
Dynamic regulation of gonadal transposon control across the lifespan of the naturally short-lived African turquoise killifish
- Bryan B. Teefy1,
- Ari Adler1,
- Alan Xu1,2,
- Katelyn Hsu1,2,
- Param Priya Singh3,7,8 and
- Bérénice A. Benayoun1,2,4,5,6
- 1Leonard Davis School of Gerontology, University of Southern California, Los Angeles, California 90089, USA;
- 2Molecular and Computational Biology Department, USC Dornsife College of Letters, Arts and Sciences, Los Angeles, California 90089, USA;
- 3Department of Genetics, Stanford University, Stanford, California 94305, USA;
- 4Biochemistry and Molecular Medicine Department, USC Keck School of Medicine, Los Angeles, California 90089, USA;
- 5USC Norris Comprehensive Cancer Center, Epigenetics and Gene Regulation, Los Angeles, California 90089, USA;
- 6USC Stem Cell Initiative, Los Angeles, California 90089, USA
Abstract
Although germline cells are considered to be functionally “immortal,” both the germline and supporting somatic cells in the gonad within an organism experience aging. With increased age at parenthood, the age-related decline in reproductive success has become an important biological issue for an aging population. However, molecular mechanisms underlying reproductive aging across sexes in vertebrates remain poorly understood. To decipher molecular drivers of vertebrate gonadal aging across sexes, we perform longitudinal characterization of the gonadal transcriptome throughout the lifespan in the naturally short-lived African turquoise killifish (Nothobranchius furzeri). By combining mRNA-seq and small RNA-seq from 26 individuals, we characterize the aging gonads of young-adult, middle-aged, and old female and male fish. We analyze changes in transcriptional patterns of genes, transposable elements (TEs), and piRNAs. We find that testes seem to undergo only marginal changes during aging. In contrast, in middle-aged ovaries, the time point associated with peak female fertility in this strain, PIWI pathway components are transiently down-regulated, TE transcription is elevated, and piRNA levels generally decrease, suggesting that egg quality may already be declining at middle-age. Furthermore, we show that piRNA ping-pong biogenesis declines steadily with age in ovaries, whereas it is maintained in aging testes. To our knowledge, this data set represents the most comprehensive transcriptomic data set for vertebrate gonadal aging. This resource also highlights important pathways that are regulated during reproductive aging in either ovaries or testes, which could ultimately be leveraged to help restore aspects of youthful reproductive function.
In much of the industrialized world, increased parental age has led to widespread fertility-related challenges associated with later-life childbearing (Kenny et al. 2013; Bertoldo et al. 2020). In humans, oocyte quality rapidly declines starting at ∼30 yr of age, whereas menopause, the irreversible loss of female fertility, occurs at ∼50 yr (Alberts et al. 2013; Finch 2014). In contrast, although male fertility gradually declines with age, the complete loss of testicular function is not a feature of male reproductive aging (Gunes et al. 2016). Mammals, as well as birds, are exceptional among vertebrates as they produce a finite supply of oocytes, ensuring that any individual that lives long enough should eventually deplete its oocytes, although the starting supply of oocytes greatly outnumbers the final number that will be ovulated (Mira 1998). However, there are many other mechanisms that influence reproductive decline, such as neuroendocrine aging, that occur in both sexes and are widely conserved among vertebrates (Perheentupa and Huhtaniemi 2009). Thus, leveraging a tractable vertebrate model organism to understand the molecular mechanisms underlying reproductive aging will provide important insights into lifelong regulation of reproductive function.
To study reproductive aging, we examined the impact of aging on testes and ovaries in the African turquoise killifish Nothobranchius furzeri, the shortest-lived vertebrate that can be bred in captivity, with a lifespan of ∼4–6 mo (Hu and Brunet 2018). The turquoise killifish evolved this short lifespan in response to their unique life cycle, which revolves around the formation of ephemeral ponds of water (Kim et al. 2016). Depending on the specific species, killifish become sexually mature as early as 2 wk after hatching, although most killifish become sexually mature ∼4–5 wk of age, the fastest known time to sexual maturation of any vertebrate (Naumann and Englert 2018; Vrtílek et al. 2018b). Killifish are asynchronous breeders, and upon sexual maturity, females continuously undergo oogenesis and lay eggs, typically on a daily basis (Terzibasi Tozzini and Cellerino 2020). Although male reproductive senescence may be negligible, females do experience an age-related decline in fecundity, although the molecular mechanisms contributing to reproductive aging in either sex have not been elucidated (Vrtílek et al. 2018a; Žák and Reichard 2021). We and others have helped establish a key functional genomics toolkit for this species, making it a uniquely tractable experimental model to decipher molecular mechanisms driving aspects of aging (Valenzano et al. 2011, 2015; Reichwald et al. 2015; Harel et al. 2016; Willemsen et al. 2020).
An emerging facet of vertebrate aging is the reactivation of transposable elements (TEs) with aging in somatic tissues (De Cecco et al. 2013; Chen et al. 2016; Simon et al. 2019). However, whether TEs also activate within the aging gonad, which is uniquely protected from spurious TE mobilization by the PIWI-piRNA pathway, has been largely unexplored in vertebrates. The PIWI pathway is an Argonaute-based small RNA pathway that maintains germline genomic integrity by antagonizing the mobilization of TEs through complementary RNA base-pairing and TE RNA target degradation during gametogenesis and embryogenesis. PIWI family proteins bind 24- to 35-nucleotide (nt) PIWI-interacting RNAs (piRNAs), which are complementary to or derived from TEs. piRNAs are expressed primarily in the gonad and are larger than miRNAs (Mani and Juliano 2013; Czech and Hannon 2016). Once bound to an RNA target, PIWI proteins cleave their target precisely 10 nt from the 5′ end of the complementary piRNA (Mani and Juliano 2013; Czech and Hannon 2016). To date, most studies examining the impact of aging on the PIWI pathway have focused on invertebrate models, for example, Drosophila melanogaster (Sousa-Victor et al. 2017; Yamashiro and Siomi 2018; Lin et al. 2020). For instance, Piwi expression is required to suppress TE expression, reduce DNA damage, and maintain intestinal stem cell lineages in the aging fly midgut (Sousa-Victor et al. 2017). In the fly ovary, the age-related decline in Piwi expression in the cells of the germline stem cell (GSC) niche results in a TE derepression and a loss of GSCs (Lin et al. 2020). In contrast to the GSC niche, the expression of PIWI pathway components in whole Drosophila egg chambers increases with age, although this does not impact global TE expression (Erwin and Blumenstiel 2019). Whether TEs are derepressed with age in the vertebrate gonad and whether TE expression is modulated by age-related PIWI dynamics remain unknown. Thus, it will be important to determine whether PIWI regulation (and concomitant TE control) in the aging gonad may influence reproductive aging.
