
Mosaic loss of Chromosome Y in aged human microglia
- 1Center for Molecular Medicine and Therapeutics, University of British Columbia, Vancouver, British Columbia V5Z 4H4, Canada;
- 2Department of Neurology, Harvard Medical School, Boston, Massachusetts 02115, USA;
- 3Paul G. Allen School of Computer Science and Engineering, University of Washington, Seattle, Washington 98195-2350, USA
Abstract
Mosaic loss of Chromosome Y (LOY) is a common acquired structural mutation in the leukocytes of aging men that is correlated with several age-related diseases, including Alzheimer's disease (AD). The molecular basis of LOY in brain cells has not been systematically investigated. Here, we present a large-scale analysis of single-cell and single-nuclei RNA brain data sets, yielding 851,674 cells, to investigate the cell type–specific burden of LOY. LOY frequencies differed widely between donors and CNS cell types. Among five well-represented neural cell types, LOY was enriched in microglia and rare in neurons, astrocytes, and oligodendrocytes. In microglia, LOY was significantly enriched in AD subjects. Differential gene expression (DE) analysis in microglia found 172 autosomal genes, three X-linked genes, and 10 pseudoautosomal genes associated with LOY. To our knowledge, we provide the first evidence of LOY in the microglia and highlight its potential roles in aging and the pathogenesis of neurodegenerative disorders such as AD.
A growing body of research has found that mosaic loss of Chromosome Y (LOY) is a common postzygotic structural mutation in males (Forsberg et al. 2014; Thompson et al. 2019). Recent estimates using the UK Biobank suggest ∼40% of men over age 70 harbor detectable LOY affecting >5% of peripheral immune cells (Thompson et al. 2019). Robust associations have been found between LOY and a diverse set of age-related diseases, including hematologic (Forsberg et al. 2014; Cáceres et al. 2020; Ouseph et al. 2021) and nonhematologic cancers (Forsberg et al. 2012, 2014; Cáceres et al. 2020), macular degeneration (Grassmann et al. 2019), cardiovascular disease (Sano et al. 2022), and Alzheimer's disease (AD) (Dumanski et al. 2016; Caceres et al. 2020). Despite these correlations, the role of LOY in disease physiology and its molecular mechanisms are not well understood. Current knowledge supports the hypothesis that hematopoietic LOY events arise through mitotic missegregation errors that increase in frequency with declining systemic genomic stability and impaired DNA repair capabilities (Wright et al. 2017; Terao et al. 2019; Thompson et al. 2019). This widespread genomic instability drives the aforementioned disease associations (Thompson et al. 2019; Guo et al. 2020), but at the same time, recent studies suggest that LOY could directly lead to disease through immune system dysfunction and tumor suppressor/oncogene dysregulation (Thompson et al. 2019; Dumanski et al. 2021; Mattisson et al. 2021; Sano et al. 2022). Further investigation is required to better understand LOY and its role in disease.
Most LOY research has been conducted using readily accessible tissues like blood, buccal mucosa, or tumor, and comparatively little is known about LOY properties in CNS tissue (Dumanski et al. 2016; Kimura et al. 2018; Graham et al. 2019). Nevertheless, early evidence suggests LOY does occur in the brain. Studies using WGS (Graham et al. 2019) and qPCR (Kimura et al. 2018) have found modest but significant indications of age-dependent LOY in human dorsolateral prefrontal cortex (DLPFC) tissue. Extreme down-regulation of Chr Y, a proxy for genomic LOY, has also been observed in multiple human brain regions and was shown to increase risk of AD development (Caceres et al. 2020). Others (Orta et al. 2021) have hypothesized that proliferative CNS cell types such as microglia (Askew et al. 2017; Réu et al. 2017) and oligodendrocyte (OL) progenitor cells (OPCs) (Fernandez-Castaneda and Gaultier 2016) could be more prone to LOY accumulation than terminally differentiated cell types (i.e., neurons, OLs). Indeed, activated microglia show a considerable proliferative capacity that could increase the likelihood of LOY-causing missegregation events during aging and neurodegeneration (Bellver-Landete et al. 2019).
