
Estimation of intrafamilial DNA contamination in family trio genome sequencing using deviation from Mendelian inheritance
- Christopher J. Yoon1,2,3,4,
- Su Yeon Kim2,
- Chang Hyun Nam3,
- Junehawk Lee5,
- Jung Woo Park5,
- Jihyeob Mun5,
- Seongyeol Park6,
- Soyoung Lee6,
- Boram Yi6,
- Kyoung Il Min3,
- Brian Wiley1,
- Kelly L. Bolton1,
- Jeong Ho Lee3,
- Eunjoon Kim7,8,
- Hee Jeong Yoo9,10,
- Jong Kwan Jun11,
- Ji Seon Choi12,
- Malachi Griffith1,4,
- Obi L. Griffith1,4 and
- Young Seok Ju3,6
- 1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri 63110, USA;
- 2Research Center for Natural Sciences, Korea Advanced Institute of Science and Technology, Daejeon 34141, Korea;
- 3Graduate School of Medical Science and Engineering, Korea Advanced Institute of Science and Technology, Daejeon 34141, Korea;
- 4McDonnell Genome Institute, St. Louis, Missouri 63108, USA;
- 5Center for Supercomputing Applications, Division of National Supercomputing, Korea Institute of Science and Technology Information, Daejeon 34141, Korea;
- 6GENOME INSIGHT Incorporated, Daejeon 34051, Korea;
- 7Department of Biological Sciences, Korea Advanced Institute of Science and Technology, Daejeon 34141, Korea;
- 8Center for Synaptic Brain Dysfunctions, Institute for Basic Science, Daejeon 34141, Korea;
- 9Department of Psychiatry, Seoul National University Bundang Hospital, Seongnam 13620, Korea;
- 10Department of Psychiatry, Seoul National University College of Medicine, Seoul 03080, Korea;
- 11Department of Obstetrics and Gynecology, Seoul National University College of Medicine, Seoul 03080, Korea;
- 12Department of Laboratory Medicine, International St. Mary's Hospital, Catholic Kwandong University College of Medicine, Incheon 22711, Korea
Abstract
With the increasing number of sequencing projects involving families, quality control tools optimized for family genome sequencing are needed. However, accurately quantifying contamination in a DNA mixture is particularly difficult when genetically related family members are the sources. We developed TrioMix, a maximum likelihood estimation (MLE) framework based on Mendel's law of inheritance, to quantify DNA mixture between family members in genome sequencing data of parent–offspring trios. TrioMix can accurately deconvolute any intrafamilial DNA contamination, including parent–offspring, sibling–sibling, parent–parent, and even multiple familial sources. In addition, TrioMix can be applied to detect genomic abnormalities that deviate from Mendelian inheritance patterns, such as uniparental disomy (UPD) and chimerism. A genome-wide depth and variant allele frequency plot generated by TrioMix facilitates tracing the origin of Mendelian inheritance deviations. We showed that TrioMix could accurately deconvolute genomes in both simulated and real data sets.
Sequencing the family members together provides additional information, such as the parental origin of genomic variants, meiotic recombination, or de novo mutations that cannot be obtained from single individual genome sequencing. Because of this benefit, there is now an increasing number of sequencing projects that have collected DNA sequences from related individuals in both normal and disease populations (Turner et al. 2017; Byrska-Bishop et al. 2022). In all sequence data, each individual genome must be checked for purity to ensure proper interpretation. Contamination would dilute true signals from the target (contaminated) DNA and introduce false signals from the source (contaminant) DNA, simultaneously generating false-negative and false-positive genotypes. Contamination of DNA can be caused by an experimental error in which samples are accidentally mixed at various stages, anywhere from sample collection to library preparation or sequence data production. Notably, there is an increased chance of sample contamination within the same sample processing batch (Zajac et al. 2019). Because familial samples are more likely to be collected and processed in parallel, DNA contamination between family members can occur more frequently than when sequenced independently.
In addition to accidental contaminations, biological “contamination” or “mixtures” within family members can also occur naturally or medically. Chimerism, the presence of two or more sets of DNA in a single individual, is sometimes reported in dizygotic twins who share a common placenta (Peters et al. 2017). Medically, an allogeneic organ transplant from a related donor introduces DNA from one family member to another (Bader et al. 2005). Correct quantification and identification of their genomic compositions would be necessary to provide appropriate medical care for such individuals.
However, detecting DNA contamination by family members is challenging because of their shared DNA sequence by inheritance. A few tools have been developed for the estimation of DNA contamination among unrelated individuals, including verifyBamID2 (Zhang et al. 2020), Genome Analysis Tool Kit (GATK) CalculateContamination (Van der Auwera and O'Connor 2020), Haplocheck (Weissensteiner et al. 2021), and Peddy (Pedersen and Quinlan 2017). These tools rely on variant allele frequencies (VAFs) and population allele frequencies of polymorphisms detected from an individual genome sequence. But their detection accuracy is compromised when intrafamilial DNA contamination is present because the distribution of genetic variants is not independent between family members. Therefore, tools for accurately quantifying and identifying the source of intrafamilial contamination are needed.
DNA contamination between family members can be quantified by considering the possible inheritance patterns for a diploid (2n) organism using Mendel's law of segregation. For example, for a single nucleotide polymorphism (SNP) that is homozygous reference A/A genotype in the mother and homozygous alternate T/T genotype in the father, an offspring would be expected to display a heterozygous A/T variant with half of the sequence reads supporting the variant allele, resulting in a VAF of ∼50%. If DNA contamination originates from family members, contamination levels can be accurately assessed as this would lead to expected changes in the VAFs at millions of SNP loci scattered throughout the genome because the genotypes of family members can be inferred. Taking advantage of VAF changes inferred from the family pedigree, we developed TrioMix by using maximum likelihood estimation on SNP read counts to accurately quantify intrafamilial contaminations.