In this study, we leverage mRNA and small RNA sequencing to capture the transcriptional trajectory of gonadal aging in a naturally short-lived vertebrate. To do so, we characterize the transcriptome of African turquoise killifish ovaries and testes in young (5-wk), middle-age (10-wk), and old (15-wk) animals in the GRZ laboratory strain. In particular, we focus on the interplay of TEs and piRNAs and assess the effect of age on the transcriptional status of these RNAs.
Results
Lifespan, reproductive, and transcriptomic landscapes of the aging gonad in the African turquoise killifish
To understand how gonads are affected during aging in a vertebrate, we decided to profile ovarian and testicular transcriptomes throughout aging in African turquoise killifish from the GRZ strain (Fig. 1A). In our housing conditions (see Methods), 15 wk corresponds to ∼90% survival for both sexes (Fig. 1B; Supplemental Table S1A), although we observed that GRZ females lived significantly longer than males (P = 0.002, log-rank test; see Methods) (Fig. 1B). To determine the effect of aging on vertebrate gonadal transcriptomes, we collected ovaries and testes from N = 5 GRZ female and male African turquoise killifish at three time points after hatching: young adulthood (5 wk; onset of fertility), middle-age (10 wk), and old age (15 wk; before any substantial population survival decrease) (Fig. 1A). These time points are consistent with accepted guidelines in aging research (see Methods). In the GRZ strain, fecundity peaks ∼10–12 wk of age (Žák and Reichard 2021), roughly corresponding to our middle-age time point. Only samples with intact RNA were processed further, yielding 14 female and 12 male samples (see Methods) (Supplemental Fig. S1A). We performed (1) mRNA transcriptome characterization (i.e., mRNA-seq using poly(A) selection), analyzing both genes and TEs, and (2) small-RNA transcriptome characterization (i.e., small RNA-seq), focusing on piRNAs (Fig. 1A; Supplemental Fig. S1A).
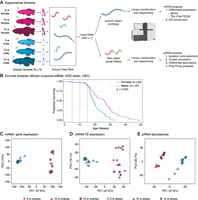
A study of gonadal aging in the naturally short-lived African turquoise killifish. (A) Experimental scheme. Killifish gonads (N = 5 per group) were harvested, and only high-quality RNA samples were further processed (see Supplemental Fig. S1A). (B) Lifespan curve for female and male GRZ strain killifish at our facility. GRZ females lived significantly longer than males (P = 0.002; log-rank test). Median lifespan: female, 26.6 wk; male, 19.4 wk. Vertical dotted lines indicate ages in this study (5, 10, 15 wk). (C–E) Principal component analyses (PCAs) for mRNA gene expression (C), TE expression (D), and TE-mapping piRNA abundance (E; see Methods).
To visualize the similarity of aging ovarian and testicular transcriptomes, we used principal component analysis (PCA) (Fig. 1C–E). PCA analysis of genic mRNA, TE mRNA, and TE-targeting piRNAs revealed that the main source of variation (PC1) corresponded to gonadal identity (i.e., ovary vs. testis), although age also weakly separated samples for mRNA and piRNA (but not TE) transcription on PC2 (Fig. 1C–E). Despite genomic TE content being fixed in the species, we observed clear sex-dimorphism in the gonadal transcriptional landscape of TEs and associated piRNAs. These observations are consistent with sex-dimorphic gonadal transcriptional programs at the level of mRNAs, TEs, and small RNAs and with a recent study in Drosophila gonads (Chen et al. 2021). Next, we analyzed the ovarian and testicular transcriptomes separately to uncover sex-specific transcriptional aging patterns (Supplemental Fig. S1B–S; see Supplemental Methods). Middle-aged ovarian expression data clustered distinctly for all analyzed RNA species by PCA (Supplemental Fig. S1B–D), which may reflect a unique transcriptional program associated with female fertility at middle-age. Female piRNA expression data segregated most strongly by age, suggesting that the small RNA program in the ovary is most sensitive to aging (Supplemental Fig. S1D). In contrast, the male transcriptomic data did not separate strongly by age (Supplemental Fig. S1E–G). Consistently, we noted stronger age clustering of ovarian versus testicular samples for gene, TE, and piRNA expression using hierarchical clustering with bootstrap resampling by “pvclust” (Supplemental Fig. S1H–M; see Supplemental Methods; Suzuki and Shimodaira 2006). Finally, we also quantified the proportion of gene, TE, and piRNA expression variance explained by age in ovaries and testes using “variancePartition” (Supplemental Fig. S1N–S; see Supplemental Methods; Hoffman and Schadt 2016). The median percentage of variance explained by age was systematically higher in ovaries than testes (genes: ∼25% vs. ∼6%; TEs: ∼15% vs. ∼6%; piRNAs: ∼49% vs. ∼2%) (Supplemental Fig. S1N–S). Together, our observations suggest that testicular identity and function are more stable with aging relative to the ovary. Our results are reminiscent of a microarray analysis of mouse gonadal aging that showed larger changes in ovarian versus testicular aging transcriptomes (Sharov et al. 2008), and of observations of negligible male reproductive aging in the killifish (Vrtílek et al. 2018a; Žák and Reichard 2021).
