
A butterfly pan-genome reveals that a large amount of structural variation underlies the evolution of chromatin accessibility
- Angelo A. Ruggieri1,
- Luca Livraghi2,3,
- James J. Lewis4,
- Elizabeth Evans1,
- Francesco Cicconardi5,
- Laura Hebberecht5,
- Yadira Ortiz-Ruiz1,6,
- Stephen H. Montgomery5,
- Alfredo Ghezzi1,
- José Arcadio Rodriguez-Martinez1,
- Chris D. Jiggins7,
- W. Owen McMillan3,
- Brian A. Counterman8,
- Riccardo Papa1,6 and
- Steven M. Van Belleghem1,9
- 1Department of Biology, University of Puerto Rico–Rio Piedras, San Juan PR 00931, Puerto Rico;
- 2Department of Biological Sciences, The George Washington University, Washington, DC 20052, USA;
- 3Smithsonian Tropical Research Institute, Apartado 0843-03092 Panamá, Panama;
- 4Department of Zoology, University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada;
- 5School of Biological Sciences, Bristol University, Bristol BS8 1QU, United Kingdom;
- 6Molecular Sciences and Research Center, University of Puerto Rico, San Juan 00926, Puerto Rico;
- 7Department of Zoology, University of Cambridge, Cambridge CB2 3EJ, United Kingdom;
- 8Department of Biological Sciences, Auburn University, Auburn, Alabama 36849, USA;
- 9Ecology, Evolution and Conservation Biology, Biology Department, KU Leuven, 3000 Leuven, Belgium
Abstract
Despite insertions and deletions being the most common structural variants (SVs) found across genomes, not much is known about how much these SVs vary within populations and between closely related species, nor their significance in evolution. To address these questions, we characterized the evolution of indel SVs using genome assemblies of three closely related Heliconius butterfly species. Over the relatively short evolutionary timescales investigated, up to 18.0% of the genome was composed of indels between two haplotypes of an individual Heliconius charithonia butterfly and up to 62.7% included lineage-specific SVs between the genomes of the most distant species (11 Mya). Lineage-specific sequences were mostly characterized as transposable elements (TEs) inserted at random throughout the genome and their overall distribution was similarly affected by linked selection as single nucleotide substitutions. Using chromatin accessibility profiles (i.e., ATAC-seq) of head tissue in caterpillars to identify sequences with potential cis-regulatory function, we found that out of the 31,066 identified differences in chromatin accessibility between species, 30.4% were within lineage-specific SVs and 9.4% were characterized as TE insertions. These TE insertions were localized closer to gene transcription start sites than expected at random and were enriched for sites with significant resemblance to several transcription factor binding sites with known function in neuron development in Drosophila. We also identified 24 TE insertions with head-specific chromatin accessibility. Our results show high rates of structural genome evolution that were previously overlooked in comparative genomic studies and suggest a high potential for structural variation to serve as raw material for adaptive evolution.
Structural variants (SVs) in genomes are a ubiquitous component of within and between species genomic variation (Mérot et al. 2020; Zhang et al. 2021). The larger size of SVs, when compared with single nucleotide polymorphisms (SNPs), may increase their likelihood of being involved in maladaptation (Collins et al. 2020). However, there are a growing number of examples of an important role of SVs in adaptive innovations (Lucek et al. 2019; Wellenreuther et al. 2019). For example, increased linkage disequilibrium and recombination suppression within large inversions can initiate co-adaptation of gene complexes in the rearranged genomic haplotype (e.g., supergenes; Jay et al. 2021; Matschiner et al. 2022). Alternatively, insertion-deletion mutations (indels) can include one or multiple functional genetic elements and studies are starting to indicate that genomic indel content might be large relative to the more commonly studied single nucleotide polymorphisms (SNPs). A study of humans found 2.3 million indels of 1–49 bp in length and 107,590 indels larger than 50 bp that accounted for up to 279 Mb in sequence differences among individuals (Ebert et al. 2021). Several studies in plants and fungi identified the widespread presence of SVs, often linked to phenotypic variation (Read et al. 2013; Plissonneau et al. 2018; Hübner et al. 2019). Aside from these studies performed on humans and nonmetazoans, a few studies in mollusks have also unveiled the possibility that gene-carrying indels may be much more widespread than originally thought (Gerdol et al. 2020; Calcino et al. 2021). Another case are Oedothorax dwarf spiders, in which a large 3 Mb indel is associated with an elaborate alternative reproductive male morph (Hendrickx et al. 2022).
A major challenge in studying the relationships between SVs and adaptive diversification has been the difficulty in characterizing the landscape of divergence in repetitive and rearranged regions of genomes. To overcome this, we here used high-quality butterfly genomes of three Heliconius species common to Central and South America and constructed a pan-genome alignment that allowed us to quantify the homologous and nonhomologous (i.e., lineage-specific insertions or deletions) portions of their genomes. Heliconius charithonia is about 11.1 (8.8–13.4) Myr divergent from Heliconius melpomene and 6.0 (4.8–7.4) Myr divergent from Heliconius erato (Fig. 1A; Kozak et al. 2015; Cicconardi et al. 2022). The three species are reproductively isolated and differ in host plant use (Brown 1981; Jiggins 2017), larval gregariousness (Beltrán et al. 2007), flight (Mallet and Gilbert 1995), pupal mating rates (Mendoza-Cuenca and Macías-Ordóñez 2010; Thurman et al. 2018), and brain structure (Montgomery and Merrill 2017).
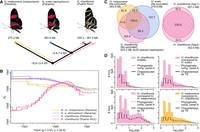
Genome divergence, lineage-specific sequence distribution, and historical demography of H. melpomene, H. erato, and H. charithonia from Panama and Puerto Rico. (A) Phylogenetic relations, genome sizes, and approximate divergence times. Colored lines indicate branches investigated in panel D. (B) Inference of historical effective population size changes using pairwise sequentially Markovian coalescent (PSMC) analysis. The PSMC estimates are scaled using a generation time of 0.25 yr and a mutation rate of 2 × 10−9. Note that the H. charithonia genome was obtained from the Puerto Rican population. (C) Venn diagrams represent homologous and nonhomologous (lineage-specific) genomic sequences (excluding Ns). Between the two pseudohaplotypes of the H. charithonia genome, we observed a total of 72.7 Mb of sequence identified as indel. Of these indels, 63.1 Mb (86.8%) were lineage-specific to H. charithonia, whereas 9.6 Mb (13.2%) were present in the H. erato genome. Consistent with divergence times, the H. charithonia genome comprised 43.5% (175.2 Mb; compared with the ∼6 Myr divergent H. erato) to 62.7% (252.3 Mb; compared with the ∼11 Myr divergent H. melpomene) of lineage-specific sequence resulting from structural variants (SVs). H. erato had 39.0% (151.2 Mb) lineage-specific sequences compared with H. charithonia and 58.0% (222.1 Mb) lineage-specific sequences compared with H. melpomene. H. melpomene had 34.5% (95.0 Mb) lineage-specific genomic sequence compared with H. erato and H. charithonia. (D) Length distribution of lineage-specific sequences. Colored histograms show the frequency of SVs for different phylogenetic comparisons (as indicated in panel A). The black line shows the frequency distribution of lineage-specific SVs that were characterized as transposable elements (TEs). Between the two H. charithonia haplotypes, indels had an average and median length of 13.5 and 2 bp. The average and median length was 34.2 and 4 bp for lineage-specific H. charithonia sequences relative to H. erato and 45.6 and 6 bp relative to H. melpomene.