Results
Overview of the tool
TrioMix requires sequence alignment files of trios (mother, father, offspring) in BAM or CRAM format and the reference genome sequence to which the sequence files are aligned (Fig. 1A). Optionally, a file of common SNP positions specified in BED format, can be supplied to reduce computation to these selected loci. TrioMix counts the read support for the reference and alternative bases for each position in the genome (or provided SNP positions) for the trios simultaneously to quantify contamination. It also produces a genome-wide VAF plot and a VAF segmentation result, which can be used collectively to determine the origins of Mendelian deviation in family trio sequencing data sets.
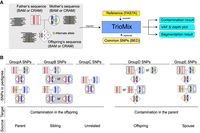
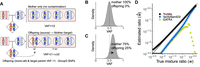
Overview of TrioMix. (A) Sequence alignment file in BAM or CRAM format for the father, mother, and offspring is required. A reference FASTA file is also required. A common single nucleotide polymorphism (SNP) position in BED format can be used as an optional input file to restrict the analysis to those sets of SNPs. Reference and alternative read counts (shown as yellow stars) at the SNP loci in the parents are used to infer the genotypes, and read counts in the offspring are used to build a maximum likelihood estimate (MLE) model to identify the source and quantity of the contamination. (B) SNPs are classified into five groups based on their genotypes. Individuals highlighted in gray are the contamination targets investigated in each SNP group. Each group is used to calculate different DNA contamination targets and sources in the mixture.
To effectively estimate DNA contamination mixtures in various contexts (e.g., parent–offspring, sibling–sibling, parent–parent, or nonfamilial contaminations), we classified SNPs into five classes (hereafter referred to as GroupA, GroupB, GroupC, GroupD, and GroupE variants) based on different genotype patterns (Fig. 1B). Each of the SNP groups is useful for the estimation of distinct origins of contaminants. For GroupA SNPs, one of the parents is a homozygous reference genotype (hereafter referred to as homo-ref), and the other parent is a homozygous alternative genotype (hereafter referred to as homo-alt). GroupA SNPs are exclusively used for estimating parent → offspring DNA contamination. For GroupB SNPs, one of the parents is a homo-ref genotype, and the other parent is heterozygous (hereafter referred to as het). GroupB SNPs are primarily used for estimating sibling → offspring DNA contamination. For GroupC SNPs, both parents are homo-ref genotypes. GroupC SNPs are used for detecting unrelated individual → offspring DNA contamination by measuring the de novo–like alteration rate (fraction of alternative reads that cannot be explained by contamination from parents or any other siblings). For GroupD SNPs, the offspring is homo-alt genotype, and the target parent is het genotype. GroupD SNPs are used for detecting offspring → parent DNA contamination. For GroupE SNPs, offspring is homo-alt genotype, one parent is homo-alt genotype, and the other parent is het genotype. GroupE SNPs are used for detecting one parent → the other parent DNA contamination. Reference and alternative read counts in the contamination target for each SNP group are then used for building statistical models for estimating the genomic compositions.
Simulation of DNA contamination
To validate the accuracy of our algorithm for estimating intrafamilial DNA contamination, we created in silico contaminated BAM files by subsampling and merging reads from two or more real BAM files from a family (mother, father, target offspring, and sibling). Reads from two or more individuals were randomly selected at known fractions. We tested the accuracy of our algorithm by comparing TrioMix's estimate with the true contamination value that was used to create the in silico contaminated BAM file.
Contamination of offspring DNA by parent DNA
Contamination of offspring DNA by parental DNA can be deduced by using the VAF deviation of GroupA variants, in which one parent is homo-ref genotype and the other parent is homo-alt genotype. Here, the offspring should have a heterozygous genotype under Mendelian inheritance (expected VAF ∼ 0.5, Fig. 2A). When maternal DNA contaminates the offspring's DNA, the VAF of such a locus would deviate from the anticipated value in the offspring. For the maternally inherited GroupA SNPs (i.e., alternative alleles inherited from the mother and paternal genotypes are homo-ref), the VAF will be increased (Fig. 2A). Simultaneously, VAFs of paternally inherited GroupA SNPs (i.e., alternative alleles inherited from the father and maternal genotypes are homo-ref) will be reciprocally decreased (Fig. 2B; Supplemental Fig. S1 for paternal DNA contamination). Of note, DNA contamination from siblings does not change the expected VAF pattern for GroupA loci because the genotypes will be het genotype for all children from the same parents.
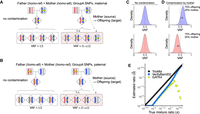
Quantification of offspring DNA contamination by parents. GroupA single nucleotide polymorphisms (SNPs) are used for quantifying parental DNA contamination in the DNA mixture. SNPs are shown as stars; each bar represents a chromosome in the diploid genomes. (A) SNP loci in which the mother has the homo-alt genotype are shown (maternally inherited GroupA SNPs). When the offspring's DNA is not contaminated, the variant allele frequency (VAF) of the offspring is 50%. When the mother's contamination level is x, the expected offspring VAF in the contaminated sample is (1+x)/2. (B) A similar analysis can be performed with SNP loci in which the father has the homo-alt genotype (paternally inherited GroupA SNPs). (C) A density plot of VAFs for maternally (blue) and paternally (red) inherited GroupA SNPs is shown when there is no contamination (offspring 100%). (D) A density plot of VAFs for maternally (blue) and paternally (red) inherited GroupA SNPs with 25% maternal contamination is shown. (E) True versus estimated value for TrioMix (black circle), VerifyBamID2 (blue square), and GATK4 CalculateContamination (green triangle) for in silico simulated DNA contamination with parental DNA. Estimated values below 10−2.5 are shown on the plot's x-axis.
We implemented a maximum likelihood estimate (MLE) framework to estimate the fraction of contamination originating from a parent using the VAF deviations measured by the reference and alternative read counts in GroupA SNPs (see “Materials and Methods” section). There are ∼130,000 GroupA SNPs genome wide from a trio family using common SNPs, although the number may vary depending on the ancestry of the family (Supplemental Table S1). In the uncontaminated offspring, VAFs of GroupA SNPs are seen at VAF = 0.5 (Fig. 2C). In contrast, a simulated BAM with maternal DNA contamination shows maternal and paternal GroupA SNP VAFs shifted reciprocally away from 0.5 (Fig. 2D). Quantitatively, TrioMix accurately estimated the fraction of parental DNA in the mixture across a wide range of contamination levels (Fig. 2E). In contrast, tools for assessing nonfamilial DNA contamination (VerifyBamID2 and GATK) underestimated the mixture ratio presumably caused by the presence of shared (inherited) DNA sequences between the parent and the offspring (Fig. 2E).