Recent studies have quantified LOY through bulk (Graham et al. 2019; Caceres et al. 2020; Cáceres et al. 2020; Dumanski et al. 2021) and single-cell RNA-seq (scRNA-seq) (Thompson et al. 2019; Dumanski et al. 2021; Mattisson et al. 2021) profiling. When using the same set of samples, LOY readouts from RNA-seq and array-based genotype technologies show robust pairwise correlation, providing confidence in the accuracy of each platform independently (Dumanski et al. 2021). Further, recent advances in single-cell multimodal profiling have enabled deeper insight into the molecular characteristics and potential disease mechanisms of LOY cells (Stoeckius et al. 2017; Reyes et al. 2019; Swanson et al. 2021). For example, a CITE-seq study investigating LOY in leukocytes found simultaneous reduction of expression and surface protein abundance of CD99, a gene commonly affected by Chr Y loss (located in the pseudoautosomal region [PAR] shared between Chromosome X and Y) (Mattisson et al. 2021).
This work aims to consolidate existing scRNA-seq and single-nuclei RNA-seq (snRNA-seq) data sets to better understand the presence of LOY in the brain, specifically in the context of neurodegenerative disease. To enable comparison between data sets, we develop a LOY adjustment method that accounts for donor and cell type–specific variation in gene expression sparsity. Using this combined data set, we assess the cell type–specific burden of LOY and its downstream impact on gene expression across five major brain cell types. Here, we provide the first evidence of LOY in the microglia and highlight its potential roles in aging and the pathogenesis of neurodegenerative disorders such as AD.
Results
Using multimodal single-cell profiling to define LOY quantification parameters
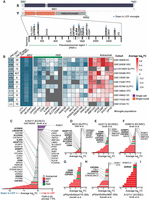
To better understand scRNA-seq LOY estimation accuracy and to define informed quality control (QC) thresholds for future analyses, we first investigated LOY using a multimodal ATAC and gene expression (GEX) snRNA-seq data set from B cell lymphoma–affected lymph node tissue (10x Genomics 2021; Methods). In total, 7283 nuclei passed initial QC (more than 1500 unique molecular identifiers [UMIs] and more than 800 genes; Methods). Nuclei were clustered and annotated with cell types using GEX levels (Fig. 1A; Supplemental Fig. S1A). As previously described (Thompson et al. 2019; Dumanski et al. 2021; Mattisson et al. 2021), the LOY status of each cell was determined using the complete lack of expression from 12 commonly expressed genes located in the male-specific Y Chromosome (MSY) region (Methods; Supplemental Fig. S2). Cells with detectable MSY expression were classified as non-LOY/normal, and those without were classified as LOY. Of all included nuclei, 25.4% (1855/7283) were classified as LOY (Fig. 1B). A vast majority of LOY nuclei (90.8%) were found in the B cell lymphoma cluster, which was foreseeable as LOY is common in many male neoplasms (Forsberg et al. 2014; Cáceres et al. 2020; Ouseph et al. 2021). Next, we tested the agreement between RNA and chromatin accessibility-based estimates of LOY. To classify LOY using snATAC-seq, we computed gene activity scores that estimate genome-wide chromatin accessibility in each nucleus by summing gene-level fragment counts (Methods; Fig. 1C). Nuclei lacking gene activity from all MSY genes were classified as LOY. When using all available data, we found LOY nuclei (classified using GEX) had significantly reduced MSY gene activity (mean = 0.005; SD = 0.231) compared with normal nuclei (mean = 0.051; SD = 0.0498; Wilcoxon P < 0.001) (Fig. 1C,E). As expected, this was a Chr Y–specific pattern that was not observed on other chromosomes (Supplemental Fig. S1B). Similar trends were observed in the isolated B tumor cluster (Fig. 1F). Overall, LOY calls agreed between snRNA-seq and snATAC-seq readouts in 88.8% of nuclei with sufficient multimodal sequencing depth (Fig. 1D). As we selected for nuclei with deeper RNA sequencing, the agreement between the multimodal assays increased and MSY accessibility scores in LOY nuclei approached zero (Fig. 2A). For example, when we selected for nuclei with more than 3000 expression UMI, mean MSY gene activity scores from LOY nuclei were 0.001 (SD = 0.011; n = 1232), whereas non-LOY MSY activity scores remained stable (mean = 0.047; SD = 0.044; n = 2602). For this reason, subsequent analyses estimating LOY frequency via the transcriptome used UMI thresholds of more than 3000 and feature thresholds of more than 1000. Consistent trends were observed for each detected MSY gene (Fig. 2B), which we visualized individually for ZFY and UTY (Supplemental Fig. S3). In each case, accessible peaks in the LOY group were largely absent, although small peaks were observed, likely representing errantly classified LOY nuclei and/or sequencing noise. Together, these findings support previous assumptions (Dumanski et al. 2021) that increasingly stringent UMI and feature thresholds reduce false-positive LOY calls as more transcriptional information is available and the effect of stochastic Chr Y gene dropout is reduced. Additionally, the agreement between multimodal assays instills confidence that 10x sc/snRNA-seq can effectively detect LOY events.