With the pan-genome alignment, we first analyzed the frequency, length distribution, and composition of lineage-specific sequences between the species. Second, we studied the evolutionary processes affecting the distribution and frequency of SVs. We expected that if SVs have a higher chance of being maladaptive, we will see a lower abundance of SVs on smaller chromosomes compared with SNPs. This expectation is derived from smaller chromosomes having a higher per base pair recombination rate that could lead them to purge maladaptive SVs more efficiently (Hill and Robertson 1966). In contrast, if SVs have a similar maladaptive load as SNPs, we expect their abundance on chromosomes to be similar to SNPs, which have a higher abundance on smaller compared with larger chromosomes in Heliconius resulting from the higher recombination rate and thus lower reduction of SNP diversity by linked selection on smaller chromosomes (Martin et al. 2019; Cicconardi et al. 2021). To further understand the maladaptive impact of SVs, we also characterized the distribution of SVs relative to gene density. Our hypothesis is that if intergenic SVs impact gene functioning negatively, then we expected to identify fewer SVs in gene-rich regions. Moreover, if SVs negatively impact gene regulation, we expect their distances from the transcription start sites (TSSs) of genes to be further compared with a random sample of genome positions.
Third, in contrast to maladaptive impacts of SVs, differences in the presence and/or accessibility of cis-regulatory loci (i.e., noncoding functional regions of the genome that influence patterns of gene expression) between divergent populations have been shown to be responsible for adaptive differences within and between species of Heliconius butterflies (Lewis et al. 2019, 2020; Livraghi et al. 2021). Therefore, to investigate the functional significance of intergenic SVs, we annotated our pan-genome with assays of chromatin accessibility, a powerful approach to identify active cis-regulatory sequences (Buenrostro et al. 2013). We focused on chromatin profiles of developing head tissue and wings as a control and observed that lineage-specific open chromatin is substantially associated with SVs. To investigate whether these lineage-specific open chromatin regions within SVs have been involved in recent adaptive evolution, we used selective sweep scans. We also correlated their abundance with gene density and TSS and compared this correlation to that of SVs that do not associate with lineage-specific changes in chromatin accessibility. Finally, using motif enrichment scans for sites with significant similarity to Drosophila transcription factor (TF) binding sites, we investigated whether these lineage-specific SVs carry a high potential for structural variation to serve as material for adaption. In summary, our work here provides a uniquely comprehensive test for the role of SVs in adaptive evolution.
Results
Genome assemblies, pan-genome alignment, and lineage-specific sequence composition
We de novo sequenced and assembled two haploid genomes from a single H. charithonia individual from Puerto Rico using 10x Chromium technology (10x Genomics). The two pseudohaploid H. charithonia genomes had a length of 355.2 Mb and 361.5 Mb. For H. erato and H. melpomene we used previously published reference genomes from individuals from Panama, which had assembly lengths of 382.8 Mb and 275.2 Mb, respectively (Davey et al. 2016; Van Belleghem et al. 2017). All assemblies had a BUSCO completeness higher than 98.9% (Supplemental Table S1).
Effective population size influences genetic diversity in SNPs (Charlesworth 2009; Leffler et al. 2012) and is thus also likely to be a major influence on indel diversity. We therefore reconstructed the historical population sizes from diversity estimates in whole-genome resequenced samples using pairwise sequentially Markovian coalescent (PSMC). These reconstructions suggest that populations from Panama have had an increase in population size over the past one Myr, with H. erato and H. charithonia having a larger population size than H. melpomene over the last 300 ky (Fig. 1B). In contrast, two H. charithonia individuals from Puerto Rico suggest a population size decline over the past 200 kyr.
For this study, we aligned the four genomes (two H. charithonia pseudohaplotypes, H. erato, and H. melpomene) into a pan-genome with a total length of 659.4 Mb. Among the three species, only 138.6 Mb (21.0%) of sequence was identified as homologous. However, this conserved sequence part retained a high BUSCO completeness of 94.9%, demonstrating it contains the highly conserved gene coding fraction of the genome (Supplemental Table S1). When investigating the proportions of nonhomologous (lineage-specific) sequences as obtained from the pan-genome, we found that the lineage-specific sequence proportion increases with phylogenetic distance (Fig. 1C). More divergent phylogenetic comparisons also had lineage-specific sequences that were generally longer (Fig. 1D), whereas less divergent phylogenetic comparisons had a higher proportion of lineage-specific sequences being accounted for by single base pair insertions (e.g., 25.7% of lineage-specific sequence between the H. charithonia haplotypes vs. 5.8% of lineage-specific sequences between H. charithonia and H. erato; Supplemental Table S2). Between the H. charithonia pseudohaplotypes we observed two genes within an indel, an endonuclease-reverse transcriptase related to a TE (evm.TU.Herato1801.176) and a zinc finger DNA-binding protein (evm.TU.Herato1104.1). Sequences specific to H. erato included 167 genes that were absent in the H. charithonia genome, and 317 genes absent in the H. melpomene genome (Supplemental Table S3). Of these, only two genes that were absent in H. charithonia were present in H. melpomene, which suggests that almost all genes unique to H. erato resulted from gene gain rather than loss in the other species. Of the lineage-specific genes, 22.3% were related to TEs, four genes were characterized to have a function in repressing TE activity and 10 genes were zinc finger proteins for which some families are involved in TE repressing (Ecco et al. 2017). Additionally, seven genes were involved in neural activity, four genes were involved in chemosensing and 33.6% were uncharacterized. In the different genome comparisons, we could further determine the identity of 43.9%–82.0% of all the lineage-specific sequences, with TE insertions being the most abundant SVs (Supplemental Table S2).
Among phylogenetic comparisons, we found generally similar patterns of TE family accumulation but observed several lineage-specific differences (Fig. 2). The most abundant elements associated with lineage-specific sequences in all genome comparisons were SINE elements (25%–41%), Rolling-circle elements (23%–35%), LINE elements (10.1%–22.4%), and DNA transposable elements (14.8 and 21.25%) (Fig. 2A). Our phylogenetic framework next allowed us to characterize the time of accumulation for TEs along the H. erato/H. charithonia branch (considering H. melpomene as the outgroup). Within the TE families, we found that Metulj-7 elements accumulated before H. erato and H. charithonia split (Fig. 2B). This was also supported by relative age of accumulation analysis based on divergence of Metulj-7_Hmel that showed accumulation was more ancient than, for example, Metulj_m51 that likely increased in number after H. charithonia and H. erato split (Supplemental Fig. S1A). Metulj-7_Hmel also accrued earlier in the H. melpomene lineage (Supplemental Fig. S1B). This implies an accumulation that preceded the split of our butterfly lineages. The reduction of Metulj-7_Hmel in more recent times supports a similar finding by Ray et al. (2019), who observed a reduction of Metulj-7_Hmel accumulation in the H. charithonia/erato lineage starting at 5 Mya (Ray et al. 2019). Between the two H. charithonia haplotypes, the two most abundant groups associated with indels were Rolling-circle (32.5%) and SINE (29.7%), with Helitron2_Hera and Metulj7_Hmel showing highest copy numbers (6.5% and 3.6% variation in activity, respectively; Fig. 2C). As higher copy numbers of Helitron2_Hera were not observed along any other parts of the phylogeny, this suggests that Helitron2_Hera accumulated more recently, causing indels. In contrast, the high copy numbers of Metulj-7_Hmel in indels indicates that these indels may persist over long timescales.