Contamination of offspring DNA by sibling DNA
To estimate sibling contamination in the target offspring sample, we use GroupB SNP loci in which one parent is homo-ref genotype and the other parent is het genotype (Fig. 1B). Here, two genotypes are possible for the offspring, homo-ref genotype or het genotype (Fig. 3A). If there is no DNA contamination, the VAF distribution of GroupB SNPs (usually n ∼ 250,000 genome wide for a trio family using common SNPs, Supplemental Table S1) in an offspring will show two distinct peaks at VAF = 0 (homo-ref) and 0.5 (het) (Fig. 3B).
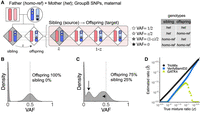
Quantification of offspring DNA contamination by a sibling. (A) GroupB single nucleotide polymorphisms (SNPs) are used for quantifying siblings’ DNA contamination in the DNA mixture. SNP variants are shown as stars; each bar represents a chromosome in the diploid genome. In the DNA mixture, the sibling's fraction is z, and the other offspring's fraction is 1−z. Meiotic recombination in one of the offspring's chromosomes is shown with alternating chromosome colors. Four possible SNP combinations between the offspring and the sibling (contamination) and their variant allele frequencies (VAFs) are shown on the right. (B) A density plot of GroupB SNP VAFs in a sample without contamination (offspring 100%) is shown. (C) A density plot of GroupB SNP VAFs in an offspring with 25% contamination from the sibling is shown. Additional peaks are shown with solid arrows. An additional peak near VAF = 0.5 is merged with the nearby heterozygous VAF = 0.5, which appears as one left-shifted peak. (D) True contaminated value versus estimated value for TrioMix (black circle), VerifyBamID2 (blue square), and GATK4 CalculateContamination (green triangle) for in silico simulated DNA contamination with sibling's DNA. Estimated values below 10−2.5 are shown on the plot's x-axis.
Because siblings will independently inherit parental alleles, there are four possible combinations of genotypes for two siblings for GroupB SNPs. When there is contamination from the sibling, VAFs for GroupB SNPs will show additional intermediate values between 0 and 0.5 at loci in which the genotypes of the two siblings differ (Fig. 3C). Similar to estimating parental DNA contamination, TrioMix estimates the sibling's DNA fraction by building an MLE model from reference and alternative read counts of GroupB SNPs in the offspring's BAM file. In our simulation studies, TrioMix estimated the fraction of sibling's DNA in the mixture accurately (Fig. 3D). Again, tools for assessing nonfamilial DNA contamination underestimated the mixture rate in the sibling contamination simulation (Fig. 3D).
Contamination of offspring DNA by nonfamilial sources
To identify offspring samples with nonfamilial DNA contamination, TrioMix calculates a de novo–like alteration rate using GroupC SNP loci, in which both parents are homo-ref genotypes (thus an offspring should be a homo-ref genotype) (Fig. 1B, Methods). Here, alternative alleles contributing to the de novo–like alteration rate are caused by nonfamilial DNA contamination or sequencing errors. Of note, intrafamilial contamination would not generate such errors in GroupC SNP loci (Supplemental Fig. S2). Samples with a de novo–like alteration rate exceeding the background sequencing error rate are thus identified, and they can be considered to have nonfamilial DNA contamination.
Contamination of offspring DNA by multiple familial sources
Thus far, we assumed only one additional individual's DNA could be contaminating the target offspring. However, more than one family member's DNA can simultaneously contaminate the offspring's DNA. Because the simultaneous presence of genetically related family members would affect expected VAF nonindependently, we jointly estimate the fraction by maximizing the likelihood for three variables (x, y, and z for contamination levels of mother, father, and a sibling, respectively) using both GroupA and GroupB SNPs (Supplemental Fig. S3, Methods). We randomly simulated several cases of contamination with multiple family members and estimated the mixture compositions (Fig. 4). In all cases, TrioMix accurately estimated the fraction of each family member in the target offspring's DNA with a mean cosine similarity of 0.99972 (95% CI = 0.99830–1.0).
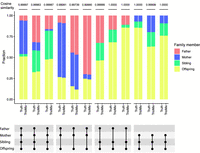
Quantification of DNA contamination in the offspring by multiple family members. We simulated DNA contaminations with various fractions of the father, mother, sibling, and offspring's BAM files. TrioMix's estimation of intrafamilial DNA fraction and the “ground truth” used for simulation are drawn next to each other. Simulations are grouped with different combinations of intrafamilial sources. Cosine similarities between the truth and TrioMix estimated fractions are shown on top of each case. Family members used for creating the simulated DNA contamination are shown with filled circles for each case.
Contamination of parent DNA by the offspring DNA
Although we have focused on Mendelian deviations that could occur when the offspring is the target, these Mendelian deviations can be found in the parents as well. TrioMix can also estimate DNA contamination in the parents’ genome sequences. To detect contamination of the parent by the offspring, we use GroupD SNPs in which the offspring is a homo-alt genotype in the autosomes. In this case, the parents can either be a het genotype or a homo-alt genotype. Another requirement for GroupD is that the VAF in the target parent is less than 1 (i.e., the contaminated parent's genotype must be het). This filtering condition removes all of the SNP loci in which the contaminated parent is a homo-alt genotype because DNA contamination by the offspring's DNA with homo-alt genotypes would still lead to a VAF = 1 (Fig. 5A). In these GroupD SNP loci, the contamination from the offspring's DNA (homo-alt) into the parent's DNA (het) would lead to an increase in the VAFs detected in the contaminated parent (Fig. 5B,C). Using simulated contaminations, TrioMix accurately estimated the contamination of parent DNA by the offspring, whereas other tools for assessing nonfamilial DNA contamination underestimated the mixture ratio (Fig. 5D).