Aging differentially impacts gene expression in turquoise killifish ovaries and testes
Because aging is often nonlinear, at least in somatic tissues (Baumgart et al. 2016; Rosenberg et al. 2021), we used the DESeq2 framework with a likelihood ratio testing (LRT) approach to identify patterns of differential gene expression with gonadal aging (Love et al. 2014). Because LRT can be overly lenient (https://hbctraining.github.io/DGE_workshop/lessons/08_DGE_LRT.html), we used a stringent false-discovery rate (FDR) threshold of FDR < 10−6. Significant transcripts were then assigned to patterns, using unsupervised hierarchical clustering to identify significant shared expression patterns among differentially expressed genes (see Methods). This unbiased approach detected four major differential patterns corresponding to transcripts whose expression is (1) transiently down at middle-age (a), (2) transiently up at middle-age (b), (3) monotonously down with age (c), or (4) monotonously up with age (d) (Figs. 2A,B, 3A; Supplemental Figs. S2A, S3A; Supplemental Table S2A,B,D). For convenience, we label these patterns hereafter as a, b, c, and d whenever they arise from unsupervised clustering. The majority of differentially expressed genes showed transient changes at middle-age in both ovaries and testes (respectively, 1931 out of 2041 and 578 out of 603 differentially expressed genes), rather than linear trends. This suggests that most gonadal transcriptional variation occurs at middle-age when fertility, at least in females, is at its peak. This observation suggests that gonadal biology may already be substantially impacted in middle-aged healthy animals.
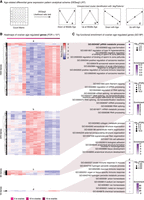
Differential gene expression analysis and GO biological process functional enrichment analysis with ovarian aging. (A) Scheme used for differential gene expression (DGE) analysis. DGE performed with DESeq2 likelihood ratio testing (LRT) revealed four groups of differentially expressed genes corresponding to expression down at middle-age (a), up at middle-age (b), down with age (c), and up with age (d). (B) Heatmaps of gene expression for genes significantly regulated with age in aging killifish ovaries by DESeq2 LRT (FDR < 10−6). (C) Functional enrichment analysis for each gene cluster in B, showing the top 10 significant GO “biological process” terms (FDR < 5%) (for complete list, see Supplemental Table S3A). The most significant term down-regulated in middle-aged ovaries is “piRNA metabolic process” (bolded). (FDR) False-discovery rate, (enrichment) fold enrichment over background.
Next, we asked which biological functions were associated with each of these gene expression patterns with gonadal aging. For this purpose, we performed Gene Ontology (GO) enrichment analysis for each pattern (Figs. 2C, 3B; Supplemental Figs. S2B, S3B; Supplemental Table S3A–F; Falcon and Gentleman 2007). First, we examined enrichment for terms in the “biological process” GO category (Figs. 2C, 3B; Supplemental Table S3A,D). Genes down-regulated in middle-aged ovaries were most strongly enriched for the “piRNA metabolic process” (i.e., pattern a; GO:0034587) (Fig. 2C; Supplemental Table S3A). Indeed, genes transcriptionally down-regulated in middle-aged ovaries include the genes encoding both effector PIWI proteins (i.e., homologs to human PIWIL1 and PIWIL2), consistent with overall decreased TE processing capacity. To note, as seen in Supplemental Table S2C, genes belonging to the “piRNA metabolic process” GO terms are, as expected, robustly expressed in killifish gonads throughout life.
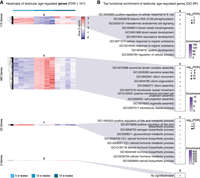
Differential gene expression and GO biological process functional enrichment analysis with testicular aging. (A) Heatmaps of gene expression for genes significantly regulated with age in aging killifish testes by DESeq2 LRT (FDR < 10−6). Groups as defined in Figure 2A. (B) Functional enrichment analysis for each cluster shown in A, showing the top 10 significant GO “biological process” terms (FDR < 5%) (for complete list, see Supplemental Table S3D). Genes up-regulated in middle-aged testes are enriched for spermatogenesis-related terms. Cluster d had too few genes for enrichment analysis. (FDR) False-discovery rate, (enrichment) fold enrichment over background.
In zebrafish, the gene encoding Piwil1, the primary catalytic protein of the PIWI pathway, is expressed most highly in developing GSCs before expression decreases with oocyte maturation (Liu et al. 2022). Thus, down-regulation of PIWI-related genes at mid-age may be related to differences in mature oocytes content in aging killifish ovaries. To test this hypothesis, we (1) performed a small-scale histological analysis of oocyte diameter in aging ovaries (because oocyte size is directly linked to maturation) (Supplemental Fig. S4A,B; see Supplemental Methods; Api et al. 2018), and (2) used a bulk transcriptome deconvolution approach using a zebrafish ovarian single-cell RNA-seq reference data set (Supplemental Fig. S4C–G; see Supplemental Methods). First, histological analysis revealed that the diameter distribution of oocytes in aging ovaries did not vary significantly with age (Supplemental Fig. S4B). Second, deconvolution analysis did not detect substantial changes in the proportion of immature germ cells in young versus middle-aged ovaries (Supplemental Fig. S4F,G). Thus, it is unlikely that observed transcriptomic changes in the middle-aged turquoise killifish ovary reflect substantial changes in the underlying ovarian cell composition. Alternatively, our observations may suggest a relaxation of piRNA-mediated TE control in ovaries at middle-aged (see Discussion). Other terms enriched for genes down-regulated in middle-aged ovaries were largely related to egg maturation and egg-specific processes (e.g., zona pellucida and regulation of acrosome reaction) (Fig. 2C), consistent with the notion that ovarian function (and oocyte quality) has already started to decline at middle-age in the African turquoise killifish.
Genes up-regulated in middle-aged ovaries were enriched for terms related to RNA metabolism, consistent with the conserved importance of maternally deposited RNAs in oocytes (i.e., pattern b) (Fig. 2C). Genes linearly down-regulated with age in the ovaries were enriched for terms relating to extracellular matrix (i.e., pattern c) (Fig. 2C). Finally, genes up-regulated with age in the ovaries included those involved in immune responses (i.e., pattern d) (Fig. 2C), which may relate to overall age-related increased inflammation observed in somatic tissues (Benayoun et al. 2019). Increased expression of inflammatory genes has also been observed in aging mouse ovaries (Sharov et al. 2008). We also performed similar analyses using terms from the “cellular component” (Supplemental Fig. S2B; Supplemental Table S3B) and “molecular function” (Supplemental Table S3C) GO categories.