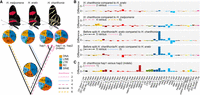
Phylogenetic dynamics of transposable elements (TEs). (A) Lineage-specific TE family accumulation. Different line types depict different branches in the phylogeny studied and allow to investigate changes in temporal accumulation of TEs. 1, TE families associated with indels between the H. charithonia haplotypes; 2, TE families accumulated in H. charithonia since the split from H. erato; 3, TE families accumulated in H. erato since the split from H. charithonia; 4, TE families accumulated in the H. erato lineage since their split from a common ancestor with H. melpomene; 5, TE families accumulated after the H. charithonia/H. erato lineage split from the common ancestor with H. melpomene but before H. erato and H. charithonia split; 6, TE families accumulated in the H. charithonia/H. erato lineage since their split from a common ancestor with H. melpomene. DNA, DNA transposons that do not involve an RNA intermediate; LINE, long interspersed nuclear elements, which encode reverse transcriptase but lack LTRs; LTR, long terminal repeats, which encode reverse transcriptase; RC, transpose by rolling-circle replication via a single-stranded DNA intermediate (Helitrons); SINE, short interspersed nuclear elements that do not encode reverse transcriptase. (B) Difference in TEs (percentage of total) between branches in the phylogeny considering the same 48 most significantly divergent TE families. Positive values indicate higher accumulation in the first branch; negative values indicate higher accumulation in the second branch of the comparison. Total TE accumulation patterns per lineage are shown in Supplemental Figure S1. (C) Difference in TEs (percentage of total) between the two H. charithonia haplotypes considering the same 48 most significantly divergent TE families.
Indel patterns and chromosome sizes
Between the homologous fraction of the genomes (i.e., subtracting lineage-specific sequence from the genome length), we calculated that the frequency of SVs between the two pseudohaplotypes of the H. charithonia individual was 0.010 per bp and slightly higher than the SNP frequency of 0.007 per bp between these haplotypes. Single bp indels were most frequent and SVs shorter than 50 bp accounted for 98.1% of all indels in H. charithonia (Supplemental Table S2). In contrast, when comparing species, substitutions were 3.7–3.8 times more frequent than SVs, with 0.030 SVs per bp versus 0.110 substitutions per bp between H. charithonia and H. erato and 0.039 SVs per bp versus 0.148 substitutions per bp between H. charithonia and H. melpomene (Fig. 3). This change in relative frequencies of SNPs and SVs could be largely ascribed to more single bp indels between the pseudohaplotypes of H. charithonia compared with interspecies comparisons (Fig. 1D; Supplemental Table S2).
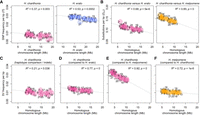
Patterns of lineage-specific sequence distribution and chromosome lengths. (A) Correlation between chromosome lengths and single nucleotide polymorphism (SNP) frequency (nucleotide diversity, π) for H. charithonia and H. erato. H. charithonia SNPs were obtained by comparing the two genome pseudohaplotypes. The H. erato SNPs were obtained from whole-genome resequence data of ten H. e. demophoon samples from Panama. Note that the higher nucleotide diversity in H. erato likely results from its larger population size. (B) Correlation between chromosome lengths when only considering homologous sequence and substitutions (pairwise nucleotide differences, DXY) averaged for each chromosome between H. charithonia and H. erato and H. melpomene, respectively. DXY was calculated from homologous sequences in the pan-genome. (C) Correlation between homologous chromosome lengths and frequency of indels in the chromosomes of H. charithonia. Correlation between homologous chromosome lengths and frequency of lineage-specific sequences in the chromosomes of (D) H. charithonia compared with H. erato, (E) H. charithonia compared with H. melpomene, and (F) H. melpomene compared with H. charithonia. Dashed lines indicate regression fit. Numbers indicate chromosome numbers. Colors refer to sequences specific to H. charithonia (pink), H. erato (blue), and H. melpomene (orange). See Supplemental Figure S2 for pattern in 1 bp indels, structural variants (SVs) between 2 and 50 bp, SVs larger than 1000 bp, and SVs characterized as TEs.
We next examined if the abundance of SVs across the genome is similarly affected by linked selection as SNP diversity. In Heliconius, there is a negative relationship between average nucleotide diversity (i.e., average pairwise nucleotide differences) and chromosome size, with larger chromosomes generally carrying lower diversity (Fig. 3A; Martin et al. 2019; Cicconardi et al. 2021). In the case of nucleotide diversity and chromosome size, this negative relationship has been explained by an increased reduction of genetic diversity at linked sites by greater background selection and genetic hitchhiking on larger chromosomes (Cutter and Payseur 2013; Campos and Charlesworth 2019; Cicconardi et al. 2021). Genetic linkage maps suggest that there is on average a single crossover per meiosis, regardless of chromosomal length (Davey et al. 2016). This results in longer chromosomes having a lower per base recombination rate, which increases the extent of linked selection and results in lower nucleotide diversity on larger chromosomes. However, if SVs have a higher maladaptive mutation load because of their size, we might expect the opposite pattern in which shorter chromosomes with higher recombination rates were able to purge SVs more easily through recombination (Hill and Robertson 1966). Thus, there might be a positive relationship between SV frequency and chromosome length. Our data are most consistent with the hypothesis that SVs are affected by linked selection in a manner similar to SNPs. Indeed, between the two pseudohaplotypes of H. charithonia, there was a significant negative relationship between the indel frequency in each chromosome and chromosome sizes (Fig. 3C). This suggests that the general SV frequency in a population may be driven by linked selection similar to SNPs. Patterns of the frequency of lineage-specific sequences may then have been largely driven by patterns of ancestral diversity, resulting in higher frequencies of lineage-specific sequences on smaller chromosomes (Fig. 3D–F), as is also observed for pairwise nucleotide divergence patterns between, for example, H. charithonia and H. erato and H. melpomene (Fig. 3B; Van Belleghem et al. 2018). This relationship between SV frequency and chromosome length holds for SVs of different size classes (1 bp indels, 2–50 bp, and >1000 bp; Supplemental Fig. S2).
