental Information.
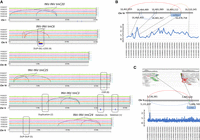
Overview of variants in CRISPR-Cas9 balancers. (A) Map of the CRISPR-ed double inversions and additional rearrangements detected. The upper tracks represent the normalized coverage for each strain. The lower track shows the position of structural rearrangements detected. The color code is green for FX30257, pink for FX30237, blue for FX30235, orange for FX30144, and blue-green for FX30133. The additional events 1–6 are detailed in Table 1. (B,C) Complex genomic rearrangements (CGRs) on Chr IV and Chr II detected in FX30144. Red link represents a copy gain. The graph on the top is a linear schematic representation of the breakpoints and junctions. Blue links represent a loss of copy. Orange link represents a gain of copy with inversion (possible inverted tandem duplication). In addition, we observed loss of copies (LOSS) by visualizing alignments using the Integrative Genomics Viewer (IGV) tool. The LOSS manifested by a complete absence of coverage (no read aligned on that segment of chromosome) but was not supported by split reads. It is possible that the loss of those regions is a consequence of the rearrangement, not a primary event. The scatter plots are a representation of the normalized coverage along the rearrangement calculated by windows of 1 kb. Both scatter plots were adapted to the size of the CGR. In C, the screenshot on the top shows how the reads aligned along the genome (IGV screenshot).
Breakpoints and genes identified in CRIPSR-Cas9 balancer strains
We found eight additional SVs and CGRs not reported before (Fig. 2A–C; Table 1), and possibly specific to these strains. Some were shared by all five strains and are therefore likely originated in the parental strain. But others were unique to only one strain, such as the two CGRs in FX30144 (Fig. 2B,C). These specific SVs were likely secondary targets of CRISPR or the result of spontaneous mutations.
For tmC20, the signature of reads for the larger inversion (I:114,579–8,210,593) is suggestive of a CGR involving a duplication and a deletion, both happening at the same breakpoint. Indeed, a usual inversion signature shows pair mate reads with the same orientation, displayed in blue by Integrative Genomics Viewer (IGV) (see Fig. 3A,B; Supplemental Fig. S2) and split reads aligned at the breakpoint junction with opposite directions. Here, we observed two distinct groups of reads. One presented characteristics of a duplication: pairs of reads (marked as green on IGV) (see Fig. 3C,D), for which the reverse read of a pair aligned before the forward read, and split reads, for which the second part of the read aligned before the first part of the read, in the same orientation (Supplemental Fig. S2). The second group of reads had characteristics of a deletion (marked as red on IGV) (see Fig. 3C,D), because the inner distance between pair mate reads is shorter than usual, and split reads for which the second part of the read was aligned further away on the genome, with the same orientation (Supplemental Fig. S2). The origin of such a signature is unclear.

Read signature when the alignments are visualized with IGV and representation of the split reads for inversions in tmC20 (FX20235). (A,C) IGV screenshots of the regions surrounding breakpoints. (B–D) Schematic representation of split read signatures. (A,B) The small inversion (4.7–8.2 Mb) shows a “normal” inversion signature: The reads in blue and blue-green are marked by IGV as the pair mates (forward and reverse read) have the same orientation instead of opposite orientation. (C,D) The larger inversion (0.1–8.2 Mb) shows a DEL-DUP read signature: Reads in green are marked as such by IGV because the reverse read aligned before the forward. Reads in red are marked as such by IGV as the inner distance between pair mates (forward and reverse reads) is longer than normal inner distance (usually 300–350 bp).
Mapping breakpoints of traditional balancers helps elucidate their effect on phenotypes
We then applied the same methods to decipher the molecular basis of 21 balancers. Four have been already published (Edgley et al. 2021; Flibotte et al. 2021; Maroilley et al. 2021), and we reported breakpoints for 17 additional traditional balancers in this study. Information regarding strains, sequencing, and experimental validation is available in the Methods and Supplemental Tables S1, S3, and S4.
For nine balancers (including eT1, previously published by Maroilley et al. 2021), our analyses only detected one large event responsible for balancing one region of the C. elegans genome (Fig. 4A; Table 2), as previously described in the literature. For most of these, we are reporting for the first time the exact genomic location, at base-pair resolution, of the breakpoints of each rearrangement and the structure at the junction of the breakpoints: strict, microduplications, or inserted sequence (Fig. 4B).