In aging testes, we observed a similar trend, with most differential genes being regulated at middle-age (i.e., patterns a and b), and few genes linearly regulated with aging (i.e., patterns c and d) (Fig. 3A; Supplemental Table S3D). Genes transiently down-regulated at middle-age were enriched for terms relating to vascular development (pattern a) (Fig. 3B). This observation suggests that the vasculature of the testis may be actively remodeled, consistent with known unique vascular requirements for testicular function (Sargent et al. 2015). Genes transiently up-regulated at middle-age included those involved in spermatogenesis, for example, “axoneme assembly,” consistent with peak spermatogenesis occurring at this stage (pattern b) (Fig. 3B). Lastly, genes down-regulated linearly with age were heavily enriched for steroid biosynthesis–related terms, suggestive of a steady decline in steroid biosynthesis (pattern c) (Fig. 3B). The down-regulation of steroid biosynthesis genes is consistent with a recent mass spectrometry study that detected significantly decreased levels of sex-steroids in aging turquoise killifish testes (Dabrowski et al. 2020). “Cellular component” analysis again corroborated the GO “biological process” analysis, especially with regards to sperm-related terms being up-regulated at middle-age (Supplemental Fig. S3B; Supplemental Table S3E). Terms down-regulated at middle-age included those associated with heterochromatin and extracellular vesicles, whereas terms down-regulated linearly with age are enriched for mitochondrial- and collagen-related terms (Supplemental Fig. S3B; Supplemental Table S3E).
Transposon expression is age- and sex-dependent in the turquoise killifish gonad
Next, we asked whether TE transcription was impacted during gonadal aging. Indeed, accumulating evidence has shown that TE expression tends to increase with age in somatic tissues across species (De Cecco et al. 2013; Chen et al. 2016; Benayoun et al. 2019; Simon et al. 2019; Bravo et al. 2020). Although TE control is especially important in the germline to maintain genome stability throughout generations, whether TE expression escapes control in aging gonadal tissues has not yet been investigated. A recent genomic analysis of several species of African killifishes suggests that the shorter-lived killifish species have accumulated TEs in their genomes, suggesting that short-lived/annual killifish species, such as the African turquoise killifish, may not efficiently repress germline TE activity (Cui et al. 2020).
To determine whether global TE expression is remodeled in aging killifish gonads, we first analyzed the proportion of mapped reads in our mRNA-seq data set derived from TE sequences (Fig. 4A,B). TE-derived sequences represented ∼7%–11% of reads in turquoise killifish ovaries and testes (Fig. 4B). Global levels of TE-derived mRNA sequences were stable between young and middle-aged ovaries but significantly dipped in old ovaries, whereas they tended to increase with age in testes (Fig. 4B). Such a pattern may reflect gametogenesis, wherein the epigenome is repatterned, resulting in TE reactivation (Ben Maamar et al. 2021), despite the expected antagonizing effects of the PIWI pathway on TE expression in the germline.
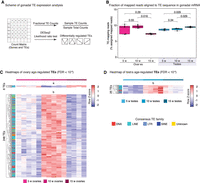
TE expression dynamics in the aging turquoise killifish gonad. (A) Scheme for TE transcriptional analysis in aging gonads. TE DGE was conducted similar to genic DGE analysis (see Methods) and labeled with nomenclature from Figure 2A. (B) Proportion of reads assigned to TEs from each library. Significance in nonparametric Wilcoxon rank-sum test. (C) Heatmaps of mRNA expression for TEs significantly regulated with age in aging killifish ovaries by DESeq2 LRT (FDR < 10−6). TE family color-coded for each row. (D) Heatmaps of mRNA expression for TEs significantly regulated with age in aging killifish testes by DESeq2 LRT (FDR < 10−6).
Next, we asked whether TE transcription was changed in aging gonads at the subfamily level, and identified differentially expressed TE transcripts (FDR < 10−6, as above) (Fig. 4A,C,D; Supplemental Fig. S5A,B; Supplemental Table S4A,B). Similar to before, we observed that most significant changes in TE expression occurred at middle-age (i.e., patterns a and b) (Fig. 4C,D; Supplemental Fig. S5A,B). In the ovary, six TEs were down-regulated only at middle-age (pattern a), and 248 TEs were specifically up-regulated at middle-age (pattern b) (Fig. 4C; Supplemental Fig. S5A). Age-regulated TEs belonged to families broadly representative of genomic TE content, with a predominance of LINE sequences, suggestive of general relaxation of TE control (Fig. 4C,D). This time point coincides with the functional enrichment for “piRNA metabolic process” for genes down-regulated in middle-aged ovaries (Fig. 2C). Thus, the transcriptional up-regulation of TEs in the middle-aged ovary may reflect the reactivation of TE species in response to a transient decline in PIWI pathway activity. Such an increase, even in mature oocytes, is expected to be deleterious (Malki et al. 2014). No significant TEs were linearly down-regulated or up-regulated with age in the ovaries (Fig. 4C; Supplemental Fig. S5A). In aging testes, only 25 TEs were significant; all up-regulated at middle-age (i.e., pattern b) (Fig. 4D; Supplemental Fig. S5B). No significant TEs were down-regulated at middle-age or linearly with age in the testes (Fig. 4D; Supplemental Fig. S5B).
piRNAs and the PIWI pathway activity are regulated throughout life in turquoise killifish gonads
Because expression of PIWI pathway components and TEs change with age, especially in ovaries, we reasoned that changes in underlying piRNA abundances may drive changes in TE transcription. To analyze PIWI pathway activity in aging ovaries and testes, we generated small RNA-seq libraries from the same samples used for mRNA-seq (Fig. 1A; Supplemental Fig. S1A). We leveraged these libraries to annotate piRNA clusters and evaluate piRNA expression. First, we computationally isolated piRNAs using a size filter of 24–35 nt, which efficiently captures piRNAs and separates them from miRNAs (Supplemental Fig. S6A; Gong et al. 2018; Huang et al. 2019). Consistent with the resulting reads deriving from piRNAs, nucleotide composition analysis showed the expected 1U bias (Fig. 5B; Thomson and Lin 2009). In addition, piRNA-derived reads arose from 222 unique piRNA clusters in the turquoise killifish genome, as identified by proTRAC (Rosenkranz and Zischler 2012). The predicted TE content of identified piRNA clusters is reflective of the annotated genomic TE content (Fig. 5C), consistent with the coevolution between the TE invasion of the genome and genomic defenses against their activity. In contrast to mice, in which ovarian piRNAs are rare and the PIWI pathway is thought to be nonessential (Watanabe et al. 2008), turquoise killifish ovaries express the components of PIWI pathway throughout life and are replete with piRNAs. This may be the result of continuous oocyte production in the killifish ovary, because PIWI is required during oogenesis (Thomson and Lin 2009; Ketting 2011; Roovers et al. 2015).