The expectation of linked selection similarly affecting SNPs and SVs is further borne out on the sex (Z) chromosome (21), where there was a reduction in SV frequency that roughly mirrored the patterns of SNP diversity. Because of its hemizygous state in females, there is a smaller effective population size (0.75 relative to autosomes) and an expected reduction in SNP diversity (Charlesworth 2001). For indels within H. charithonia, we found a 0.77 ratio of indel frequency on Chromosome 21 compared with the autosomes, suggesting that indels are subject to differences in effective population size similarly to SNPs.
We next characterized the distribution of SVs relative to genes to further explore the potential maladaptive impact of SVs. TEs, the most abundant SVs, are argued to most often have a neutral or negative impact and end up silenced by genome defense mechanisms (Okamoto and Hirochika 2001; Rigal and Mathieu 2011). If intergenic TEs impact gene functioning negatively, we expected to identify fewer TEs in gene-rich regions. Moreover, if TEs negatively impact gene regulation, we expected their distances from the 5′-end of genes (as a proxy for the TSS) to be further compared with a random sample of genome positions. In agreement with the former expectation, the frequency of lineage-specific TEs correlated negatively with gene frequency (R2 = −0.27, P < 0.001; Fig. 4A), suggesting a general purifying selection against SVs and TEs in gene-dense regions. The distance distribution of TEs to TSS was significantly higher than random expectations although visually similar (Fig. 4B), which may reflect their tendency to randomly insert in the genome in terms of genomic position.
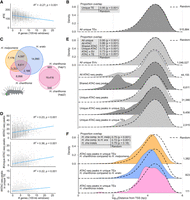
Lineage-specific sequences and their relationship with chromatin accessibility and gene distribution. (A) Correlation of gene density in 100-kb windows with frequency of transposable elements (TEs). (B) Density plot of distance of lineage-specific TEs to closest transcription start site (TSS) pooled over all species genome comparisons. (C) Lineage-specific and shared open chromatin signals (ATAC-seq peaks) found in head tissue of 5th instar caterpillars in each species. Peaks are considered shared (homologous) when they overlap at least 50% reciprocally. (D) Correlation of gene frequency in 100-kb windows with frequency of all lineage-specific structural variants (SVs), all ATAC-seq peaks, lineage-specific ATAC-seq peaks, and lineage-specific TE insertions with ATAC-seq peaks. (E) Density plot of distance of lineage-specific sequence features to closest TSS pooled over all species genome comparisons. We found the distribution of lineage-specific structural variants (SVs) was most similar to a random distribution of positions in the genome (overlapping index = 95%), with a median/mean distance of 21,701/40,790 bp of a lineage-specific sequence and 20,801/39,908 bp of any random position to a TSS. (F) Density plot of distance of lineage-specific TEs with ATAC-seq peaks in H. charithonia to closest TSS. Dashed lines show the distance distribution to TSS of 100,000 randomly selected positions. Tables at the top left in panels B, E, and F report overlapping indexes and pairwise Wilcoxon test P-values between the distributions of lineage-specific sequence features and the random positions. Numbers on the right indicate the number of the respective sequence features.
Genomic landscape of DNA accessibility and functional potential of TEs
Although the genome-wide distribution patterns of SVs and TEs seem to be affected by linked selection, we next wanted to investigate the functional and adaptive significance of lineage-specific intergenic SVs. TEs, for example, have been suggested to be important genomic material for cis-regulatory element evolution (Pontis et al. 2019; Branco and Chuong 2020; Fueyo et al. 2022). To test this, we studied the genomic distribution of potential cis-regulatory elements (CREs) using Assays for Transposase-Accessible Chromatin using sequencing (ATAC-seq) (Buenrostro et al. 2013). We obtained ATAC-seq data for head tissue from fifth instar caterpillars, a tissue labile to adaptive change (Montgomery and Merrill 2017; Montgomery et al. 2021) and a developmental stage that can be confidently timed (Reed et al. 2007) to minimize differences in developmental rates between species that could otherwise also cause differences in ATAC-seq profiles. In H. melpomene, H. erato, and H. charithonia, we counted, respectively, 21,708, 28,264, and 21,097 ATAC-seq peaks that significantly represented open chromatin (Fig. 4C). Of these peaks, 6611 (13.8%) of the total recorded peaks were identified as homologous (overlapped at least 50% reciprocally between all three species), whereas 31,066 were lineage-specific. Although some of the lineage-specific chromatin accessible peaks may result from differences in developmental timing between the three species and some signals may not have reached ATAC-seq peak calling thresholds in one of the species (i.e., false negatives), we find that out of these 31,066 lineage-specific peaks, 9456 (30.4%) were within SVs of which 2915 (9.4%) could be annotated as TEs.
If open chromatin indeed correlates with active gene regulation, we expected to find more ATAC-seq peaks in gene-dense regions of the genome. In agreement with such active gene regulation, ATAC-seq peaks were indeed enriched in regions of the genome with higher gene density (R2 = 0.23, P < 0.001; Fig. 4D). This positive correlation with gene density was also observed for ATAC-seq peaks that were lineage-specific (R2 = 0.36, P < 0.001), and ATAC-seq peaks that were within lineage-specific SVs and TEs (R2 = 0.22, P < 0.001), which supports that they may also have cis-regulatory activity. Moreover, these ATAC-seq peaks were closer to TSS than random and 180 were within 500 bp from a gene's TSS (Fig. 4E; Supplemental Table S4).
We next investigated if the distribution of lineage-specific ATAC-seq peaks within TEs closer to TSS may have been caused by inserting in open chromatin, or whether these TE insertions may have caused the open chromatin and have been selectively retained at these positions. For this, we need to consider that the ATAC-seq peaks identified in the head tissue could also be accessible in the germline in which TE insertions must occur to be heritable. We looked for chromatin signals in homologous sequences flanking the TEs in the other species and found that 395 (13.5%) out of 2915 lineage-specific TEs with ATAC-seq peaks had a significant ATAC-seq signal in the other species within 2000 bp of homologous sequence flanking the insert. This was higher than an expected 2% obtained from 1000 random permutations of an equal number of TEs that did not associate with ATAC-seq peaks. Nevertheless, 2520 (86.4%) did not have any ATAC-seq signal in the other species. To further test whether these TEs have been selectively retained closer to TSS, we performed a TSS distance distribution comparison of SVs within ATAC-seq peaks specific to H. charithonia (Fig. 4F). A comparison relative to H. erato and H. melpomene showed significantly closer TSS distances of lineage-specific sequences with ATAC-seq peaks compared with random (Wilcoxon P-value < 0.001), whereas the distribution of indels with ATAC-seq peaks within the single H. charithonia individual was not statistically different compared with a random distribution of positions in the genome (Wilcoxon P-value = 0.18).
Although the distribution of ATAC-seq peaks within TEs can fit selective retention of these SVs, we wanted to directly test for the influence of selection using selective sweep analysis. Given the demographic history of our taxa and using an effective population size of two million individuals (Moest et al. 2020), it is important to recognize that our ability to identify signals of adaptation is restricted to selection acting within the past 80,000 yr (0.6% of the studied evolutionary timescale). Under these restricted conditions, we did not find a pattern of recent adaptive evolution (Supplemental Fig. S3). We did observe that TE insertions associated with open chromatin were more fragmented compared with other TEs in the genome (Supplemental Fig. S4).