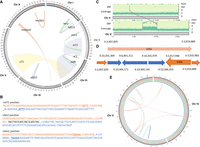
Overview of balancers. (A,B) Single event rearrangements. (A) Circos plot representing the regions affected by single-rearrangement balancers. Green links represent inversions (hIn1, mIn1). Blue links represent deletions (nDf6, sDf22, hDf15). Red links represent duplications (translocated duplication mnDp33 and free duplication mnDp3). Ribbons represent translocations (eT1, mT1). Scatter plots in the outer layer represent the read coverage calculated by window of 1 kb and normalized for KR2839's Chr I, MT690's Chr III, BC1217's Chr IV, and SP123's Chr X. It shows an increase in coverage in case of duplication and a decrease in case of deletion. (B) Breakpoints’ junctions for mT1, hIn1, and mIn1, as confirmed by Sanger sequencing. Orange and blue sequences are from different sides of the junction (different chromosomes for a translocation or different regions of one chromosome in case of inversion). Sequence in black is an inserted sequence at the junction. Bold underlined sequences are potential microduplications at the junction as those bases aligned on both sides of the junction. (C–E) Complex rearrangements. (C,D) stDp2. (C) The graph of the upper layer represents by a dot a single-nucleotide variant (SNV) detected in the RW6002 genome. Dots on the higher layer are homozygous SNVs with two copies of the alternate allele. Dots on the middle layer are heterozygous SNVs with only one copy of the alternate allele. Stretches of heterozygous SNVs suggest that the region is balanced by a complex rearrangement. The coverage graph is a scatter plot representing the read coverage of the genome along the chromosome, calculated by window of 1 kb. The graph shows an increase of read coverage of a short region of Chr X. The purple lines represent the breakpoints detected. (D) Interpretation of the structure of stDp2 based on srWGS analyses. Gain and loss events were detected by coverage analysis only and not supported by split reads, suggesting that they are consequences of the rearrangement, and not the main event. (E) Circos plot representing breakpoints for nT1 (red), eDf43 (blue), and e917 (green).
Breakpoints and structure of one-event traditional balancers
Often, we determined that balancers are larger than expected (Fig. 1A). For instance, mT1 was described as a translocation (II;III) balancing the right portion of Chr II from the right end through dpy-10 (mapped at II:6,710,149–6,712,227) and the right portion of Chr III from the right end to between daf-2 (III:2,994,514–3,040,846) and unc-93 (III:3,644,375–3,648,822). We analyzed the genome of SS746 [klp-19(bn126)/mT1[dpy-10(e128)] III]. We detected and confirmed the translocation breakpoints at II:6,296,872 and III:3,635,354, expanding the balanced region of Chr II by ∼400 kb.
The balancer sC1 is described as effectively balancing a 15-map unit portion of Chr III from unc-45 (III:491547–502625) to near daf-2 (III:2994514–3040846) (Edgley et al. 2006). However, the structure was yet to be described. We identified a large inversion on Chr III from III:0.3 Mb to III:4.6 Mb. Our analyses of srWGS data showed the right breakpoint at the position III:4,641,137. However, the left breakpoint falls into a large repetitive region, and two breakpoints are equally plausible—III:323,321 or III:330,985 (Supplemental Fig. S2), perhaps explaining why the balancer was never molecularly characterized.
We found lesions at some rearrangement breakpoints that could be responsible for observed phenotypes. For example, mT1 homozygotes are sterile, and we found that the translocation disrupts linc-95 (Table 2), a long intervening noncoding RNA that may be important for fertility. In addition, the mIn1 inversion disrupts the essential gene tbc-17 (Table 2), which may explain why homozygous mIn1 animals do not survive freezing well (Edgley and Riddle 2001), although mIn1 also carries dpy-10(e128), and dpy-10 homozygotes were already known to poorly survive freezing (Levy et al. 1993).
Uncovering CGRs in balancers
The structure of 10 of the balancing rearrangements (including hT2 [Flibotte et al. 2021], sC4 [Maroilley et al. 2021], and qC1 [Edgley et al. 2021]) was revealed to be more complex than expected (Table 3).