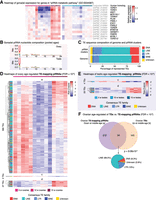
PIWI pathway activity and piRNA abundance characterization in aging turquoise killifish gonads. (A) Heatmaps of normalized gene expression from ovaries and testes with aging in the GO term “piRNA metabolic pathway.” Killifish gene names and their best human homologs are indicated. Rows were normalized by the median expression level in the cognate young gonad to facilitate visualization of age-related changes. (B) Sequence logo plots of piRNA nucleotide composition from ovaries and testes. As expected, killifish piRNAs show a strong 1U bias. Note a slight 10A bias, consistent with active ping-pong biogenesis. (C) Stacked barplots depicting the TE family composition of piRNA clusters and reference genome, as a percentage of identified elements. (D) Heatmaps of TE-targeted piRNA abundance for TEs significantly regulated with age in aging killifish ovaries by DESeq2 LRT (FDR < 10−6). TE family is color-coded for each row. (E) Heatmaps of TE-targeted piRNA abundance for TEs significantly regulated with age in aging killifish testes by DESeq2 LRT (FDR < 10−6). (F) Venn diagram of overlap between TE-mapping piRNAs significantly up-regulated in middle-aged ovaries (pattern a; see D) and TEs significantly down-regulated in middle-aged ovaries (pattern b) (see Fig. 4C). Significance for larger overlap than expected calculated using Fisher's exact test, compared with TEs detected in both analyses. A pie chart of the composition of “consistent” TEs is also reported.
Because piRNAs align to TE sequences, we modeled piRNA abundance dynamics by performing differential expression analysis using counts from the piRNAs mapping to genomic TE sequences. This approach enables us to distinguish which TE species are differentially targeted by the PIWI pathway in turquoise killifish gonads as a function of age. As piRNA biogenesis is expected to occur as a response to TE activity, TE-specific piRNA counts should positively correlate with expressed TE species. Indeed, we observed a strong positive correlation between the TE mRNA levels and cognate piRNAs abundance (Spearman's rank correlation Rho ≥ 0.66) (Supplemental Fig. S6B,C). Similar to transcriptional patterns observed in our genic and TE data, the bulk of differential piRNA expression occurred at middle-age in ovaries (i.e., pattern a) (Fig. 5D; Supplemental Fig. S6D; Supplemental Table S5A). However, mirroring the TE transcriptional changes where many TE species were up-regulated in the middle-aged ovaries (Fig. 4C), the majority of differentially abundant piRNAs were down-regulated in middle-aged ovaries (Fig. 5D). Reduced piRNA abundance is consistent with down-regulation of PIWI-related genes (Figs. 2C, 5A). To note, TEs up-regulated in middle-aged ovaries and TEs targeted by piRNAs with decreased abundance in middle-aged ovaries significantly overlap (P = 9.08 × 10−3 in Fisher's exact test) (Fig. 5F), consistent with relaxation of piRNA-mediated TE repression in middle-aged ovaries. Testis piRNA expression showed only few age-regulated piRNA species, most of which showed increased expression (i.e., pattern d) (Fig. 5E; Supplemental Fig. S6E).
Ping-pong activity declines with age in oocytes, remains stable in testes
In the presence of a particular TE mRNA, the PIWI pathway initializes catalytic degradation of the TE transcript guided by TE-complementary “primary” piRNA sequences, which triggers the production of “secondary” piRNA sequences targeting active TE sequences through a process known as “ping-pong” biogenesis (Fig. 6A; Czech and Hannon 2016). Effectively, ping-pong biogenesis is an adaptive mechanism by which the PIWI pathway defends the germline against active TE threats (Czech and Hannon 2016). Because of the annealing requirements between primary piRNA sequences, target TE mRNAs, and TE-derived secondary piRNAs, a signature of ping-pong biogenesis is a 10-bp overlap between the 5′ ends of opposite orientation piRNAs (Fig. 6A; Supplemental Fig. S7A,B; Wang et al. 2015). Ping-pong biogenesis can be measured globally or at the level of a specific TE.
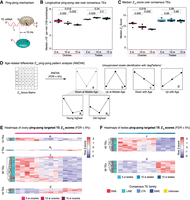
Analysis of ping-pong biogenesis in aging turquoise killifish gonads. (A) Explanatory diagram of ping-pong biogenesis. PIWI1 (the PIWI protein loaded with “primary” piRNAs complementary to TEs) binds to and cleaves TE mRNA. The cleaved TE mRNA is loaded into PIWI2 (the PIWI protein loaded with cleaved TEs, or “TE-derived secondary” piRNAs). PIWI proteins always cleave targets 10 bp from the 5′ end of the piRNA. This mechanism, known as ping-pong biogenesis, will generate complementary piRNAs with a 10-bp overlap. (B) Boxplot showing the median frequency of 10-bp overlaps for 1 million piRNA reads over 100 bootstraps per sample generated by PPmeter. (ppr-mbr) Ping-pong reads per million bootstrapped reads. Nonparametric Wilcoxon rank-sum tests were used to test for differences between age groups. (C) Boxplot showing median Z10-scores over consensus TEs for each sample. Z10-scores are an alternative ping-pong measure. Nonparametric Wilcoxon rank-sum tests were used to test for differences between age groups. (D) Schematic for analysis of differential Z10-score patterns with gonadal aging. Z10-scores were generated for each consensus TE in FishTEDB in each sample. ANOVA was run to detect significant differences in Z10-score between ages within each sex (FDR < 5%). Significant TEs were assigned to patterns based on expression dynamics aligning to the broad groups defined in Figure 2A, and two new subgroups showing minimal expression at middle-age (a1 and a2), with the highest Z10-scores in the young samples and old samples, respectively. (E) Heatmaps of differential Z10-scores with age in aging killifish ovaries by ANOVA (FDR < 5%). (F) Heatmaps of differential Z10-scores with age in aging killifish testes by ANOVA (FDR < 5%). Cognate TE family is color-coded by row.