In several studies, TEs have been correlated to evolutionary changes in chromatin state, gene expression, and adaptive evolution at a genome-wide scale (Bourque et al. 2018; Liu et al. 2019; Diehl et al. 2020; Ohtani and Iwasaki 2021). The TE family composition of TEs that associated with open ATAC-seq peaks was markedly different between the three Heliconius species (Supplemental Fig. S5). To infer the evolutionary potential of the accumulated TE families, we next identified enrichment of sequence motifs in ATAC-seq peaks that are within lineage-specific TE insertions and investigated their potential as TF binding sites. TF binding motif enrichment analysis on the 2915 lineage-specific ATAC-seq peaks within TE insertions showed that each genome has unique signals of binding site enrichment with significant similarity to binding sites of TFs in Drosophila (Supplemental Fig. S6). Moreover, nine of the identified 21 enriched binding motifs resembled binding sites of TFs with known functions in nervous system development in Drosophila (Supplemental Fig. S6).
Finally, by comparing the head ATAC-seq data to that of developing wing tissue, we looked for head-specific chromatin changes within lineage-specific TE insertions. The tissue-specific accessibility of these TE insertions would provide indications that these SVs interact with tissue-specific factors and could provide strong candidates as targets of adaptive evolution. We identified 24 head-specific ATAC-seq peaks within a lineage-specific TE insertion that were not accessible in wing tissues (Fig. 5; Supplemental Table S5). Of these, 2, 4, and 18 were specific to H. charithonia, H. erato, and H. melpomene, respectively. Five were located <50 kb from genes with known functions in nervous system development in Drosophila. In H. melpomene, this included the gene sloppy paired 2 (slp2) that also showed TF binding site enrichment in lineage-specific ATAC-seq peaks within a TE (Supplemental Fig. S6).
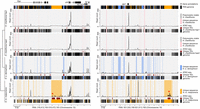
Example intervals of the pan-genome assembly of H. charithonia (pink), H. erato (blue), and H. melpomene (orange) with alignment of lineage-specific genome sequences, transposable element (TE) annotations, and ATAC-seq profiles in the pan-genome coordinate space. The plots show an illustrative interval of the pan-genome assembly near the gene chiffon (chif) and sloppy paired 2 (slp2) that highlights sequences present in each of the genomes relative to the pan-genome (black shading underneath each of the graphs), lineage-specific sequences in each of the genomes (pink, blue, and orange shading in graphs), TEs that overlap with lineage-specific sequences (dark red), and ATAC-seq profiles for head tissue (average of two biological replicates). Gray shading in the H. charithonia haplotype 2 (hap2) graph indicates an indel in the genome of a single H. charithonia individual. Red stars indicate ATAC-seq peaks with head-specific accessibility (compared with wing tissue) that intersect with a lineage-specific TE insertion. See Supplemental Figure S7 for additional examples of intervals around tropomodulin (tmod) and Mitofilin (Mitofilin). The Supplemental Material provides code to reproduce similar plots for any region in the pan-genome.
Discussion
In Heliconius, the extent of SV within and between species has been previously limited to studies of the repetitive sequence content within individual reference genomes (Lavoie et al. 2013; Ray et al. 2019), collinearity of genomes (Davey et al. 2017; Cicconardi et al. 2021), structural rearrangements in a “supergene” related to a color pattern polymorphism (Joron et al. 2011; Edelman et al. 2019), and duplications that likely underestimated the extent of SV as a result of stringent confidence cutoffs needed when using short-read sequences (Pinharanda et al. 2017). Our approach combined four high-quality Heliconius genome assemblies, including two pseudohaplotypes, with a pan-genome alignment to quantify the extensive uniqueness between these genomes owing to SVs. For example, genome-wide nucleotide diversity (π) obtained from SNPs was 0.007 within H. charithonia and DXY (average pairwise nucleotide differences) ranged from 0.11 between H. charithonia and H. erato to 0.15 between H. charithonia and H. melpomene. This suggests an average sequence divergence of 0.7% between the haplotypes of H. charithonia and 11%–15% of sequence divergence between homologous parts of the genomes of these species. In contrast, SV analysis showed that an additional 18.0% of the genome of H. charithonia included hemizygous indel sequences and up to 43.5% and 62.7% of additional genomic differences between H. charithonia and H. erato and H. melpomene, respectively, resulted from SVs.
In contrast to Heliconius populations from Panama, we observed a population size decline over the past 200 ky for H. charithonia from Puerto Rico, which fits with divergence time estimates from mitochondrial DNA (mtDNA) of the Puerto Rican population (Davies and Bermingham 2002). This implies that the indel diversity as estimated from the pseudohaplotypes of the single Puerto Rican H. charithonia individual in this study may be a general underestimate of indel proportions in other species or populations such as those from Panama. These populations may thus carry a high genomic fraction that is subject to presence/absence variation. Although the total SV and SNP frequencies (estimated per bp) were similar between the H. charithonia pseudo-haplotypes, substitutions were 3.7–3.8 times more frequent than SVs when comparing species. Notably, this change in relative frequencies of SNPs and SVs could be largely ascribed to more single bp indels between the pseudohaplotypes of H. charithonia compared with interspecies comparisons and may indicate that as the length distribution of SVs shifts to larger sizes in interspecies comparisons, negative selection against SVs may become stronger compared with SNPs. Despite this marked difference in frequencies, linked selection seems to similarly affect SNPs and SVs, indicating that most SVs are similarly affected by genetic drift and that many may be selectively neutral.
Next, using ATAC-seq data, we assessed the extent to which differences in chromatin accessibility resulted from SVs and TE insertions. We observed that out of the 515,884 SVs identified as lineage-specific TE insertions, only 0.56% were associated with changes in chromatin accessibility between species. However, out of the 31,066 identified lineage-specific changes in chromatin accessibility, 30.4% were within SVs and 9.4% were characterized as lineage-specific TEs. We also note that the absolute number of functional elements within SVs and TEs may be much higher than what is described in our study because we restricted our chromatin data to only one tissue type and developmental time point. As a comparison, a genomic study across 20 mammalian genomes spanning 180 Myr of evolution identified roughly half of all active liver enhancers specific to each species, but argued that most of these lineage-specific enhancers evolved through redeployment of ancestral DNA and that a significant contribution of repeat elements to enhancer evolution was only found for more recently evolved enhancers <40 Myr old (Villar et al. 2015).