Breakpoints (BKPs) of high-complexity traditional balancers
The balancer stDp2 (X;II) has been described as a translocated duplication, effectively balancing a small region of the center of Chr X, from around unc-58 to around unc-6 (Meneely and Wood 1984; Edgley et al. 2006). Our genomic analysis of the strain RW6002 [stDp2 (X;II)/ + II; unc-18(e81) X] revealed a more complex crossover suppressor structure, with 13 breakpoints localized on Chr II and Chr X (Table 3; Fig. 4C,D). To summarize, the region on Chr X from around X:6.8 Mb to 10.3 Mb is represented by four copies, instead of two, with an inversion at one of the extremities of the duplicate. Those two additional copies were inserted in Chr II ∼3.9 Mb, in tandem, with one copy inverted. This caused rearrangements on Chr II, including the complete loss of the region from II:3,907,835 (first breakpoint linking Chr II and Chr X) to II:3,910,889, with an inversion from II:3,910,889 to II:3,910,983 and a gain of copy of the region until II:3,917,999 (second breakpoint between Chr II and Chr X).
The nT1 balancer was previously described as a stable reciprocal translocation, effectively balancing the right end of Chr IV through unc-17 and the left portion of Chr V through unc-76 (Ferguson and Horvitz 1985; Clark et al. 1988; Rogalski and Riddle 1988; Edgley et al. 2006). We detected a more complex type of rearrangement resembling chromoplexy, with four pairs of breakpoints linking Chr IV and Chr V (Table 3; Fig. 4E), suggesting a shuffling of several segments of those two chromosomes, as well as additional rearrangements at the breakpoints’ junctions.
The balancer mnC1 still harbored an unknown structure, probably because its complexity had made the characterization of its structure challenging with classical molecular biology techniques (FISH, G-band, and aoCGH). mnC1 has been reported as a very effective balancer for the right portion of Chr II from around dpy-10 to around unc-52 (Herman 1978). Overall, we identified five pairs of breakpoints (Table 3), suggesting a complex inversion (three inversions and a deletion) to be responsible for suppressing crossover from II:4,904,692 to II:14,909,258, a region larger than previously described with the gene dpy-10 being localized at II:6,710,149–6,711,866 and unc-52 at II:14,647,325–14,684,456 (Supplemental Fig. S3).
The hDf8 balancer has been described as a deficiency of Chr I and balancing probably from around unc-13 to at least dpy-5, extending to the left end of the chromosome (McKim et al. 1992; Edgley et al. 2006). Clucas et al. (2002) located the left breakpoint of hDf8 between the predicted genes R10A10.1 and rbx-2. By the coverage analysis, it seems that the left end of the Chr I is indeed deleted (Supplemental Fig. S4) up to ∼800 kb; however, our deeper analysis has also revealed 12 pairs of breakpoints. The structure of this rearrangement involves complex inversions and large copy number gains (Supplemental Fig. S5).
The balancer mnDp3 is reported as a free duplication of the right end of the Chr X (Herman et al. 1976, 1979; Herman 1984; Edgley et al. 2006). Analyzing the coverage of the SP123 strain (unc-3(e151) X; mnDp3 (X;f)) showed an increase at the right end of the Chr X, with a coverage ratio of 1.2, no abrupt increase in coverage, and no split reads around the edges, suggesting indeed a free duplication (Supplemental Figs. S6, S7). By further analyzing the duplication, we retrieved 14 pairs of breakpoints (Table 3; Supplemental Fig. S8). Among them, we identified two junctions between Chr I and Chr X, including one confirmed by PCR and Sanger sequencing. This could suggest that the duplicate region is in fact inserted around the right end of Chr I (Supplemental Fig. S8). However, mnDp3 has been previously examined and observed by immunofluorescence as free duplication (Lamelza and Bhalla 2012). In addition, both breakpoints fall into a repetitive and telomeric Chr I region and at the extremity of the free duplicate of Chr X. So, such breakpoints could be an artifact owing to the repetitive character of telomeres and the telomeric-like region that allows structural stability to the duplicate without being physically linked to any chromosome.
srWGS uncovers balancer derivatives
For the strains RM2431 and CB3475, the interpretation of their sequenced genome differs from the expected genotype, suggesting that such strains might present a derivative of traditional balancers.