We first used PPmeter to measure ping-pong biogenesis rates globally in each group (see Methods) (Fig. 6B; Jehn et al. 2018). In parallel, we used an independent method in which, for each consensus TE sequence, we computed Z10-scores, a standard measure of ping-pong activity, defined as the Z-score of the 10-bp overlap frequencies between opposite orientation piRNAs within a 20-bp window (see Methods) (Fig. 6C; Han et al. 2015; Vandewege et al. 2022). Both methods revealed a steady decrease of the piRNA population participating in ping-pong in aging ovaries, whereas this fraction was relatively stable in testes (Fig. 6C; Supplemental Table S6A,B). Age-related decline in ovarian ping-pong may be related to decreased expression of piRNA processing components at middle-age (Fig. 5A). This trend could result in a decrease in the ability of the PIWI pathway to control TE transcription with ovarian aging. Alternatively, steady decrease in ovarian ping-pong biogenesis may reflect the interplay of (1) decreased PIWI pathway efficiency at middle-age (Figs. 2C, 5A) and (2) overall decrease of TE transcription in old ovaries (Fig. 4B). Meanwhile, the observed steady global ping-pong rates in testes are consistent with relatively stable TE and piRNA expression dynamics (Figs. 4D, 5E).
To determine whether ping-pong biogenesis may be differentially regulated for each TE sequence, we examined our Z10 data at the consensus TE level. We used an ANOVA-based test to determine which TEs have differential levels of ping-pong activity, as measured by Z10-scores (Fig. 6D–F; Supplemental Fig. S7C,D). Then, TEs with significant age-related changes in ping-pong biogenesis were clustered into patterns, similar as before. This analysis identified ping-pong biogenesis patterns corresponding to previously described patterns b, c, and d (Figs. 2A, 6D). In addition, it identified two distinct patterns for TEs with decreased ping-pong biogenesis at middle-age (the pattern previously designated as a): maximal ping-pong biogenesis in young gonads (a1) or maximal ping-pong biogenesis in old gonads (a2). Most TEs showing differential ping-pong levels in the ovaries showed significantly decreased ping-pong with aging (pattern c) (Fig. 6E; Supplemental Fig. S7C), consistent with global trends (Fig. 6B,C). In the testes, 82 TEs showed differential ping-pong scores but without a clear global age-related trend, possibly reflecting sustained robust PIWI activity (Fig. 6F; Supplemental Fig. S7D). In summary, multiple methods of analysis show ping-pong activity progressively wanes in the aging ovary but remains steady in the testes, which may be reflective of respective gametogenesis rates in the female versus male gonads throughout life.
Discussion
A resource for the study of reproductive aging in vertebrates
The germline is considered immortal, but the somatic cells that comprise and support the germline are affected by aging. In this study, we examined how aging affects the female and male gonads of a naturally short-lived vertebrate species, the African turquoise killifish. To our knowledge, this data set is the largest “omic” data set evaluating vertebrate gonadal aging and will represent a unique resource for reproductive aging research. To note, we performed this study using fish from the GRZ inbred strain, and it is possible that other strains may show different gonadal aging trajectories. For instance, some laboratory strains are longer lived than the GRZ strain (Kim et al. 2016) and could therefore show different reproductive aging patterns. Further, laboratory strains represent only a fraction of the genetic diversity available in the wild for this species. Indeed, GRZ and wild-derived turquoise killifish reared under the same conditions experience different fecundity and fertility rates during aging (Žák and Reichard 2021), but consistent aging trends have been reported (albeit at different timescales). Although African turquoise killifish may not experience significant age-related decline in reproduction over their short lifespan in the wild, a decline in relative fecundity (i.e., egg generation controlled for total body mass) has been reported in wild females (Vrtílek et al. 2018a). Thus, future studies incorporating more genetically diverse strains (and in their natural habitat) may reveal variation in the molecular regulation of reproductive aging in this species.
With our inclusion of a middle-aged time point, this data set enables the discovery of nonlinear patterns of regulation, thus helping explore gonadal aging trajectories in addition to just gonadal aging end points. Indeed, we found that many key events, including those relating to TE regulation and piRNA pathway regulation, are most salient in middle-aged ovaries, rather than undergoing linear changes with age. Our observations differ from reports in Drosophila egg chambers, in which PIWI-related genes were up-regulated during aging although global TE expression levels remained largely stable (Erwin and Blumenstiel 2019). Importantly, our observations that the transcriptome of ovaries is more strongly impacted by aging than that of testes are consistent with previous microarray studies of mouse gonadal aging (Sharov et al. 2008). To note, in long-lived species (e.g., humans), male germ cells can accumulate mutations from repeated divisions, which can impact offspring fitness and disease risk: the “father's age effect” (Kong et al. 2012). Because of the relatively short lifespan of vertebrate models (e.g., turquoise killifish, mouse), it is unlikely that the lifetime impact of repeated male germ cell divisions can be appropriately modeled in these species, potentially explaining the paucity of changes in aging testes.
A unique cross talk between piRNA biogenesis and TE activity in the aging vertebrate gonad
We characterized TE expression and PIWI pathway behavior throughout life in turquoise killifish gonads and found that the PIWI pathway controls TE expression throughout life. Age-related fertility decline is common across metazoans (Jones et al. 2014). The turquoise killifish is no exception to this rule, with fertility generally declining after middle-age, especially in females (Žák and Reichard 2021). For both sexes, fertility peaks around the middle-age time point used in this study (Žák and Reichard 2021). This time point coincided with the largest changes in TE- and piRNA-related events in both sexes, albeit more pronounced in ovaries compared with testes. This may correspond to a period of increasing gametogenesis in which epigenetic modifications are globally removed from germ cells before repatterning. Such epigenetic erasure would provide a means of “escape” for TEs in the form of transcription. Because females are asynchronous spawners and continuously produce oocytes, we might expect that, like in males, TE transcription may reflect gamete production output. To note, our observations may capture differential amplitudes of reproductive aging between the sexes, with more differential increases in TE transcription in ovaries, which undergo a clearly measurable reproductive senescence, compared with more modest changes in testes, which are thought to undergo near negligible reproductive senescence (Vrtílek et al. 2018a; Žák and Reichard 2021).