Although we did not find any indication of recent adaptive evolution using a selective sweep analysis, our observations indicate an important potential role of TEs in generating genetic variation with functional effects through changes in chromatin state and potentially the regulation of nearby genes. First, even if SVs are mostly neutral or deleterious, their shear abundance and association with chromatin accessibility differences between species underscores their adaptive potential. Second, we observed a pattern in which TE insertions associated with open chromatin were closer to TSS only in interspecies comparisons, not among the H. charithonia haplotypes. This pattern could have potentially arisen over time if TE insertions closer to TSS have a higher chance of affecting gene expression and being involved in adaptive changes between species. Third, we observed that TE insertions associated with open chromatin were more fragmented compared with other TEs in the genome, which may suggest stronger selection for immobilization or adaptive change of these TE insertions (Joly-Lopez and Bureau 2018). Fourth, lineage-specific TEs that underlie changes in chromatin accessibility included 21 enriched motifs with significant similarity to Drosophila TF binding sites. These included lola, Dref, shn, Hr51, slp2, wor, esg, Btd, and Fer1 with functions in neural development (Wimmer et al. 1993; Ashraf et al. 1999, 2004; Sato and Tomlinson 2007; Iyer et al. 2013; Kozlov et al. 2017; Guo et al. 2019). Three other TF motifs have been previously linked to wing or color pattern development in Lepidoptera. Mad is a TF linked to wing development in H. melpomene (Baxter et al. 2010). Mitf has been associated with color pattern development in other animals (Poelstra et al. 2015; Mallarino et al. 2016), and in Heliconius butterflies potentially interacts with aristaless (Westerman et al. 2018). Finally, dsx controls sex-limited mimicry patterns in Papilio polytes and Zerene cesonia butterflies (Nishikawa et al. 2015; Rodriguez-Caro et al. 2021). Moreover, 24 TE insertions had head-specific accessibility compared with wing tissues and provide strong candidates as targets of adaptive evolution.
In conclusion, our comparative genome-wide quantification strategy for SVs showed they can underlie more than 10-fold sequence differences compared with SNPs between two haploid genomes of a single individual. Such remarkable differences in genome content are also becoming more obvious in other comparative genome studies that incorporated SVs in their analysis, including comparisons between humans and chimpanzee for which genome similarity is much lower than the 99% estimated from the first comparative genomic studies that only considered SNPs and small indels (The Chimpanzee Sequencing and Analysis Consortium 2005; Suntsova and Buzdin 2020). Similar to many other organisms, the biggest proportion of these genomic differences is mainly explained by TE accumulation (Garcia-Perez et al. 2016; Cerbin and Jiang 2018). Moreover, examples are accumulating of SVs and, in particular, TE insertions as the mutational changes underlying adaptive phenotypic variation (Schrader and Schmitz 2019). For example, in the bird genus Corvus, adaptive evolution of plumage patterning, a premating isolation trait, was found to be the result of a TE insertion that reduced the expression of the NDP gene (Weissensteiner et al. 2020). Several examples also come from the genomes of Lepidoptera. In the classic example of industrial melanism of the peppered moth, a novel 21-kb TE insertion that impacts the function of the gene cortex is responsible for the development of the different color morphs (Van't Hof 2016). Another TE insertion has been linked to the silencing of a cortex regulatory region and may be responsible for the yellow band on the hindwing in geographic variants of H. melpomene butterflies (Livraghi et al. 2021). In Colias butterflies, an alternative life history strategy that involves resource allocation to reproductive and somatic development and wing color polymorphism was mapped to a TE insertion near the homeobox transcription factor gene BarH-1 (Woronik et al. 2019). In a pair of Papilio species, a female-limited mimetic polymorphism has been linked to a supergene including doublesex (dsx) and recombination suppression in this supergene has been suggested to result from TE accumulation (Iijima et al. 2018). In Lycaeides butterflies, SVs have been shown to be strongly selected in hybrid zones and contribute to hybrid fitness and reproductive isolation (Zhang et al. 2022). Altogether, these examples and our pan-genome study suggest that TE insertions coupled to gene regulation may be an underappreciated source of variation for natural selection to act on. We expect that the accumulation of high-quality genome assemblies generated by long-read sequencing technologies will continue to improve the identification of SVs and highlight their importance in generating adaptive genetic variation.
Methods
Heliconius charithonia haploid genome assemblies
For H. charithonia, we extracted high-molecular-weight DNA from a flash frozen pupa obtained from a wild-caught female sampled in San Juan, Puerto Rico using QIAGEN Inc. Genomic-tip 100/G. Library preparation using 10x Chromium technology for linked reads (10× Genomics) and Illumina sequencing was performed by Novogene Co., Ltd., which generated 44.9 Gb for a target coverage of 100×. We assembled the linked-read sequencing data using the Supernova 2.1.1 assembler (Weisenfeld et al. 2014) using the default recommended settings and a maximum number of reads of 200 million. Raw assembly outputs were transformed to FASTA format using the pseudohap2 option to generate two parallel pseudohaplotypes from the diploid genome. Quality control of the H. charithonia genome was performed using genome-wide statistics calculated on the phase blocks, synteny with the H. melpomene v2.5 genome using Tigmint v1.2.3 (Jackman et al. 2018), and using benchmarking universal single-copy ortholog (BUSCO) analysis with the lepidoptera_odb10 database to assess genome assembly and annotation completeness (Simão et al. 2015). Fragmented H. charithonia scaffolds were ordered with Tigmint using synteny with the H. melpomene v2.5 genome.
Pan-genome alignment
In comparison to using a single genome as a reference, a pan-genome represents a composite of different genomes and serves as a global reference with which to make comparisons between genomes (e.g., conservation and unique sequences) or genome features (e.g., gene and TE annotations). We aligned the two newly assembled haploid H. charithonia genomes with the H. e. demophoon and H. m. melpomene genome using seq-seq-pan (Jandrasits et al. 2018). Seq-seq-pan extends the functionality of the multiple genome aligner progressiveMauve (Darling et al. 2010) by constructing a composite consensus or pan-genome that includes both homologous sequences or locally collinear blocks (LCBs) as well as lineage-specific (nonhomologous) sequences in each of the genomes. This pan-genome is then used as the reference coordinates space for the multi genome alignment, which can then include sequences specific to any of the genomes. We used the H. e. demophoon v1 reference genome as the first genome in the genome list so that the resulting pan-genome alignment would be ordered according to the H. e. demophoon reference. This resulted in a pan-genome sequence with a total length of 659,350,588 bp. To avoid spurious feature mappings (i.e., TEs and ATAC-seq peaks), we excluded scaffolds that have not been linked to chromosome positions in H. e. demophoon in further analyses by cutting the pan-genome alignment at the end of Chromosome 21 (position 578,665,626 in the alignment). The absence and presence of genome sequences in each of the genomes relative to the pan-genome was assessed with a custom Python script that generates a BED file of start and end positions of LCBs and nonhomologous sequences. These BED files were used to identify lineage-specific or homologous sequences between genomes using BEDTools v2.27.1 (Quinlan and Hall 2010). Lineage-specific sequences were obtained by first recording sequence coordinates of each genome relative to the pan-genome using a custom Python script and intersecting these coordinates of each genome against a merged library of sequence coordinates of all other genomes using BEDTools.