The rearrangement szT1 balances the left portion of Chr I through unc-13 and nearly all of Chr X from the right end to around dpy-3. It was previously described as a reciprocal translocation (I;X), with rare exceptional progeny carrying one half-translocation as a complex free duplication (Fodor and Deàk 1985; McKim et al. 1988; Howell and Rose 1990; McKim and Rose 1990; Edgley et al. 2006). We analyzed the genome of the strain CB3475 (mcm-4(e1466)/szT1 [lon-2(e678)] I; +/szT1 X). Our initial analysis showed a signature of a translocation (I;X) with breakpoints at I:7,631,470 and X:2,314,605 and an inversion X:2,314,606–2,597,434 (Fig. 5A–C; Table 3).
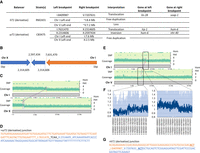
Overview of potential derivative balancers. (A) Breakpoints of hT1 and szT1 derivatives. If the breakpoints are localized in a gene and disrupting it, we report such gene (“gene at left breakpoint” for a left breakpoint mapping in a gene and “gene at right breakpoint” for a right breakpoint mapping in a gene). (B–D) szT1 derivative. (B) Interpretation of the structure of szT1 derivative in CB3475 based on srWGS analyses. (C) In the graph of the upper layer, SNVs detected in the CB3475 genome are denoted by dots. Dots on the higher layer are homozygous SNVs with two copies of the alternate allele. Dots on the middle layer are heterozygous SNVs with only one copy of the alternate allele. Stretches of heterozygous SNVs suggest that the region is balanced by a complex rearrangement. The coverage graph is a scatter plot representing the read coverage of the genome along the chromosome, calculated by window of 1 kb. The graph shows an increase in read coverage of left parts of Chr I and X. The purple lines represent the breakpoints detected. (D) Sequence at the junction of the breakpoint (I;X) obtained by Sanger sequencing. (E,F) hT1 derivative. (E) In the graph of the upper layer, SNVs detected in the RM2431 genome are denoted by dots. Dots on the higher layer are homozygous SNVs with two copies of the alternate allele. Dots on the middle layer are heterozygous SNVs with only one copy of the alternate allele. Stretches of heterozygous SNVs suggest that the region is balanced by a complex rearrangement. The coverage graph is a scatter plot representing the read coverage of the genome along the chromosome, calculated by window of 1 kb. The graph shows an increase in read coverage of left parts of Chr I and X. The purple lines represent the breakpoints detected. (F) Scatter plots focused on the regions where the ratio of coverage changes on Chr I and Chr V. The normalized coverage along the rearrangement calculated by windows of 1 kb. The darker line represents the moving average (trend line, calculated over 20 points). (G) Sequence at the junction of the breakpoint (I;V) obtained by Sanger sequencing.
By coverage analysis (Fig. 5C), we determined that both segments of Chr I (from left end to ∼I:7.6 Mb) and Chr X (from left end to ∼X:2.5 Mb) were duplicated, with a coverage ratio of around 1.2, which suggests that both can occur as free duplications. It is supported also by the stretch of heterozygous variants on the left end of Chr I with unbalanced representation of the alternative allele. Indeed, heterozygosity is usually determined as ∼50% reads supporting the reference allele; ∼50%, the alternative allele. Visual inspection of the alignment for variants on the right ends of Chr I and X (listed in Supplemental Table S7) showed a proportion of reads supporting the reference allele different from the expected 50/50. That reflects the presence of an additional mosaic copy of the region.
After validation of the breakpoints by PCR and Sanger sequencing (Fig. 5D), we then interpreted this structure as both duplicates being attached together, with a 280-kb inversion at the extremity of Chr X (Fig. 5A–C). However, this structure does not corroborate that szT1 balances Chr X from the right end. Indeed, our data showed no stretches of heterozygous SNVs from the right end of Chr X to the translocation breakpoint (Supplemental Table S7). In addition, a derivative of szT1, called szDp1, has been previously reported (Edgley et al. 2006), with one of the half translocations szT1(X) carrying a portion of Chr I that was present as a duplication in addition to two normal copies of Chr I and two normal copies of Chr X. It is then possible that the genome of CB3475 sequenced in this study carries a derivative of szT1.