Quantification of parent DNA contaminated by offspring. (A) GroupD single nucleotide polymorphisms (SNPs) are defined as homo-alt genotypes in the offspring and variant allele frequency (VAF) <1 in the contaminated parent. Here, contamination of the mother by the offspring is shown. The other parent's (father in this case) genotype can either be het or homo-alt. The fraction of offspring contamination is w. (B) A density plot of GroupD SNP VAF in the mother is shown when there is no contamination. (C) A density plot of GroupD SNP VAF in the mother when there is 25% contamination by the offspring's DNA is shown. The peak shift is shown with an arrow. (D) True versus estimated value for TrioMix (black circle), VerifyBamID2 (blue square), and GATK4 CalculateContamination (green triangle) for in silico simulated parent DNA contaminated by the offspring DNA. Estimated values below 10−2.5 are shown on the plot's x-axis.
Contamination of one parent's DNA by the other parent's DNA
The DNA of one of the parents could contaminate the DNA of the other parent as well. In a common nonconsanguineous marriage, the two parents would be unrelated in their DNA sequences. But, in the presence of the offspring's DNA, the genotypes of the two parents are no longer independent of each other. Here, we use GroupE SNPs defined as homo-alt genotype in the offspring, VAF in the target (contaminated) parent is less than 1, and homo-alt genotype in the source (contaminating) parent. This allows the genotype of the target parent to be fixed to the het genotype in GroupE SNPs (Fig. 6A). For these GroupE SNP loci, the contamination from the source parent's DNA (homo-alt) into the target parent's DNA (het) would lead to an increase in the VAFs detected in the target parent (Fig. 6B,C). Using simulated contaminations, TrioMix accurately estimated the contamination of one parent's DNA by the other parent's DNA. VerifyBamID2 and GATK4 also performed well because the two parents are unrelated individuals, which is what these tools were designed for (Fig. 6D).

Quantification of parent DNA contaminated by the other parent. (A) GroupE single nucleotide polymorphisms (SNPs) are defined as homo-alt genotypes in the offspring and variant allele frequency (VAF) <1 in the contaminated parent and homo-alt genotype in the contaminating parent. Here, contamination of the mother's DNA by the father's DNA is shown. The fraction of contaminating parent's DNA is v. (B) GroupE SNP VAF density plot in the mother is shown when there is no contamination. (C) GroupE SNP VAF density plot in the mother when there is 25% contamination by the father's DNA. The peak shift is shown with an arrow. (D) True versus estimated value for TrioMix (black circle), VerifyBamID2 (blue square), and GATK4 CalculateContamination (green triangle) for in silico simulated parent DNA contaminated by the other parent's DNA. Estimated values below 10−2.5 are shown on the plot's x-axis.
Visualization of supporting evidence
To support the analysis, TrioMix provides read-depth and VAF plots for the offspring in the trio, for GroupA and GroupB SNPs of the offspring with their respective parent of origin. In an uncontaminated sample, all GroupA SNPs show a VAF = 0.5 for het genotypes (Fig. 7A). GroupB SNPs exist at VAF = 0 (homo-ref) and VAF = 0.5 (het) at approximately equal counts (Fig. 7A). In the case of offspring DNA contaminated by the mother, maternally inherited GroupA SNPs show increased mean VAF and paternally inherited GroupA SNPs show decreased mean VAF as described earlier (Fig. 7B). In addition, we also observe low VAF SNPs in maternal GroupB SNPs. Offspring DNA contaminated by the father (Fig. 7C) shows a converse pattern compared with contamination by the mother (Fig. 7B), with decreased VAF of maternally inherited GroupA SNPs and increased VAF of paternally inherited GroupA SNPs. Offspring DNA contaminated by the sibling would show no change in GroupA SNPs (because both children are het genotype). However, it would display a segmented GroupB SNP VAF pattern that reflects the meiotic recombination patterns in the parental homologous chromosomes (Fig. 7D).

Visualization of variants in the offspring with intrafamilial contamination. Variant allele frequencies (VAFs) of each variant type in the offspring, based on the parent of origin and parental genotype combinations (GroupA and GroupB), are shown with the total depth plot on the genomic coordinate. Blue and red dots represent maternally and paternally inherited variants, respectively. (A) Offspring without DNA contamination. VAF of GroupA SNPs are at 0.5. VAF of GroupB SNPs are at either 0 or 0.5. (B) Offspring (75%) contaminated by the mother (25%). (C) Offspring (75%) contaminated by the father (25%). (D) Offspring (75%) contaminated by a sibling (25%). The segmented pattern of GroupB SNPs represents meiotic recombination between the parental homologous chromosomes. The gray dashed line on the depth plot represents the mean autosomal depth. The black dashed lines on the VAF plots represent VAF = 0.5. Up and down arrows indicate an increase or decrease in VAF from 0.5. The left arrows indicate the presence of variants with low VAF because of contaminations.
Decomposing maternal DNA contamination in placental tissue
As a proof-of-concept study, we applied TrioMix to placental samples, in which the mother's cells frequently contaminate the fetal tissue. In a sample of placenta tissue, TrioMix identified ∼6.6% maternal DNA contamination (Supplemental Fig. S4A). Contamination from the mother's DNA in the placenta was further supported by the reciprocal peak shift of the GroupA SNP VAF density plot (Supplemental Fig. S4B,C).
Decomposing sibling DNA contamination in monochorionic dizygotic twins
We applied TrioMix to a pair of known chimeric monochorionic dizygotic twins (Chung et al. 2018). These twins are chimeras resulting from two genetic siblings (dizygotic twins) exchanging cells with each other during their development in utero. TrioMix successfully quantified DNA chimerism from the two dizygotic twins at 16.1% and 21.8%, respectively. GroupB SNP VAFs show segmented patterns across different regions, which reflect the meiotic recombination patterns between homologous chromosomes in the parents that are differentially inherited between the siblings (Supplemental Fig. S5).