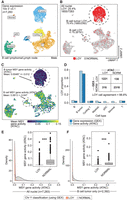
snRNA-seq agrees with snATAC-seq when classifying Y Chromosome loss using a multimodal data set. (A) UMAP projection and cell type identities after clustering 7283 thyroid nuclei (snRNA-seq) from a male donor with non-Hodgkin's lymphoma. (B) snRNA-seq clustering of LOY nuclei (red) and non-LOY/normal nuclei (gray; more than 3000 nUMI and more than 1000 nGene). (C) UMAP projection (snRNA-seq) colored by snATAC-seq-derived gene-activity scores of male-specific Y genes using Signac. (D) LOY estimates from ATAC and gene expression assays for each cell type. Inset table shows total nuclei arranged by LOY classification and assay. ATAC and gene expression LOY classification agreed in 88.8% of nuclei. (E,F) Density of ATAC gene activity in LOY (red) and normal (gray) nuclei for all nuclei (E) and B cell cancer nuclei only (F). Wilcoxon, one-sided: (***) P < 0.001.
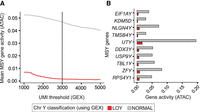
Setting UMI depth quality-control thresholds using multimodal assays. (A) Mean gene activity (snATAC-seq) of LOY and non-LOY/normal nuclei (classified using snRNA-seq) as the minimum UMI count threshold is increased (snRNA-seq). Black line indicates the UMI depth threshold used in subsequent LOY analyses (more than 3000 UMI). (B) Mean gene activity of each detected MSY gene for LOY (red) and non-LOY/normal nuclei (gray).
Microglia show elevated LOY frequencies
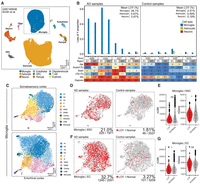
To characterize cell type–specific LOY frequencies in the human brain, we processed 30 publicly available scRNA-seq and snRNA-seq data sets (Fig. 3A; Supplemental Table S1). For simplicity, we will refer to both single-nuclei and single-cell transcriptomes as cells. Our combined data set yielded 873,742 male cells (more than 3000 nUMI; more than 1000 nGene) from 310 donors. After QC filtering (Methods; Supplemental Fig. S4), 851,674 cells from 259 donors and 13 brain regions were included for LOY analysis (Fig. 3B). Donors included subjects with AD (n = 60), frontotemporal/Lewy body dementias (other dementias; n = 17), Parkinson's disease (PD; n = 12), Huntington's disease (HD; n = 11), amyotrophic lateral sclerosis (ALS; n = 11), and multiple sclerosis (MS; n = 4). The control population consisted of subjects lacking a neurodegenerative diagnosis (n = 144). Age data were available for 250 donors (mean age = 64.03, age range = 4–96) (Supplemental Fig. S5). For further details, see Supplemental Figure S2 and Methods.
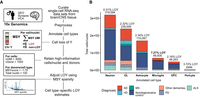
Summary of brain LOY analysis workflow and combined sc/snRNA-seq brain data set. (A) Summary of methods used to estimate LOY in the brain. For additional information, see Supplemental Figure S2 and Methods. (B) Total number of included cells/nuclei annotated to each major brain cell type, colored by neurodegenerative diagnosis. Total nuclei/cell sample size and net LOY frequency are listed above each bar. Only droplet-based 10x Genomics sc/snRNA-seq data sets were considered for the study. Cells/nuclei with more than 3000 UMI counts and more than 1000 genes were included in LOY frequency analysis.