The balancer hT1 was described as balancing the left portion of Chr V from the left end through dpy-11 (V:6511583–6515240) (McKim et al. 1988; Howell and Rose 1990). By analyzing the strain RM2431 [unc-13(md2415)/hT1 I; +/hT1 V], we showed that the breakpoint is localized ∼0.8 Mb further away (V:7,207,631) in the soap-1 gene. In addition, hT1 was described as balancing the left portion of Chr I from the left end through let-80, a gene with no known genomic position. Such lack of information could restrict the usage of this balancer. Now, we have completed the description by mapping the second breakpoint of the reciprocal translocation in the lin-28 gene at the position I:8,409,987.
In our hands, the strain selected for sequencing (RM2431) also shows on the left end of Chr I an increase in read coverage until around I:8,735,001 (coverage ratio of 1.2). Ratio of coverage was calculated by dividing the read coverage by window of 1 kb, divided by the average of coverage of the entire genome, and normalized against the ratio in control (N2 strain; see Methods). Usually, for regions with no variations of copy number, the ratio equals one. For deletion, the ratio equals zero (homozygous) or 0.5 (heterozygous; loss of one copy). And for tandem duplications, the ratio is 1.5 (heterozygous) or two (homozygous). A ratio of 1.2 is only observed in case of free duplications and translocated duplications, which is, in this case, probably suggestive of a free duplication (Fig. 5E,F). It also showed a decrease in coverage for the left end of Chr V (Fig. 5E,F). With the experimental validation of the inter-chromosomal breakpoint junction (Fig. 5G), our data show that the strain RM2431 is not a clean translocation but presents a free duplication. The rearrangement hDp133, a derivative of hT1, has been previously reported to produce rare exceptional progeny that could carry one half-translocation as a complex free duplication (Edgley et al. 2006). Altogether, this suggests that the genome of RM2431 sequenced in our study carries a derivative of hT1.
Unsuspected complex variation of C. elegans balancer genomes beyond balancing variants
Besides balanced regions previously partially characterized, we explored the entire genomes of all the strains (for complete list and phenotypes, see Supplemental Information). Overall, we found an average of six additional pairs of breakpoints per strain (range 1–17), suggesting the existence of large variation in C. elegans genomes owing to SVs and CGRs being understudied. We ignored breakpoints related to insertion events (e.g., mobile element insertions).
Earlier in this paper, we described eight SVs and CGRs in the five CRISPR-Cas9 strains (Fig. 2; Table 1). In addition, beyond the regions balanced by the traditional balancers they carry, we explored the entire genomes of 20 strains (18 with one balancer, two with two balancers) (Supplemental Table S1). We report tandem duplications (direct and inverted), deletions, inversions, and CGRs of all sizes and on all chromosomes. Table 4 displays a subset of SVs and CGRs detected in traditional balancer strains, further validated by PCR and Sanger sequencing (PCR gels are available in Supplemental Information, and Sanger sequences in Supplemental Table S5). The complete list of the 96 additional rearrangements detected by RUFUS (Ostrander et al. 2018) is available in Supplemental Table S5 (32 homozygous deletions, 10 heterozygous deletions, four homozygous tandem duplications, 22 heterozygous tandem duplications, six inverted tandem duplications, three inversions, and 19 CGRs).
Subset of SVs and CGRs detected in C. elegans genomes and validated by PCR and Sanger sequencing
Of note, we have only reported SVs that were not present in our N2 reference strain and CB4856 strain. Still, some SVs were found in several strains such as the tandem duplication I:14573853–14580325 detected in KR2839 and VC528, or the homozygous deletion II:10440527–10440558 present in the genomes of the strains SP123, KR1233 and VC528.