Overall, the PIWI pathway seems to broadly keep gonadal TE transcription stable at the global transcriptomic level (Fig. 4B), although there is variation depending on specific TE sequences. This relatively controlled transcriptional output for TE loci contrasts with reports of global transcriptional derepression of TE sequences in aging somatic tissues (De Cecco et al. 2013; Bravo et al. 2020). When expressed, TEs can cause DNA damage and/or initiate innate immune responses owing to their intrinsic similarity to viruses (Simon et al. 2019). This contrast between TE activity in germline/somatic tissues is especially stark considering that nonaging taxa (e.g., Hydra, Planaria) can express PIWI in somatic stem cells (Schaible et al. 2015; Sahu et al. 2017; Teefy et al. 2020). This suggests that looser TE control may drive aspects of aging, especially in the soma, whereas tighter TE control is crucial to allow the emergence of nonaging states, such as those found in the germline.
Dynamic TE control in the aging African turquoise killifish ovary
If piRNA metabolism dynamics in the turquoise killifish ovary are similar to those of zebrafish (with Piwil1 expression largely confined to immature germ cells) (Liu et al. 2022), then down-regulation of PIWI-related genes in middle-aged ovaries could merely reflect a lower proportion of immature oocytes. However, our analyses suggest that large-scale changes in oocyte maturation are unlikely to underlie changes in PIWI pathway activity during ovarian aging (Supplemental Fig. S4). Increased TE expression in middle-aged ovaries (and decreased TE control) may also be a mere symptom of decreased oocyte quality before fertility starts to decline, suggesting that reproductive aging may precede organismal decline. Specifically, decreased TE transcription and increased PIWI pathway components expression in old ovaries may constitute a delayed response to the inverse scenario at middle-age.
Alternatively, it is possible that transient relaxation of TE control in middle-aged ovaries may have adaptative value in the turquoise killifish. Indeed, parental age has been proposed to convey environmental information for evolutionary benefit in this species; embryos of old breeders are more likely to enter a state of suspended animation (i.e., diapause), which killifish use to persist under dry conditions, than are embryos of young breeders (Api et al. 2018). Within a single wet season, the offspring of older mothers are more likely to immediately encounter a dry season, whereas those of younger mothers are more likely to propagate over multiple generations (Api et al. 2018). Likewise, allowing age-associated TE mobilization in the germline could help ensure adequate levels of genetic diversity in a species with naturally small populations owing to its unique ecology. Consistently, annual killifishes have much higher genomic TE content than that of nonannual species that do not experience extreme seasonal population bottlenecks (Cui et al. 2020). TEs have long been appreciated in plant biology for their contribution to genetic diversity and are increasingly appreciated sources of diversity and drivers of speciation in animals (McClintock 1984; González et al. 2010; Belyayev 2014; Catlin and Josephs 2022). In line with the hypothesis that relaxed germline TE restriction may enhance population-level genetic diversity is our observation that ping-pong rates in killifish ovaries steadily decrease with age. This age-related PIWI behavior could support a population facing severe bottlenecks by first generating many healthy (but less genetically diverse) progeny during young adulthood. Subsequently, with older females still breeding, reduced PIWI pathway activity could help generate a more genetically diverse pool of progeny, thus improving genetic diversity for the long-term adaptability of the local population.
In summary, our data support the notion that even a short-lived species such as the African turquoise killifish can maintain adequate TE silencing in germline-containing tissues throughout life, likely owing to the activity of the PIWI pathway. The impact of fluctuations in TE transcription and PIWI pathway activity, especially in ovarian tissues, remains an open question. Understanding disruptions to gonadal genomic regulatory networks during aging will be crucial to define strategies to preserve or prolong reproductive fitness with aging.
Methods
African turquoise killifish husbandry
African turquoise killifish were raised according to standard husbandry procedures (see Supplemental Methods; Dodzian et al. 2018). Fish were euthanized between 2:00 p.m. and 4:00 p.m. by immersion in 1.5 g/L of tricaine MS-222 dissolved in system water followed by decapitation. All procedures were approved by the University of Southern California IACUC under protocols 20879 and 21023.
Lifespan data collection
Lifespan data were derived from historical colony survival, collected between 2019 and 2022. The time from hatching to humane end point or natural death was recorded. All reported data are derived from single-housed fish to limit the impact of death or injuries from frequent fighting in this species.
RNA extraction and sequencing
Gonads were dissected from 5-, 10-, and 15-wk-old turquoise killifish (N = 5 per group) and flash-frozen on dry ice. Biological replicates for each group are in the recommended range for differential gene expression analysis (Lamarre et al. 2018). Age groups were defined based on generally accepted guidelines: (1) the “young” adult time point represents sexual maturity (i.e., the age at which individuals are able to reproduce); (2) the “old” time point represents ∼90% population survival in our colony (Fig. 1B), allowing sampling of aged animals while minimizing survivorship bias (Flurkey et al. 2007); and (3) the “middle-aged” time point is the midpoint between these extremes.
Total RNA was extracted from flash-frozen gonads by homogenizing using TRIzol (Thermo Fisher Scientific 15596018) and lysing Matrix D tubes (MP Biomedicals 11422420) on a BeadBug 6 microtube homogenizer (MilliporeSigma Z742683) for two rounds of 30 sec at 3500 rpm. RNA quality was assayed using the Agilent Bioanalyzer total RNA assay, and only high-quality samples (RIN > 7) were further processed to avoid biases related to RNA degradation. Because of poor RNA quality, samples for three males and one female were eliminated at this stage (Supplemental Fig. S1A). Samples were sent to Novogene for library construction and sequencing, where mRNA and small RNA libraries were generated in parallel from the same samples. mRNA libraries were prepared by enrichment with oligo(dT) beads and sequenced on an Illumina NovaSeq 6000, generating 150-bp paired-end libraries. Small RNA-seq libraries were generated by 3′ adapter ligation, 5′ adapter ligation, reverse transcription, PCR amplification, and gel purification and size selection. Small RNA samples were sequenced on an Illumina NovaSeq 6000 generating 50-nt single-end read libraries.
mRNA-seq data preprocessing and genomic alignment
FASTQ reads from mRNA libraries were hard-trimmed using fastx_ trimmer (fastx_toolkit 0.0.13) (http://hannonlab.cshl.edu/fastx_toolkit/) to remove 5′ adapter fragments and low-quality bases at the 3′ end of the reads using parameters “-f 16,” “-l 100,” and “-Q33.” Illumina adapters were removed using Trim Galore! 0.6.7 (supported by cutadapt 3.3) (https://zenodo.org/record/5127899#.Y789dOzMJUM). We used the most contiguous genome reference available for the African turquoise killifish, obtained from the NCBI Genome database (https://www.ncbi.nlm.nih.gov/data-hub/genome/) under accession GCA_014300015.1 (Willemsen et al. 2020). To enable repetitive sequence analysis, we performed soft-masking with RepeatMasker 4.1.2-p1 (Smit et al. 2013–2015) and a turquoise killifish specific consensus TE library from FishTEDB (downloaded on February 5, 2021) (Shao et al. 2018). RNA-seq reads were aligned to the soft-masked genome using STAR 2.7.0e, allowing 200 multimappers to accommodate TE alignment (Dobin et al. 2013; Jin et al. 2015). Gene and TE counts were generated using TEtranscripts 2.2.1 (Jin et al. 2015).