We tested each cell in our combined brain data set for Y Chromosome presence (Fig. 3A). We found LOY cells represented 1.73% (n = 14,775) of the total population and were observed in 253 of 259 donors (Supplemental Table S2). When LOY was quantified across five major brain cell types, we observed elevated LOY in microglia populations. Of 69,835 tested microglia, 7.21% were classified as LOY, greater than OPCs (2.16%, n = 48,293), OL (2.34%, n = 239,009), astrocytes (1.11%, n = 170,097), and neurons (0.31%, n = 319,594) (Fig. 3B; Table 1). When LOY frequencies were summarized for each cell type within each donor, enrichment was again observed in microglia (Supplemental Fig. S6A; Supplemental Data S2).
Summary of Y Chromosome loss estimation across major brain cell types using scRNA-seq
After our initial analysis, we noticed MSY expression sparsity remained a significant confounder despite filters applied on donor/cell type populations. When MSY expression was sparse in the Y-expressing cells of a population, LOY estimates were often elevated (Supplemental Fig. S7A). To adjust donor-level LOY estimates, we developed a method that uses the positive residuals of an exponential decay model fit between LOY percentage and MSY expression sparsity (Methods; Supplemental Fig. S7A,C). After applying the adjustment, LOY enrichment in microglia became more pronounced (Fig. 4A; Supplemental Fig. S6B), and correlations with technical confounders were reduced (Supplemental Fig. S7B). In microglia populations, individuals displayed a mean adjusted LOY frequency of 5.14% (range = 0%–56.4%, n = 94), which was significantly greater than all other brain cell types (Wilcoxon, P < 0.05) (Fig. 4A).
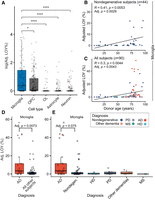
Loss of Chromosome Y (LOY) is common in human microglia and is associated with aging and AD. (A) Mean adjusted LOY percentages for each major brain cell type in each subject. Each point represents a cell type–specific LOY estimate in an individual donor (more than 100 cells). Wilcoxon: (*) P < 0.05, (***) P < 0.001. (B,C) Association between donor age and microglia LOY percentage in nondisease control subjects (B) and all available subjects (C). Points are colored by neurological diagnosis. Black lines indicate linear regression correlation (Pearson's correlation). Adjusted P-values are derived from a multiple linear regression model that accounts for nUMI, 10x chemistry, and MSY sparsity (Methods). (D) Adjusted LOY percentage in AD microglia (n = 24) compared with all other subjects (n = 64). (E) Adjusted LOY percentages contrasted between AD, nondisease control, and other neurodegenerative diseases. (E,D) Adjusted P-values are from a multiple linear regression model that accounts for age.
In control donors, LOY increased significantly with age in microglia (P = 0.0029) and OPCs (P = 0.049) but not astrocytes (P = 0.23) or OLs (P = 0.286) (Fig. 4B; Supplemental Fig. S8B), consistent with significant LOY beyond noise level only in microglia and OPCs. When including all donors (control and neurodegerative), age-LOY associations remained significant in microglia populations (P = 0.0043) (Fig. 4C). All LOY associations with age were also observed using unadjusted LOY values (Supplemental Fig. S8A).
Next, we examined the association between LOY and neurodegenerative disease status, adjusting for sample size, MSY sparsity score, mean UMI depth, 10x chemistry, and donor age (Methods; Supplemental Fig. S9). We found LOY frequency was significantly elevated in AD donor microglia (mean = 11.87%, n = 26) compared with all other donors (mean = 2.72%, n = 64, P = 0.00739) (Fig. 4D). LOY enrichment remained modestly significant when AD microglia were tested against control donors alone (P = 0.075) (Fig. 4E; Supplemental Fig. S10A,C). Furthermore, despite limited sample size, we observed increased LOY frequency in HD donor OPCs (n = 8, P = 0.00054) (Supplemental Fig. S10B,D). Although further analysis is required to validate these findings, it suggests LOY may accumulate within several brain cell types in a disease-specific context.