Transposable element (TE) annotation and analysis
To identify TEs, we used a two-stage strategy combining the programs RepeatModeler2 (Flynn et al. 2020) and RepeatMasker (Tarailo-Graovac and Chen 2009) using available curated TE libraries as well as novel TE discovery. In the first stage, RepeatModeler 2.0.1 was run on the four genomes for de novo identification of TEs, to classify them into families, and merge the results into a single library. We used the Perl script “cleanup_nested.pl” from the LTR_retriever package (Ou and Jiang 2018) with default parameters to reduce redundant and nested TEs. The TE library was then filtered to eliminate all sequences shorter than 200 bp and all sequences that matched any non-TE-related genes using a Blast2GO homology search (Conesa et al. 2005) with the insect-only default library (nonredundant protein sequence nr v5). Finally, the filtered TEs were matched with the Heliconius specific TE library from Ray et al. (2019) using Blast2GO. This library was produced with de novo TE annotations of 19 Heliconiinae, including H. erato and H. melpomene. The remaining sequences with a TE annotation from RepeatModeler that did not match the Heliconius specific TE library from Ray et al. (2019) were analyzed with different strategies appropriate for the transposon type. First, the putative autonomous elements (DNA, LTR, and LINE) were analyzed with Blast2GO against the insect-only default library. DNA and LTR elements had to have at least a TE-derived transposase and/or match with other DNA/LTR elements. The LINE required the presence of a reverse transcriptase. Second, the putative SINEs were searched in SINEbase (Vassetzky and Kramerov 2013) and accepted only if at least one of their parts (head, body, tail) matched with a SINE element in the database. Third, the putative Helitrons were identified using DeepTE with the parameters -sp M -m M -fam ClassII (Yan et al. 2020). TEs identified as Helitrons were then scanned with CENSOR (Kohany et al. 2006) to confirm their origin. From these analyses we annotated an additional 93 TEs compared with Ray et al. (2019). These TEs were labeled as “putative TEs” and were added to the library from Ray et al. (2019) to obtain the final library.
In the second stage, we used the nonredundant library as a custom library in RepeatMasker 4.1.0 to annotate the TEs within our genomes. The RepeatMasker results were cleaned with “one code to find them all” (Bailly-Bechet et al. 2014). This script combines fragmented RepeatMasker hits into complete TE copies and solves ambiguous cases of nested TE. We identified TE families that have been differentially active between phylogenetic branches using a chi-square test with false discovery rate correction. We characterized temporal variation of Metulj-7 and Metulj-m51, two TEs that showed the strongest temporal changes in activity, using the percent of divergence compared with the TE library reference sequence obtained from RepeatMasker, corrected with the Jukes-Cantor model. Finally, TE fragmentation was calculated based on the total length of each element recovered from the reference library.
ATAC-seq library preparation
ATAC-seq libraries were constructed as in Lewis and Reed (2019), a protocol modified from Buenrostro et al. (2013), with minor modifications. H. melpomene rosina and H. erato demophoon butterflies were collected in Gamboa, Panama; H. charithonia butterflies were collected in San Juan, Puerto Rico. Two caterpillars of each species were reared on their respective host plants and allowed to grow until the wandering stage at 5th instar. Live larvae were placed on ice for 1–2 min and then pinned and dissected in 1× ice cold PBS. Using dissection scissors, the head was removed, and incisions were performed between the mandibles and at the base of the vertexes. Fine forceps were then used to remove the head cuticle to expose the tissue below. The brain and eye-antennal tissue was subsequently dissected out, by removing the remaining cuticle still attached to the tissue. Similarly, developing wings were dissected from the 5th instar caterpillars and the left and right forewing and left and right hindwing were pooled, respectively.
The tissues were then submerged in 350 µL of sucrose solution (250 mM D-sucrose, 10 mM Tris-HCl, 1 mM. MgCl2, 1× protease inhibitors) inside 2 mL dounce homogenizers for tissue homogenization and nuclear extraction. After homogenizing the tissue on ice, the resulting cloudy solution was centrifuged at 1000 rcf for 7 min at 4°C. The pellet was then resuspended in 150 µL of cold lysis buffer (10 mM Tris-HCl, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630 [Sigma-Aldrich], 1× protease inhibitors) to burst the cell membranes and release nuclei into the solution. Nuclear concentration in each sample was checked using a microscope with a counting chamber. This concentration was used to assess the number of nuclei, and therefore DNA, to be exposed to the transposase. This number was fixed on 400,000, as it is the number of nuclei required to obtain the same amount of DNA from an ∼0.4 Gb genome, such as that of H. erato and H. charithonia, as is contained in 50,000 human nuclei—the amount of DNA for which ATAC-seq is optimized (Buenrostro et al. 2013). For H. melpomene this number was 500,333, where the genome size of H. melpomene is 0.275 Gb. For this quality control, a 15-µL aliquot of nuclear suspension was stained with trypan blue, placed on a hemocytometer and imaged at 64×. After confirmation of adequate nuclear quality and assessment of nuclear concentration, a subsample of the volume corresponding to 400,000 nuclei (H. erato and H. charithonia) and 500,333 (H. melpomene) was aliquoted, pelleted 1000 rcf for 7 min at 4°C and immediately resuspended in a transposition mix, containing Tn5 enzyme (Illumina DNA Prep) in a transposition buffer. The transposition reaction was incubated at 37°C for exactly 30 min. A PCR Minelute Purification Kit (Qiagen) was used to interrupt the tagmentation and purify the resulting tagged fragments, which were amplified using custom-made Nextera primers and a NEBNext High-fidelity 2× PCR Master Mix (New England Labs). The amplified libraries were quantified on a Qubit, visualized on an Agilent Bioanalyzer 2100 and sequenced as 37–76 bp paired-end fragments with NextSeq 500 Illumina technology (Supplemental Table S6).
ATAC-seq data analysis
Raw Illumina reads were filtered for adapters and quality using Trimmomatic v0.39 (Bolger et al. 2014). Filtered reads for each sample were then mapped to their respective reference genome using Bowtie 2 v2.2.6 (Langmead and Salzberg 2012) using default parameters. We used SAMtools v1.2 (Li et al. 2009) to sort mapped reads and only retained reads with a mapping Phred score higher than 20 (-q 20) and that were uniquely mapped and properly oriented (-f 0 × 02). PCR duplicates were identified and removed using Picard-tools v2.5 (http://picard.sourceforge.net).
ATAC-seq peak intervals were called on the mapped reads (BAM files) of each sample using the MACS2 “callpeak” command with –g set to the respective reference genome size and –shift set to −100 and –extsize set to 200 (Zhang et al. 2008). Peaks were only retained if they occurred in both replicates with a reciprocal minimal 25% overlap, as determined with BEDTools intersect function. The function “multicov” from BEDTools was used to obtain read counts within ATAC-seq peaks. These read counts were used to obtain library size scaling factors using the function “estimateSizeFactors” from the R package DESeq2 (Love et al. 2014; R Core Team 2018). Next, BAM files were converted to BEDGraphs using the BEDTools function “genomecov” and scaled using the size scaling factors. Mean ATAC-seq traces for each species were obtained from the two replicate samples using WiggleTools (Zerbino et al. 2014). Differential accessibility between head and wing tissues was tested in each species using DESeq2 (Love et al. 2014) with an adjusted P-value smaller than 0.05 and fold change larger than 1.