Discussion
In this study, we have generated the first molecular map of C. elegans balancers with genomic locations of their breakpoints and characterized for the first time the structure of a subset of these balancers. We also elucidated higher complexity for other balancers, which is an unsuspected finding based on previous analysis with low-resolution genetic techniques. Accumulation of spontaneous short mutations has been previously observed in balanced regions (Rosenbluth et al. 1983), and here, we generated the first atlas of molecularly dissected and experimentally confirmed genomic variations based on SVs and CGRs, still understudied in model organisms.
By applying our sequence analysis protocol published by Maroilley et al. (2021), we successfully used the short-read WGS approach to map breakpoints for all types of SVs (inversions, deletions, tandem duplications, inverted tandem duplications, translocated duplications, reciprocal translocation, insertions) and even complex rearrangements (chromoanagenesis-like). In addition, we were able to detect such rearrangements as a free duplication, which would translate in human studies as a mosaic complex rearrangement. We then showed that short reads with tailored bioinformatic downstream analyses can contribute more to sequence analysis than what is appreciated in most previous studies, which tend to focus srWGS applications to SNVs, short insertions, and deletions (Maroilley and Tarailo-Graovac 2019). With such an approach, whole-genome sequencing based on short reads constitutes an interesting way to explore, in a high-throughput manner, the entire spectrum of genomic variability in C. elegans and other model organisms.
In some instances, the analyses of the genomes of strains selected for specific balancers, such as szT1 but also hT3 and mnDp1 (for discussion, see Supplemental Information), showed a different structure than previously described and, more importantly, a discrepancy regarding the regions balanced. It has been well described that some balancers are unstable (Edgley et al. 2006). Our work further supports this and advocates that balancer strains be either sequenced by the laboratories or at least PCR-tested (facilitated by primers designed in our study) to validate the expected strains and the balanced region when using the balancer strains.
Despite the successful application of the srWGS in our work to detect a diverse spectrum of SNVs as well as SVs and CGRs, we acknowledge that identification of certain types of complex variants is quite challenging using short reads, considering the known limitations of the technology. First, one read cannot overlap the entire rearrangement. Detection and interpretation of breakpoints rely then on the presence of split reads at breakpoint junctions and the ability of bioinformatics processes to recognize split reads, reliant on tailored algorithms but also the coverage and the quality of sequencing data (Supplemental Fig. S9). Second, the alignment of the reads is influenced by the aligner used and the genome reference that might not be ideal for every sample. For instance, the C. elegans reference genome was based on N2 strain, which may only partially represent the variability of other strains and create challenges during read alignment. Third, the resolution of the structure of complex rearrangements with short reads is subjected to one's interpretation of the variation of coverage and split reads at breakpoint junctions, which may be challenging. Finally, there is a possibility that some breakpoints located in the highly repetitive regions may be missed (Supplemental Fig. S10; Supplemental Table S8). Therefore, in more complex genomes, such as those of wild isolates (Lee et al. 2021) or even those of some challenging rearrangements in our study (e.g., szT1), it may be useful to apply long reads to resolve the complex variants that are within highly variable and repetitive regions.
We detected several rearrangement breakpoints mapping in genes, including some essential genes (e.g., soap-1), and even in potentially essential, previously unexplored long intervening noncoding RNAs (linc-95). It is challenging to interpret the effect of such disruption on the expression of genes and resulting phenotypes. Indeed, although various tools that interpret the effects of single-nucleotide variants and indels in coding regions have been developed, only a few tools exist that predict in silico the effect of SVs. It is also important to consider the impact of breakpoints in noncoding regions, as they could disrupt genes by falling in introns or disrupt regulatory mechanisms by falling into a topological associated domain, thereby disrupting the link between a gene and an enhancer.