Transcriptional impact of LOY
We next investigated differentially expressed (DE) genes between LOY and non-LOY cells: (1) as a secondary validation for LOY estimation and (2) to identify transcriptional events that help explain LOY processes. First, we focused on the diploid PARs (PAR1 and PAR2) that are shared between the distal ends of Chromosomes X and Y (Fig. 5A). LOY causes hemizygous loss of the PAR, which can induce expression reduction of several dose-sensitive genes in the region (Raznahan et al. 2018; Zhang et al. 2020; Astro et al. 2022). Thus, a significant reduction in PAR expression can act as an independent transcriptional biomarker of LOY, accompanying our main approach, which classifies LOY using null Chr Y expression. In agreement, microglia showed greater PAR expression than other brain cell types, and microglia LOY populations had significantly reduced PAR expression (P = 3.6 × 10−12; paired Wilcoxon) (Supplemental Fig. S11A). General PAR expression loss was also observed in OPCs (P = 7.5 × 10−8). When the data were stratified by cohort, we again found a significant reduction of cumulative PAR gene expression in 12 microglia and 10 OPC LOY populations (P < 0.05) (Supplemental Fig. S11B).
Decreased PAR gene expression is common in LOY microglial populations. (A) Schematic of the human sex chromosomes and pseudoautosomal regions (PARs). The PARs are homologous sequences shared between Chromosomes X and Y. Blue triangles indicate PAR genes with significant expression reduction in at least one donor microglia LOY population (FDR < 0.05). (B) Heatmap of microglia DE genes across several single-cell/nuclei cohorts with at least 100 LOY cells/nuclei (average logFC). If multiple brain regions were sampled within a given data set, they were split by brain region and labeled accordingly. The total number of significant, non-MSY DE genes (P < 0.1) detected per data set is provided on the left. The second column displays cumulative logFC of all expressed PAR genes. Rows are colored based on source of nucleic acid (whole cell or nuclei). Bonferroni significance is provided within each cell: (*) P < 0.1, (**) P < 0.01, (***) P < 0.001. (C–I) Average log fold change (logFC) of DE genes between LOY and non-LOY cells/nuclei within microglia from seven subjects, including the following: (C) A163/17 (AD; GSE160936); (D) MCI3 (MCI; syn12514624); (E) A277/12 (AD; GSE160936); (F) A096/14 (AD; GSE160936); (G) pPDsHSrSNxi4775 (PD; GSE178265); (H) pPDsHSrSNxi3887 (PD; GSE178265); and (I) A127/11 (control; GSE160936) (more than 2000 UMI and more than 1000 nGene; FDR < 0.05).
We next focused our DE analysis on 15 subjects (19 samples) with sizeable microglial LOY populations (more than 50 LOY cells). Of the included samples, eight showed independent overrepresentation of PAR genes in their LOY populations (hypergeometric test, FDR < 0.1) (Fig. 5C–I; Supplemental Table S4). To improve statistical power for genome-wide differential gene expression analysis, microglia transcriptomes from included donors were pooled together, normalized, and tested for LOY DE. From the pooled analysis, we identified 193 microglial LOY associated transcriptional event (mLATE) genes (P < 0.1; Bonferroni) (Supplemental Fig. S12A). Of these, 41 autosomal and six PAR genes were significantly dysregulated independently in multiple subjects, suggesting a potential LOY-specific signature (Fig. 5B; Supplemental Fig. S11C; Supplemental Table S3).
Autosomal mLATE genes displayed diverse functions in CNS development, neurodegeneration, and cancer-promoting processes, including roles in axon guidance (ROBO1, DOCK1, PTPRM), immune cell and glioma migration (ROBO1, SLIT2), EMT-associated splicing regulation (RBFOX2), phagocytosis (DOCK1, CD163), neuron–glia interaction (ST6GALNAC3), cell division and polarization (PARD3B), cell adhesion (DSCAM), and lipoprotein metabolism (APOE, APOC1, ABCA1). Roundabout guidance receptor 1 (ROBO1), part of the SLIT/ROBO signaling pathway, was the most overexpressed mLATE gene (avg log2FC = 0.75; P = 7.42 × 10−69) and was significantly up-regulated independently in eight subjects. SLIT/ROBO genes were rarely expressed in non-LOY microglia in any brain data set (Supplemental Fig. S13). Additionally, SLIT2 was observed as a significant mLATE gene (avg log2FC = −0.17; P = 0.022). The SLIT2/ROBO1 signaling pathway is an interesting target for future LOY studies as it is known to influence tumor-associated microglia/macrophage (TAM) behavior in glioblastoma (GBM) (Geraldo et al. 2021), and overexpression is observed in ∼20% of male GBMs (Gao et al. 2013).