Feature mapping to pan-genome coordinates and comparisons
Features, including genome sequences that are lineage-specific, TE annotations from RepeatMasker, gene annotations (obtained from H. e. demophoon), and ATAC-seq peaks from MACS2, were compared after converting their genome coordinates to pan-genome coordinates. This was performed by first using the “map” utility of the seq-seq-pan software (Jandrasits et al. 2018) and custom scripts. Features that overlapped with scaffold starts or ends in any of the genomes were masked using BEDTools “subtract” (-A) to avoid including results from fragmented or missing sequences. Next, lineage-specific sequences were intersected with TE annotations and ATAC-seq peaks using BEDTools “intersect”. We only considered ATAC-seq peaks (with an average size of 500.45 bp [standard deviation = 283.57]) that were completely within an SV to be considered resulting from SV. ATAC-seq analyses are thus performed on the faction of SVs larger than 50 bp. Lineage-specific sequences in one of the genomes that did not match a TE annotation were identified as duplications when identifying a BLAST hit with a similarity higher than 70% elsewhere in the genome using BLAST v2.10.0.
Feature distribution
We measured the genomic distance along the pan-genome of lineage-specific sequences, TEs, and ATAC-seq peaks from the closest TSS of a gene using the function “annotatePeaks” from the software suite HOMER (Heinz et al. 2010). Each distribution was compared with that of 100,000 random positions with a pairwise Wilcoxon test. For each distribution pair an overlapping index was measured, using the R package overlapping v1.6 (Pastore 2018).
Motif enrichment
Differential motif enrichment analysis was performed for ATAC-seq peaks that overlapped with lineage-specific TEs using the STREME tool from the MEME suite (Machanick and Bailey 2011; Bailey 2021). This was performed for four phylogenetic comparisons: H. charithonia compared with H. erato, H. charithonia compared with H. melpomene, H. erato compared with H. melpomene, and H. melpomene compared with H. erato. As a background model, we constructed a custom data set including a combined set of lineage-specific TEs without ATAC-seq peaks from the phylogenetic comparisons. Motifs with a P-value smaller than 0.001 were analyzed with Tomtom from the MEME-suite to identify motifs similar to transcription factor binding sites in Drosophila melanogaster (Gupta et al. 2007).
Historical population demography
Changes in historical population sizes from individual genome sequences were inferred using the pairwise sequentially Markovian coalescent (PSMC) as implemented in MSMC (Schiffels and Durbin 2014). Genotypes were inferred using SAMtools v0.1.19 (Li et al. 2009) from reads mapped to the respective reference genomes using BWA v0.7 (Li and Durbin 2010). This involved a minimum mapping (-q) and base (-Q) quality of 20 and adjustment of mapping quality (-C) 50. A mask file was generated for regions of the genome with a minimum coverage depth of 30 and was provided together with heterozygosity calls to the MSMC tool. MSMC was run on heterozygosity calls from all contiguous scaffolds longer than 500 kb, excluding scaffolds on the Z Chromosome. We scaled the PSMC estimates using a generation time of 0.25 yr and a mutation rate of 2 × 10−9 as estimated for H. melpomene (i.e., spontaneous Heliconius mutation rate corrected for selective constraint [Keightley et al. 2015; Martin et al. 2015]). We obtained whole-genome resequencing reads for H. e. demophoon and H. m. melpomene from two individuals each from Panama obtained from the NCBI BioSample database (https://www.ncbi.nlm.nih.gov/biosample/) under accession numbers SAMN05224182, SAMN05224183, SAMEA1919255, and SAMEA1919258 from Van Belleghem et al. (2018). For H. charithonia, we obtained resequencing data for one sample from Panama (BioSample: SAMN05224120 from Van Belleghem et al. [2017]) and two samples from Puerto Rico (BioSample: SAMN05224121 from Van Belleghem et al. [2017] and one using the 10x linked-read sequencing data used for the genome assembly from the Puerto Rican population).
Signatures of selective sweeps
SweepFinder2 (Degiorgio et al. 2016) was used to detect signatures of selective sweeps in genomic regions with ATAC-seq peaks with lineage-specific TEs. Genotypes from 10 H. erato demophoon and 10 H. melpomene rosina individuals from Panamanian populations were obtained from Van Belleghem et al. (2018). Allele counts for biallelic SNPs were generated using a custom Python script. SNPs were polarized using H. hermathena and H. numata for the H. erato and H. melpomene population, respectively. SweepFinder2 was run using default settings and set to test SNPs every 2000 bp (-sg 2000).
Data access
The 10x Chromium sequencing and ATAC-seq raw read data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA795145 (SAMN24661992 and SAMN24689923-SAMN24689940). The H. charithonia pseudohaplotypes have been submitted to DDBJ/ENA/GenBank under accession number JAKFBP000000000. Code for the analyses is available as Supplemental Material and at GitHub (https://github.com/StevenVB12/Genomics).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Christine Jandrasits for advice in using seq-seq-pan, Markus Möst for help with running SweepFinder2, and Simon H. Martin for help in interpreting chromosomal indel diversity patterns. We also thank Silvia Planas from the Sequencing and Genotyping Facility of the University of Puerto Rico-Rio Piedras for assistance with genome and ATAC-seq library preparation and sequencing. This work was supported by a National Institutes of Health-4 NIGMS COBRE Phase 2 Award—Center for Neuroplasticity at the University of Puerto Rico (Grant No. 1P20GM103642) to S.M.V.B., a Puerto Rico Science, Technology & Research Trust Catalyzer Award (#2020-00142) to S.M.V.B. and R.P., and a National Science Foundation (NSF) EPSCoR RII Track-2 FEC (Grant No. OIA 1736026), an NSF IOS 1656389, and a Fondo Institucional para la Investigación (FIPI), Universidad de Puerto Rico–Recinto de Río Piedras, Decanato de Estudios Graduados e Investigación to R.P. For sequencing and computational resources, we thank the University of Puerto Rico Sequencing and Genotyping Facility IDeA Networks of Biomedical Research Excellence (INBRE) Grant No. P20 GM103475 from the National Institute for General Medical Sciences (NIGMS), a component of the National Institutes of Health (NIH), and the Bioinformatics Research Core of the INBRE. The contents of this work are solely the responsibility of the authors and do not necessarily represent the official view of NIGMS or NIH.
Author contributions: This study was conceived and designed by S.M.V.B. with contributions from A.A.R., R.P., J.J.L., L.L., B.A.C., W.O.M., C.D.J., J.A.R.-M., A.G., and S.H.M. S.M.V.B. and A.A.R. analyzed the data. L.L., L.H., Y.O.-R., E.E., and S.M.V.B. collected ATAC-seq data. Y.O.-R. and S.M.V.B. performed genome sequencing. F.C. performed genome quality analyses. A.A.R. and S.M.V.B. wrote the initial manuscript with input and edits from all authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276839.122.
- Received April 16, 2022.
- Accepted September 13, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