Detection of uniparental disomy
Because TrioMix uses Mendelian inheritance patterns of SNPs to detect contamination, it can also identify other genomic abnormalities that deviate from Mendelian inheritance patterns, such as uniparental disomy (UPD). UPD occurs when an offspring receives two homologous chromosomes from only one of the parents rather than inheriting one copy from each parent (Benn 2021). Variants deviating from Mendelian inheritance caused by UPD will be restricted to these localized regions, which can be differentiated from the genome-wide deviations seen with parental DNA contamination. Localized homozygosity of GroupA SNPs inherited from one parent would suggest a UPD event. When UPD is present, GroupB SNPs can further distinguish between a uniparental isodisomy event and a uniparental heterodisomy event because GroupB SNPs track the heterozygous variants of two homologous chromosomes separately in the parents. We applied TrioMix on a sample with whole-chromosome UPD of Chromosome 4 (Kim et al. 2022) (Fig. 8). Here, copy-number neutral loss-of-heterozygosity (LOH) was shown for Chromosome 4. The possibility of monosomy was ruled out by the lack of drop in sequencing depth. Maternal GroupA variants were all homo-ref genotypes (VAF = 0), suggesting a complete loss of the maternal Chromosome 4 in the offspring. In contrast, paternal GroupA variants were all homo-alt genotypes (VAF = 1). Paternal GroupB SNPs were either homo-ref genotypes (VAF = 0) or homo-alt genotypes (VAF = 1) in the offspring, suggesting that the two homologous chromosomes in the offspring are derived from a single paternal chromatid (paternal uniparental isodisomy, UPiD).
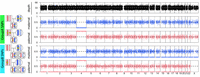
Detection of uniparental disomy (UPD). Variant allele frequencies (VAFs) of each variant type in the offspring, based on the parent of origin and parental genotype combinations (GroupA and GroupB), are shown with the total depth plot on the genomic coordinate. Whole-chromosome level UPD was identified with TrioMix, revealing a paternal isodisomy pattern. The gray dashed line on the depth plot represents the mean autosomal depth. The black dashed lines on the VAF plots represent VAF = 0.5.
Detection of sample swaps between family members
In family sequencing, pedigree information may be swapped or mislabeled (Manichaikul et al. 2010). TrioMix can detect these sample swaps by plotting the genome-wide VAFs to show deviation from the expected inheritance patterns (Supplemental Fig. S6).
Speed and memory requirements of TrioMix
Parallel computing options were implemented to allow faster computing. Performance was assessed on Amazon Web Services on an Ubuntu 20.04 LTS operating system built with an Intel(R) Xeon(R) CPU E5-2666 v3 @ 2.90 GHz (max 16 CPU) processor. The run time was ∼60 min for a single core and 20 min for four cores on a standard 30× BAM file from a trio (Supplemental Fig. S7).
Discussion
We created a bioinformatics software tool that can detect and quantify intrafamilial contamination in sequencing data of parent–offspring trios. TrioMix can accurately quantify the DNA mixture level when genetically close individuals (family members) are the source of the additional DNA. TrioMix uses genotypes directly inferred from the family relationship to estimate the DNA mixture fraction. We are aware of only one other tool that estimates maternal contamination for trio data, which also uses Mendelian inheritance to estimate contamination levels for adjusting the genotype calls in the presence of maternal DNA contamination (Nabieva et al. 2020). In TrioMix, we implemented a comprehensive and robust statistical framework using maximum likelihood to estimate contamination between family members for single-source and multiple-source contamination cases.
We also show that the TrioMix can be extended to detect rare genomic abnormalities that deviate from Mendel's law, such as chimerism and UPD. The true frequency of biological chimerism in the human population has not yet been studied for a lack of systematic screening of large populations (Peters et al. 2017). Recent sequencing studies have shown that UPD is detected in as much as 0.05% of the population and 0.3% in a clinical sequencing cohort (King et al. 2014; Nakka et al. 2019; Scuffins et al. 2021). With the increasing application of family cohort sequencing and systematic analyses of “contamination,” we may uncover more chimeras and UPDs in the general population. Accurately estimating the genomic composition of such individuals will be critical as they may need more careful consideration for their health and family planning than unaffected individuals.
With the increasing use of family trio sequencing, TrioMix can facilitate quality control by identifying potential intrafamilial contamination that could be missed or underestimated by other contamination detection tools. Furthermore, TrioMix can also identify chimeras and UPDs that would otherwise be discarded as “contamination.” We believe that TrioMix is a user-friendly tool that provides a quick and easy discovery of contamination and genomic abnormalities from the BAM or CRAM files of trios.
Methods
Creation of in silico BAM with a known mixture ratio
BAM files were subsampled with the following SAMtools (Li et al. 2009) commands: “samtools view ‐‐subsample $subsample_ratio1 -hbo $subsampled_bam1 $bamfile1 ; samtools view ‐‐subsample $subsample_ratio2 -hbo $subsampled_bam2 $bamfile2”. The subsample fractions were adjusted for the total autosomal reads in each input BAM file. Subsampled BAM files were merged with “samtools merge -o $merged_bam $subsampled_bam1 $subsample_bam2”. A custom script to generate synthetic DNA mixture is provided as Supplemental Code. Three 1000 genome families with four members (father, mother, offspring, and sibling) were used for contamination simulations (family IDs: M004, M008, SH074) (Byrska-Bishop et al. 2022).
Detection of offspring's DNA contamination by the parent
To estimate parental DNA contamination in the offspring, we use autosomal SNPs that are homo-ref genotype in one of the parents and homo-alt genotype in the other parent (Fig. 1B, GroupA SNPs). Mendelian inheritance from the parents will yield a het genotype in the offspring. For these GroupA SNP loci, we know the parent of origin of each alternative allele in the offspring.