Pathway enrichment analysis using Metascape (Zhou et al. 2019) identified 44 significantly overrepresented pathways (P < 0.01; Bonferroni) (Supplemental Table S5), including cholesterol and phospholipid efflux, lipoprotein assembly, regulation of proliferation, inflammatory response, and nervous system development (Supplemental Fig. S12B). These results allude to the possibility of increased LOY in activated or inflamed microglia. Inflamed, phagocytic microglia clearing injured tissue, amyloid beta, and other debris proliferate rapidly, potentially leading to increasingly frequent LOY events. We additionally compared our mLATEs with 489 LATEs observed in the peripheral leukocytes of aging men (Dumanski et al. 2021) and found significant overlap (Fisher's exact test P = 0.01) (Supplemental Table S3). Overlapping autosomal LATE genes included TMEM176B, S100Z, TMEM71, CD226, B2M, SCMH1, LITAF, and IL15, again highlighting roles in immune function and inflammation.
LOY is observed across all microglia subtypes
The Smith et al. (2022) data set contained deeply sequenced nuclei transcriptomes in AD and control donors from paired brain regions. This provided a quality opportunity to compare regional LOY in glial cells at higher resolution (Fig. 6A,C; Supplemental Fig. S14A,B; see Supplemental Results). Consistent with our initial analysis, (1) LOY cells were frequent in microglia and rare in neurons and astrocytes (Fig. 6B; Supplemental Fig. S15), and (2) LOY frequency was elevated in AD microglia (28.1%) compared with controls (2.51%). Furthermore, LOY microglia displayed similar sequencing metrics to normal microglia, providing confidence that LOY calls are representative of the underlying biology and are not a result of gene dropout (Fig. 6E,G). Interestingly, we found microglia LOY was consistently elevated in entorhinal cortex (EC) samples compared with paired somatosensory cortex (SSC) samples (Fig. 6B). In the SSC, 21.0% of AD microglia were classified as LOY compared with 1.81% in controls (Fig. 6D), whereas in the EC, LOY frequency was 32.7% in AD and 3.27% in controls (Fig. 6F). Elevated LOY in the EC of AD donors is of interest as evidence suggests the EC is uniquely prone to proteopathies (Kaufman et al. 2018) and is thought to be the first brain region affected by AD (Gómez-Isla et al. 1996; Kobro-Flatmoen et al. 2021).
LOY frequency is elevated in AD microglia across the entorhinal cortex (EC) and somatosensory cortex (SSC). (A) UMAP projection and cell type annotations using snRNA-seq from 48,748 AD and nondisease control brain tissue nuclei (GEO: GSE160936) (Smith et al. 2022). (B) LOY frequency in each brain sample for microglia, astrocytes, and neurons. Donors are ordered by AD diagnosis, AD (left) and nondisease control (right). Each donor was sampled from both the EC and SSC. (Bottom) Heatmap displaying additional phenotypic information of each donor/sample. Provided measures of amyloid percentage, Braak score, and pTau percentage were all determined using quantitative image analysis. (C) Microglia nuclei from the SSC (top) and EC (bottom) were integrated and clustered using Seurat. Subclusters were annotated using microglia gene panels (Supplemental Table S8). Homeostatic subclusters (H) are colored in various blues; inflammatory subclusters (I) are colored in oranges; and disease-associated microglia (DAM) clusters are colored in magentas. (D,F) UMAP reductions of SSC (D) and EC (F) showing LOY nuclei clustering patterns facetted by AD diagnosis. Nuclei are colored by LOY (red) and non-LOY/normal (gray). LOY nuclei frequencies are provided at the bottom of each UMAP plot. (E,G) Comparison of UMI counts (nUMI) and detected genes (nGenes) between LOY and normal nuclei. Sequencing attributes do not differ significantly between microglial LOY and normal populations. Wilcoxon test: (ns) P > 0.1, (*) P < 0.1.