We have also reported additional SVs and CGRs identified with srWGS in the strains included in this study. Our findings revealed that most of these balancing strains are carrying various types of large events, some of them apparently owing to mutagenesis but others likely as part of the spontaneous genetic variation in C. elegans. Like Newman et al. (2015) have reported, we found translocated duplications and free duplications to be quite rare compared with tandem duplications. In addition, we observed that CGRs, resulting from several overlapping SVs, are more frequent in these balancer genomes than previously appreciated. It is tempting for investigators to assume that balancer strains behave essentially as wild type in all aspects except their balancing behavior. However, by showing that balancer strains also carry various SVs, beyond the expected rearrangement that results in balancing, our study indicates that some SVs might cause additional phenotypes or interact with the balanced mutations. Then, variations of genetic background in balancer strains should be considered, and balanced mutations should be analyzed in more than one background. Beyond the mutagen-induced balancers, our analysis of CRISPR-Cas9-induced balancers revealed some interesting findings. First, some inversions induced by CRISPR-Cas9 showed a signature different from the typical inversion signature. The nature and origin of this signature will need further investigation. Furthermore, we identified additional SVs that were not previously known. Some were shared by all five strains and thus were probably inherited from the background genome used to create those balancers. But other SVs were only present in one or two of the CRISPR-Cas9 balancer genomes. It would be interesting to investigate whether either of these variants occurred naturally or whether they could be off-site targets of the RNA guide.
Overall, this atlas of molecular structure of genetic balancers is very important for the C. elegans community because balancers are widely used genetic tools. Indeed, by describing their structures, we gain a better understanding of how the balancers are working. It also facilitates balancer usage by mapping exactly their breakpoints for an easy confirmation of their presence in strains and better awareness of the capabilities of each balancer (specifically, which exact region can be balanced by each rearrangement) (Fig. 1A; Supplemental Table S6). Finally, the description of the exact position of breakpoints disrupting genes in rearrangements reported to be recessively lethal could be very important for C. elegans genetic studies of currently understudied genes. On a larger scale, this study clearly shows the power of srWGS in resolving genomic complexity and paves the way to tailored bioinformatic workflows for both model organism and human studies. Such workflows would better take advantage of srWGS data sets, reduce cost, and reduce the multiplication of techniques necessary to explore the whole complexity of the genomes.
Methods
CGC data
For each rearrangement as balancer, we used the CGC database to retrieve the list of strains carrying the balancer of interest. We used the CGC query system “search strain” and filtered by phenotype. We manually reviewed the final lists to avoid picking up genotypes that would contain a marker/gene with an identification similar enough to the balancer to be picked up by the query. We then sent those lists of strains to the CGC to obtain the number of orders of each strain over time. We calculate the number of orders by balancer by adding up order for each stain carrying each balancer.
Worm maintenance
All worm strains in this study were obtained from the CGC. All strains were maintained at 16°C and kept on standard NGM plates streaked with OP50.
Sequencing
We sequenced the genomes of 25 strains, for a total of 30 genomes: genomes from homozygous individuals of the strains BC1217, CB3475, CB4281, CZ1072, DP246, FX30133, FX30144, FX30235, FX30237, FX30257, GE2722, KR1233, KR1876, KR2839, MT690, RM2431, RW6002, SP123, SP127, SP309, SP423, SP998, VC1771, VC471, VC528; genomes from heterozygous worms of the strains DP246, SS746, VC1771, VC471, and VC528. We used the sequencing data of N2 and CB4856 strains published by Maroilley et al. (2021). The strains FX30133, FX30144, FX30235, FX30237, and FX30257 were engineered by CRISPR-Cas9 (Dejima et al. 2018). The strain BC1217 was used to explore sDf22 and nT1. The strain GE2722 was used to retrieve qC1 breakpoints published by Edgley et al. (2021). The strain VC471 carried hT2, already described by Flibotte et al. (2021). The strain KR2839 was used to describe hDf15 and hIn1. The strains KR1876 and SP423 were originally selected to study hT3 and mnDp1, respectively, but the sequencing data did not allow the resolution of those two balancers (see Supplemental Information). All genomes sequenced in this study were used to build the catalog of structural variations.
DNA extraction and library preparation
Genomic DNA was collected from ∼100 mg of worm tissue using the Qiagen blood and tissue kit (13323) following the manufacturer's recommendations. DNA was eluted with 10 mM Tris–HCl (pH 8.0). Samples were quality-checked to ensure a minimum quantity of 1500 ng and a 260/280 ratio of 1.8 before submitting for sequencing. Paired-end short-read WGS were obtained for all strains with PCR-free library preparation protocol and NovaSeq 6000 Illumina sequencing technology.