Let us assume that the mother's DNA is mixed with the offspring's DNA with x and 1 − x ratios, respectively. If the alternative (variant) allele is inherited from the mother (i.e., maternal GroupA SNP), the expected
variant allele frequency (VAF) can be expressed with x,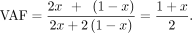 The likelihood for a maternal GroupA SNP loci (Lm) is as follows. At loci j, let the read depth be Nj, and the alternative allele counts be nj, and then the likelihood of a given observation can be modeled as a binomial sampling,
The likelihood for a maternal GroupA SNP loci (Lm) is as follows. At loci j, let the read depth be Nj, and the alternative allele counts be nj, and then the likelihood of a given observation can be modeled as a binomial sampling,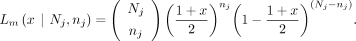 Similarly, when the alternative allele is inherited from the father (i.e., paternal GroupA SNP), VAF and the likelihood (Lp) can be calculated,
Similarly, when the alternative allele is inherited from the father (i.e., paternal GroupA SNP), VAF and the likelihood (Lp) can be calculated,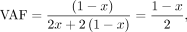
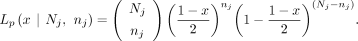 To estimate the mother's DNA contribution, we find the
To estimate the mother's DNA contribution, we find the 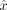 that maximizes the sum of the log-likelihood of both maternally and paternally inherited GroupA SNPs,
that maximizes the sum of the log-likelihood of both maternally and paternally inherited GroupA SNPs,
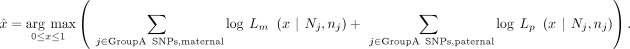 Similarly, the father's DNA contribution can be solved by finding the
Similarly, the father's DNA contribution can be solved by finding the  that maximizes the log-likelihood of both paternally and maternally inherited GroupA SNPs,
that maximizes the log-likelihood of both paternally and maternally inherited GroupA SNPs,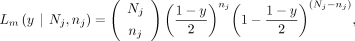
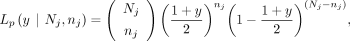
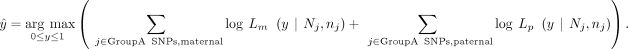 The mle2 function “bbmle” R package (https://cran.r-project.org/web/packages/bbmle/index.html) using the “Brent” method was used to estimate the parent's contamination level
The mle2 function “bbmle” R package (https://cran.r-project.org/web/packages/bbmle/index.html) using the “Brent” method was used to estimate the parent's contamination level  or
or  in the offspring's DNA.
in the offspring's DNA.
Detection of offspring's DNA contamination by the sibling
To estimate the sibling's DNA contamination in the offspring, we identify autosomal SNP loci in which one of the parents is
a homozygous reference genotype (homo-ref; VAF = 0) and the other parent is a heterozygous genotype (het; VAF is between 0.4 and 0.6) (Fig. 1B, GroupB SNPs). Under Mendel's law of segregation, the offspring has a 1:1 chance to have the homo-ref and het genotype in each SNP locus. When the DNA of an offspring is contaminated with its sibling at z fraction, there are four possible genotype combinations of the offspring and the sibling (homo-ref/homo-ref, het/het, het/homo-ref, homo-ref/het, shown in offspring/sibling order), in each locus (Fig. 2A). We calculate the expected allele frequencies for each of the four possible genotype combinations. When both the offspring
and sibling are heterozygous (het/het), the two alleles will exist at 0.5 regardless of x (VAF = 0.5). Likewise, when both offspring and sibling inherit the homozygous reference alleles (homo-ref/homo-ref), only the reference allele will exist (VAF = 0). When the sibling inherits the alternative allele from the heterozygous
parent, and the offspring inherits the reference allele from the heterozygous parent (homo-ref/het), the VAF in the mixture DNA can be expressed with z,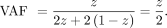 Similarly, if the offspring inherits the alternative allele and the sibling inherits the reference allele (het/homo-ref), the VAF will be
Similarly, if the offspring inherits the alternative allele and the sibling inherits the reference allele (het/homo-ref), the VAF will be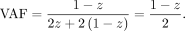 Therefore, DNA with sibling contamination will have four possible VAFs (0,
Therefore, DNA with sibling contamination will have four possible VAFs (0,  ,
, 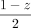 , and 0.5) instead of just two possible VAFs (0 and 0.5) in an uncontaminated DNA of an offspring.
, and 0.5) instead of just two possible VAFs (0 and 0.5) in an uncontaminated DNA of an offspring.
We built a likelihood model for each identified GroupB SNP locus based on the reference and alternative base read counts with
all four possible genotype combinations,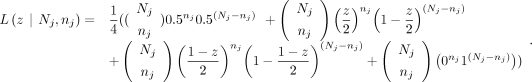 We then calculate the log-likelihood for all loci j and find the
We then calculate the log-likelihood for all loci j and find the  that maximizes the sum of the log-likelihood of the observed data,
that maximizes the sum of the log-likelihood of the observed data,
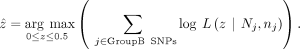 The mle2 function “bbmle” R package was used with the “Brent” method to estimate the sibling's contamination level
The mle2 function “bbmle” R package was used with the “Brent” method to estimate the sibling's contamination level  , in the offspring's DNA.
, in the offspring's DNA.
Detection of offspring's DNA contamination by nonfamilial DNA
In the intrafamilial DNA contaminations, whereas the VAF may deviate from the expected pattern, all of the variants can be explained by their presence in the parents. When nonfamilial DNA contaminants are present in the offspring's DNA, this will be shown as a “de novo” or new variants that were not present in the parents. We look at SNPs in which both parents are homo-ref genotypes (Fig. 1B, GroupC SNPs) to detect these de novo–like alterations. Because the offspring has to inherit both reference alleles from the parents (thus a homo-ref genotype), any alternative alleles in the offspring are either a sequencing/mapping error or contamination by nonfamilial DNA. TrioMix reports the de novo–like alteration rate, the total alternative read fraction in GroupC SNPs in the offspring. Contribution from true de novo variants will be negligible because there are less than 100 de novo mutations per child from the entire genome (or common SNPs), which is vastly greater (Kong et al. 2012).
Detection of offspring's DNA contamination by mother, father, and sibling
The MLE framework can be extended to predict the DNA contamination in the offspring when multiple family members could be
the contaminating sources (Supplemental Fig. S3). The fraction of mother, father, sibling, and offspring is x, y, z, and 1–x–y–z, respectively. We first use GroupA SNP loci in which one of the parents is a homo-alt genotype, and the other is a homo-ref genotype (Supplemental Fig. S3A). For the maternally inherited GroupA SNPs, the VAF is as follows: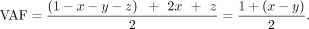 Similarly, for the paternally inherited GroupA SNPs, VAF is as follows:
Similarly, for the paternally inherited GroupA SNPs, VAF is as follows: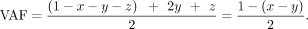 For both paternal and maternal GroupA SNPs, the sibling's contribution (z) cancels out in the equation because both offspring and sibling are of identical genotype (het). Therefore, the equation becomes a function of the difference between the mother's and father's fractions (x–y). Using GroupA SNPs, we first estimate the x–y value with MLE from a range of −1 to 1 using the “Brent” method of the mle2 function. We will refer to the x–y value as k.