Lastly, to further investigate LOY microglia subtype specificity, we focused on nine data sets containing subjects with elevated LOY (>10% LOY). Microglia and other brain macrophage transcriptomes were computationally isolated, reclustered, and annotated for subtypes using published gene sets (Supplemental Fig. S16A–I). In most data sets, LOY proportions were similar across microglia subtypes, although we did observe examples of data set–specific LOY subtype enrichment (Supplemental Fig. S17A,B; Supplemental Table S6). For example, in the Olah et al. (2020) data set, LOY proportion was elevated in the proinflammatory cluster (34.1%, 85/249) compared with all other microglia (18.8%, 488/2583; Supplemental Fig. S17C). In agreement, PAR expression was significantly reduced in this proinflammatory cluster (P < 0.001; Wilcoxon) (Supplemental Fig. S17E). LOY was also elevated in blood-derived tumor-associated macrophage (BDTAM) clusters of the Sayed et al. (2021) data set, which were defined using the overexpression of known BDTAM markers including KYNU and TGFBI as well as published BDTAM gene modules (Supplemental Fig. S17H,J; Müller et al. 2017). In response to brain trauma and disease, peripheral monocytes are known to cross the BBB and populate the CNS (Varvel et al. 2016). BDTAMs showed 22.6% (207/708) LOY compared with 7.9% (628/7914) in microglia (Supplemental Fig. S17I). Accordingly, we tested all other microglia data sets for LOY enrichment in cells expressing peripheral monocyte markers (VCAN, CCL2, and FCN1) (Supplemental Table S7). However, with the exception of the BDTAMs from the Sayed et al. (2021) data set, transcriptomes with monocyte markers were rare and did not commonly show elevated LOY frequencies compared with putative microglia. In the Smith et al. (2022) data set, we observed similar LOY proportions between microglia and a peripheral monocyte cluster (Supplemental Fig. S18). Based on these data, we conclude that brain LOY events likely occur more frequently among brain-resident microglia and macrophages, but blood-derived myeloid cells appear to provide an alternative input of LOY into the CNS. Additionally, LOY appears to affect all types of microglia but may become more subtype specific in response to donor-specific microenvironments and disease states. We note that partitioning data sets by subtype limits effective sample size and increases LOY estimation volatility, which limits the interpretability of these results. LOY-focused sequencing studies are required to fully understand the origin and mechanisms of LOY in microglia and other brain macrophages.
Discussion
Mosaic LOY has been associated with age-related degenerative CNS diseases (Dumanski et al. 2016; Kimura et al. 2018; Graham et al. 2019; Caceres et al. 2020); however, its incidence and mechanisms in the brain are not well understood. To better characterize LOY-affected CNS cell types and to inform future LOY studies, we present an extensive analysis of LOY in the brain using single-cell and single-nuclei transcriptomes. Importantly, we provide evidence that among CNS cell types, LOY occurs most frequently in microglia and causes a transcriptomic signature significantly overlapping that found in peripheral leukocytes with LOY. Moreover, we provide additional evidence that LOY prevalence is enriched in the brain tissue of male AD donors, specifically within the microglia population. We found microglial LOY induces a transcriptional signature involving the dysregulation of 193 genes with roles in aging, glioma biology, and inflammation. Our data support the hypothesis that LOY-causing missegregation events in microglia occur locally in the CNS; however, an isolated case of LOY deriving from infiltrating VCAN+/FCN1+ myeloid cells suggests that multiple mechanisms are likely involved. Although future studies are required to replicate mLATEs and characterize their roles in disease physiology, we postulate that somatic LOY accumulation in the microglia could represent an additional process in age-related dysfunction, leading to chronic inflammation and neurodegeneration.