Primary data analyses
We checked the quality of the FASTQ files using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). The reads were on average 151 bp long. We trimmed the reads and removed the adapters using Trimmomatic v0.36 (Bolger et al. 2014). For each sample, we aligned between 16 million and 34 million reads using the BWA-MEM v0.7.17 (Li 2013) algorithm to the C. elegans reference genome WS265. It resulted in a 31× read coverage per strain on average (Supplemental Table S3). We then sorted the reads according to their coordinates with “samtools sort” (SAMtools v1.5, Li et al. 2009).
For all genomes, we performed an analysis of coverage by calculating the read depth by window of 1 kb. We normalized the average of read coverage in each 1-kb window by dividing it by the average of read depth across the entire genome and by the read depth of the same window in the reference strain (N2). If N2 ratio was equal to zero in a specific window (no reads in the N2 strain sequenced in our lab), the ratio reported for further analyses was then only normalized by the average coverage of the genome. The ratio obtained was then plotted according to its genomic position on a graph using the R package “karyoploteR” (Gel and Serra 2017; https://bernatgel.github.io/karyoploter_tutorial/; https://github.com/bernatgel/karyoploteR). The Circos plots on Figure 4 were realized using Circos (Krzywinski et al. 2009; http://circos.ca/).
Breakpoints were identified as in the work of Maroilley et al. (2021). Briefly, we used the k-mer approach implemented in RUFUS (https://github.com/jandrewrfarrell/RUFUS) to detect de novo breakpoints, using N2 and Hawaiian strains as “parents.” Additional breakpoints were detected using GRIDSS (https://github.com/PapenfussLab/gridss) when a CGR was suspected. All breakpoints were assessed visually using IGV, which allows the visualization of the reads aligned along the reference genome (see Supplemental Information; Robinson et al. 2011). We performed a read inspection for each breakpoint. We extracted reads around each breakpoint using “samtools view” (SAMtools v1.5) and realigned them using BLAT (UCSC, portable version). The presence of several split reads supporting the breakpoints was decisive whether or not to send the breakpoint for experimental validation. We extracted the sequence of one split read for each breakpoint to design primers for PCR and Sanger sequencing. In the absence of split read at the breakpoint, we looked for discordant pair of reads.
Independent analyses
For the balancers nT1 and mIn1, an independent analysis was performed as in the work of Flibotte et al. (2021) and Edgley et al. (2021). In both cases, the same breakpoints were retrieved.
Experimental validation
We confirmed breakpoints of SVs and complex rearrangements by PCR and Sanger Sequencing. Primers were design with PrimerQuest IDT tool (https://www.idtdna.com/PrimerQuest). All primers and sequences are available in Supplemental Tables S4 and S5.
Data access
The data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA838950. The codes for Figures 1A, 2A, 4C, 5C, and 5E are available in the Supplemental Material and at GitHub (https://github.com/MTG-Lab/C-elegans-balancers).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by funding from Alberta Children's Hospital Research Institute Foundation, Canadian Institutes of Health Research, CIHR-Project grant number PJT-156068, Eyes High Postdoctoral Fellowship, Natural Sciences and Engineering Research Council of Canada (NSERC) Postdoctoral Fellowship, and NSERC undergraduate research awards. The funding sources did not have a role in the design of the study, collection and interpretation of data, or writing of the manuscript. The research was enabled by using the Compute Canada (www.computecanada.ca) computing resources and in part by support provided by the Research Computing Services group at the University of Calgary. All strains were provided by the CGC, which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD01440). We want to thank Aric Daul, who helped with extracting information from the CGC regarding strain use and shipments; Jasmine Shi, Matthew Oldach, Catherine Diao, and Susan J. Stasiuk for their support in initial stages of this project; and Dr. Mains and the members of Hansen and Tarailo-Graovac laboratories for suggestions and support throughout this project.
Author contributions: Research design and conceptualization were by T.M., S.F., M.E., D.M., and M.T-G.; data analysis and interpretation were by T.M., S.F., A.G., A.R.C., F.C., D.M., and M.T-G.; molecular analysis and confirmation of the variants were by T.M., F.J., V.R.A.B., X.L., and L.O.; and original draft preparation was by T.M., S.F., M.E., D.M., and M.T-G. All authors have read and approved the submitted manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276988.122.
- Received June 1, 2022.
- Accepted November 29, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