For both paternal and maternal GroupA SNPs, the sibling's contribution (z) cancels out in the equation because both offspring and sibling are of identical genotype (het). Therefore, the equation becomes a function of the difference between the mother's and father's fractions (x–y). Using GroupA SNPs, we first estimate the x–y value with MLE from a range of −1 to 1 using the “Brent” method of the mle2 function. We will refer to the x–y value as k.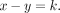 The likelihood function of maternal GroupA SNPs can now be expressed with a variable k,
The likelihood function of maternal GroupA SNPs can now be expressed with a variable k,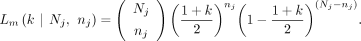 The likelihood function of paternal GroupA SNPs can also be expressed similarly,
The likelihood function of paternal GroupA SNPs can also be expressed similarly,
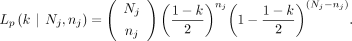
Note that the above likelihood functions would be reduced to the same equation in the single parental contamination cases
if we set either x = 0 or y = 0. Using these likelihood functions for each SNP in GroupA, we can find the  that maximizes the sum of the log-likelihood of the observed data,
that maximizes the sum of the log-likelihood of the observed data,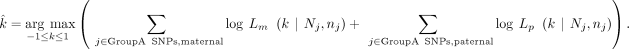 Now we use GroupB SNPs to optimize the remaining variables (Supplemental Fig. S3B). There are a total of four possible genotype combinations between the sibling and offspring. VAFs of GroupB SNPs are expressed
in x, y, and z. Using the
Now we use GroupB SNPs to optimize the remaining variables (Supplemental Fig. S3B). There are a total of four possible genotype combinations between the sibling and offspring. VAFs of GroupB SNPs are expressed
in x, y, and z. Using the 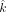 estimated from above, now we substitute
estimated from above, now we substitute 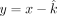 into all of the VAFs. Therefore, we have reduced the likelihood estimate into a function of two variables (x and z). For the maternally inherited SNPs, the likelihood function Lm is given as follows:
into all of the VAFs. Therefore, we have reduced the likelihood estimate into a function of two variables (x and z). For the maternally inherited SNPs, the likelihood function Lm is given as follows: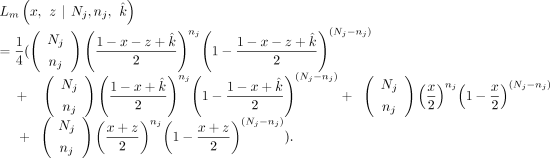 For the paternally inherited SNPs, the likelihood function Lp is given as follows:
For the paternally inherited SNPs, the likelihood function Lp is given as follows: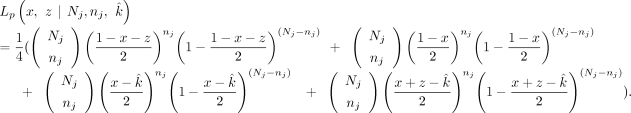 For estimating two variables (x and z) with constraints, we use the mle2 function with the “L-BFGS-B” method (Byrd et al. 1995). Initial guesses are max(0,
For estimating two variables (x and z) with constraints, we use the mle2 function with the “L-BFGS-B” method (Byrd et al. 1995). Initial guesses are max(0, 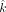 ) and 0 for x and z, respectively,
) and 0 for x and z, respectively,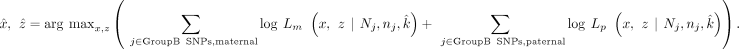 The boundary conditions of a positive variable x is as follows, constrained by the value of
The boundary conditions of a positive variable x is as follows, constrained by the value of 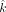 so that the sum of all individual fractions is equal to 1:
so that the sum of all individual fractions is equal to 1: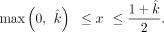 The offspring's fraction (1–x–y–z) can be expressed as
The offspring's fraction (1–x–y–z) can be expressed as 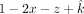 , which is greater than or equal to 0,
, which is greater than or equal to 0,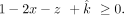 In addition, we set the contaminating sibling's contribution (z) to be smaller than the offspring itself
In addition, we set the contaminating sibling's contribution (z) to be smaller than the offspring itself 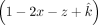 ,
,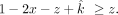 If MLE optimization fails to converge with our initial starting values, we then use a grid-based approach, in which we calculate
the log-likelihood of all combinations of x and z within the boundary conditions with a small incremental change (e.g., 0.01). We use the combination of x and z that maximizes the log-likelihood as the new initial guesses for the MLE.
If MLE optimization fails to converge with our initial starting values, we then use a grid-based approach, in which we calculate
the log-likelihood of all combinations of x and z within the boundary conditions with a small incremental change (e.g., 0.01). We use the combination of x and z that maximizes the log-likelihood as the new initial guesses for the MLE.
Cosine similarity of reconstructed DNA contamination versus ground truth
We measured the cosine similarity between each family member's original ground truth fraction and the TrioMix's joint estimation module's results. The “cosine” function of the lsa package (https://cran.r-project.org/web/packages/lsa/index.html) in the R programming language (v3.6.0) (R Core Team 2019) was used.