Differential gene and TE expression analysis
TEtranscripts’ count matrices for genes and TEs were analyzed in R 4.1.2 (R Core Team 2021) using DESeq2 v1.34.0 (Love et al. 2014). We used LRT separately for each sex with each age as its own group, and significant genes were defined as those with adjusted P-value < 10−6. Genes and TEs were then analyzed separately. Once significant genes and TEs were identified, differentially expressed clusters were unbiasedly identified using “degPatterns” from “DEGreport” v1.30.3 (http://lpantano.github.io/DEGreport/).
Functional enrichment analysis
African turquoise killifish protein sequences from the GCA_014300015.1 genome version (Willemsen et al. 2020) were aligned using BLASTP (NCBI BLAST v2.13.0) against Ensembl release 104 human protein sequences. For each query killifish sequence, only human best hits with an E-value < 10−3 were retained, and these homologs were used for functional enrichment. GO enrichment analysis was performed with GO terms downloaded using “biomaRt” 2.50.3 (Ensembl release 104) using “GOstats” 2.60.0 (Durinck et al. 2009) and a FDR < 5%.
piRNA selection, annotation, and piRNA cluster analyses
FASTQ reads from small RNA libraries were adapter-trimmed while keeping a length of ≥18 nt using Trim Galore! 0.6.7 (https://zenodo.org/record/5127899#.Y789dOzMJUM) with the commands “‐‐length 18,” “-a GTTCAGAGTTCTACAGTCCGACGATC,” and “-a GTTCAGAGTTCTACAGTCCGACGATC.” piRNAs were size-selected for 24–35 nt from trimmed small RNA libraries (Gong et al. 2018; Huang et al. 2019). Length distribution and sequence logos were derived from these sequences (see Supplemental Methods). piRNA clusters were identified using proTRAC 2.42 and standard parameters (see Supplemental Methods; Rosenkranz and Zischler 2012).
piRNA differential expression and ping-pong analyses
piRNAs were mapped to the soft-masked reference genome using Bowtie 1.2.3 (Langmead et al. 2009), allowing three mismatches (Juliano et al. 2014; Zhang et al. 2015; Teefy et al. 2020) using “-v 3 -a ‐‐best ‐‐strata -S” before conversion to BAM format with SAMtools 1.10 (Danecek et al. 2021). We used “featureCounts” from Subread 2.0.2 to quantify piRNAs using fractional count attribution for multimapping reads (Liao et al. 2014), as well as a custom GTF consisting of genes, TEs, piRNA clusters, and rRNA repeats. rRNA-mapping reads were discarded before normalization, and piRNA counts to genes, TEs, and piRNA clusters were normalized together with DESeq2. DGE analysis was performed as above.
To analyze global ping-pong biogenesis, we used PPmeter 0.4 (Jehn et al. 2018). For statistical robustness, we used 100 bootstraps to estimate the ping-pong rate per million bootstrapped reads (ppr-mbr) in each library. Bootstrap values were combined per replicate, and the median value was taken (see Supplemental Methods). For TE-specific ping-pong levels, we used custom scripts to calculate Z10-scores for each consensus TE sequence, as defined previously (see Supplemental Methods; Han et al. 2015; Vandewege et al. 2022). For some TE sequences, incomplete coverage prevented derivation of Z10-scores, and these sequences were discarded from consideration. Group-level Z10-scores were tested for significant differences by one-way ANOVA, and only TEs with an adjusted P-value < 0.05 were considered significantly regulated with aging. Significant TEs were classified into clusters using “degPatterns” as above.
Data access
Raw sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA854614. All scripts used in this study are available at GitHub (https://github.com/BenayounLaboratory/Killifish_reproductive_aging_resource) and as Supplemental Code. Ovarian histology pictures are available at Figshare (https://figshare.com/articles/dataset/Killifish_ovarian_ histology_with_aging/21572727) and as Supplemental Data.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
Some panels were made with BioRender (https://biorender.com). We thank Suchi Patel of the USC Genome Core for running RNA Bioanalyzer. We thank Jomille Jerez, Isabel Ollerton, and Rajyk Bhala for assistance with killifish husbandry. We thank Dr. Carolyn Phillips, Dr. Minhoo Kim, Dr. Itamar Harel, Justin Gilmore, Juan Bravo, Casandra McGill, and Rajyk Bhala for insights on the study. We thank Drs. Ryo Sanabria and Gilberto Garcia for advice on histological image analysis. This work was supported by a National Institute on Aging (NIA) T32 AG052374 postdoctoral training grant fellowship to B.B.T., NIA R21 AG063739, National Institute of General Medical Sciences R35 GM142395, a pilot grant from the NAVIGAGE Foundation, and a Hanson-Thorell Family award to B.A.B. We acknowledge the Center for Advanced Research Computing (CARC) at USC for providing computing resources that contributed to the results reported within this publication (https://carc.usc.edu). Ovarian histological analysis was performed by the Translational Pathology Core at the USC Norris Comprehensive Cancer Center (supported by the National Cancer Institute, P30 CA014089).
Author contributions: B.B.T. and B.A.B. designed the study. B.B.T. and A.A. performed fish husbandry. A.A. dissected killifish gonadal tissues. B.B.T. performed RNA extraction of samples and computational analyses, with assistance from A.X., P.P.S., and B.A.B. B.B.T. and B.A.B. wrote the manuscript with input from all authors. K.H. captured microscope images of aging ovaries; B.B.T., A.X., K.H., and B.A.B. conducted ovarian histology analysis. All authors edited and commented on the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277301.122.
- Received September 9, 2022.
- Accepted December 23, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