Detection of parent's DNA contamination by the offspring
To estimate parent DNA contaminated by offspring's DNA, we identify autosomal SNP loci in which the offspring is a homo-alt genotype (VAF = 1) and the contaminated parent is a het genotype (Fig. 1B, GroupD SNPs). When the offspring is a homo-alt genotype, the parents are either het genotype or homo-alt genotype. Homo-alt genotype loci need to be removed to identify het genotype loci in the contaminated parent. This is achieved by filtering SNP loci with VAF = 1 because contamination of the
homo-alt genotype by another DNA with homo-alt genotype would still result in a VAF = 1. We also remove SNP loci with only one read supporting the reference allele, as
these are likely from sequencing error. Thus, the genotypes of the offspring (homo-alt) and the contaminated parents (het) are fully defined, which can be used to build a likelihood model. Let us assume that the contamination parent's DNA is contaminated
by w fraction of the offspring. In GroupD SNPs, the VAF in the contaminated parent is as follows: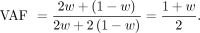 Then, the likelihood in the contaminated parent (Ld) and
Then, the likelihood in the contaminated parent (Ld) and 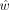 that maximizes Ld can be calculated,
that maximizes Ld can be calculated,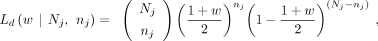

Detection of the parent's DNA contamination by the other parent
To estimate parent DNA contaminated by the other parent's DNA (i.e., the father's DNA contaminating the mother's DNA), we
identify autosomal SNP loci in which the offspring is a homo-alt genotype (VAF = 1), the contaminated parent is a het genotype, and the contaminating parent is a homo-alt genotype (Fig. 1B, GroupE SNPs). Similar to the approach with GroupD, we remove SNP loci in which the contaminated parent has VAF = 1 to restrict
the genotype of the contaminated parent to be a het genotype. We also remove SNP loci with only one read supporting the reference allele, as these are likely from sequencing
error. Thus, the genotypes of the contaminating parent (homo-alt) and the contaminated parents (het) are fully defined, which can be used to build a likelihood model. Let us assume that the contamination parent's DNA is contaminated
by v fraction of the offspring. In GroupE SNPs, the VAF in the contaminated parent is as follows: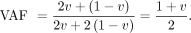 Then, the likelihood in the contaminated parent (Le) and
Then, the likelihood in the contaminated parent (Le) and  that maximizes Le can be calculated,
that maximizes Le can be calculated,
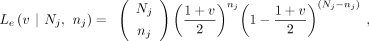

Selection of common SNPs from gnomAD database
We selected common SNPs from the gnomAD database (Karczewski et al. 2020). For GRCh37 we used gnomAD v2, and for GRCh38, we used gnomAD v3. The raw VCF files were downloaded, and common biallelic SNPs with a population allele frequency (AF) greater than 0.3 with root mean square mapping quality (MQ) greater than 50 were selected for analysis. There are 2,855,456 SNPs for GRCh37 and 1,907,413 SNPs for GRCh38 after filtering. The BED files of the selected SNP loci are provided with the source code. Users can also specify their own SNP sets. Providing the SNP BED files is only optional but results in faster computation because it will only identify informative SNPs from the given BED file rather than scanning the whole genome.
Running the tool
TrioMix takes in four required inputs (father's BAM, mother's BAM, offspring's BAM, and reference FASTA file; Fig. 1A). The following command will compute the DNA mixture in the offspring by comparing its sequence to the parental DNA. “python3 triomix.py -f father.bam -m mother.bam -c child.bam -r reference.fasta”. An optional argument “-s common_snp.bed” can be provided to narrow the pileup to common SNP regions only. A “-t” argument allows parallel computing of mpileup generation to increase the speed. If UPD is present, it may appear as contamination of the father or mother's DNA, even when there is no contamination, because of the variants that deviate from Mendelian inheritance pattern. If the user wants to only detect contamination but not UPD, then the “-upd 0” argument can be specified to filter GroupA variants with VAF = 0 or 1, leading to a better estimation of sample contamination. A “‐‐parent” argument will estimate the intrafamilial DNA contamination in the sequence of the parents.
Visualization and segmentation of VAFs for detecting UPD
TrioMix produces a visualization plot by default. In addition, we use VAFs of GroupA SNPs to identify UPD segments. PSCBS (Olshen et al. 2011) and DNAcopy (https://bioconductor.org/packages/release/bioc/html/DNAcopy.html) libraries from the R programming language were used with a minimum segment size of 106 bases.
Computational performance evaluation
Computational performance was evaluated on an AWS instance (c4.4xlarge) with an Intel(R) Xeon(R) CPU E5-2666 v3 @ 2.90 GHz (max 16 CPU) processor built with an Ubuntu 20.04 LTS operating system. Dockerized TrioMix was tested with varying numbers of CPUs (range: 1–16). A 1000 genome family (family ID: M008, father: NA19661, mother: NA19660) was used for the performance analysis (Byrska-Bishop et al. 2022). A simulated target offspring (NA19662, female) with 25% contamination from a sibling (NA19685, male) and uncontaminated offspring (NA19662 100%) was used as the input.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board of KAIST (KH2019-174), International St. Mary's Hospital (IS19TIME0070), Seoul National University Hospital (H-9712-038-002), and Seoul National University Bundang Hospital (B-2204-748-301).
Example data
Sequence data used in the study were retrieved from the European Genome-phenome Archive (EGA; https://ega-archive.org) under the following accessions (Maternal contamination of the placenta: EGAS00001006155. Monochorionic dizygotic chimera: EGAS00001005997. UPD: EGAS00001006154). 1000 Genomes Trio data were retrieved from the International Genome Sample Resource portal (https://www.internationalgenome.org/data-portal/sample).
Software availability
TrioMix is written in Python 3 (v3.5) and R (v3.6.0) programming languages. Full source code is available at GitHub (https://github.com/cjyoon/triomix). A docker image is available at Docker Hub (https://hub.docker.com/r/cjyoon/triomix). A detailed user manual is available at http://triomix.io. The software is also available as Supplemental Code.
Competing interest statement
Y.S.J. is a founder and chief executive officer of GENOME INSIGHT, Inc.
Acknowledgments
This work has been supported by the National Institutes of Health grants (F30HD106744, T32GM007200 to C.J.Y.). This work was also supported by the National Research Foundation of Korea funded by the Korean government, Ministry of Science and ICT (Leading Researcher Program NRF-2020R1A3B2078973 to Y.S.J.), and KREONET (Korea Research Environment Open NETwork), which is managed and operated by the Korea Institute of Science and Technology Information.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276794.122.
-
Freely available online through the Genome Research Open Access option.
- Received March 28, 2022.
- Accepted October 31, 2022.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