The importance of microglia in age-related neurodegenerative processes is rapidly emerging (Olah et al. 2020; Pan et al. 2020). Age-related alterations in gene expression lead to dystrophic microglia that are less ramified (Streit et al. 2020), have a reduced ability to phagocytose debris (Gabandé-Rodríguez et al. 2020), and produce greater amounts of proinflammatory cytokines (Wong 2013; Niraula et al. 2017). Further, microglia are long-lived cells (Réu et al. 2017) that are derived from EMPs and turnover locally in the CNS, largely isolated from the periphery (Ajami et al. 2007; Ginhoux et al. 2013). These properties make microglia prone to mutation and selective pressures. Moreover, during the progression of many neuropathologies such as AD, resident microglia rapidly proliferate, providing an increased opportunity for missegregation errors leading to Y loss (Gómez-Nicola et al. 2013). Similar processes occur in aged microglia, where a hallmark, “primed,” proinflammatory profile develops, resulting in heightened proliferation (O'Neil et al. 2018; Costa et al. 2021). Both mechanisms could explain elevated LOY occurrence in microglia and in AD. At the same time, spontaneous, somatic mutations in microglia—made increasingly likely by age-related genomic instability—could directly contribute to the pathogenesis of neurodegenerative disease (Mass et al. 2017). For example, an induced somatic BRAFV600E mutation in murine HPSCs leads to hematological malignancy, but when the same mutation is induced in yolk-sac EMPs, it results in a late-onset neurodegenerative disorder (Mass et al. 2017). As microglia turnover, the mosaic population of BRAFV600E-affected, dysfunctional microglia have a selective advantage leading to dystrophy, chronic inflammation, and ultimately neurodegeneration. Further research is necessary to show similar processes can be induced by LOY in the brain, but the cell type–specific lineage properties of microglia afford plausibility.
Several dysregulated genes in LOY microglia could lead to pathological CNS conditions through processes including immune dysfunction, disruption of homeostatic function, and uncontrolled proliferation. Some potential candidates include the PAR genes CSF2RA and CD99. Within our data set, microglia and CNS-associated macrophages uniquely express CSF2RA (Supplemental Fig. S19). CSF2RA is a receptor subunit for colony stimulating factor 2 (CSF2), which is dysregulated in AD and has known roles in CNS development and microglia homeostasis maintenance (Chitu et al. 2020). Imbalance of CSF1R-CSF2 ratios through CSF2RA deficiency could contribute to chronic inflammation and/or senescence, leading to proneurodegenerative conditions (Chitu et al. 2020). CD99 is a cell-surface glycoprotein that has multifaceted functions in leukocyte cell adhesion, trans-endothelial migration, MHC class I transport, and apoptosis (Pasello et al. 2018). CD99 shows contradicting roles in various cancers, acting as both a oncosuppressor (osteosarcoma, Hodgkin's lymphoma) and oncogene (Ewing sarcoma, malignant glioma) (Manara et al. 2018). Furthermore, CD99 surface protein abundance is reduced in leukocyte LOY populations, establishing a link between mRNA and protein dysregulation (Mattisson et al. 2021). Loss of Y-linked genes could also lead to LOY-associated dysfunction. For example, KDM5D and UTY are histone H3 remodelers regarded as tumor suppressors in prostate cancer and clear-cell renal cell carcinoma (Arseneault et al. 2017) and expressed in microglia (Supplemental Fig. S20). Loss of KDM5D expression and subsequent H3K4me dysregulation has been associated with expediated cell cycle and mitotic entry (Komura et al. 2018). The Y-linked lncRNA LINC00278 is also of interest. Through the disruption of AR signaling pathways, down-regulation of LINC00278 can affect the progression of esophageal squamous cell carcinoma (Wu et al. 2020).
Our study also highlights 172 autosomal genes and three Chr X genes dysregulated in LOY microglia that could plausibly link LOY with immune and CNS dysfunction. For instance, ROBO1, encoding a major receptor in the SLIT/ROBO signaling pathway, was up-regulated in LOY microglia populations from eight subjects (FDR < 0.05). In addition to important roles in neural development, metastasis, and inflammatory cell chemotaxis, SLIT/ROBO signaling displays contradicting tumor-promoting and tumor-suppressing properties in various cancers (Zhao et al. 2018; Jiang et al. 2019; Geraldo et al. 2021). In low-grade glioma (LGG) and GBM, ROBO1 and SLIT2 are up-regulated and are associated with PIK3CG activation, poor survival, and resistance to inhibitor therapy (Geraldo et al. 2021). Despite intriguing roles in glioma metastasis, links to Chromosome Y loss are unknown. However, bulk RNA sequencing in individuals with sex chromosome disorders provides additional evidence that ROBO1 expression is associated with sex chromosome ploidy. Compared with the controls, ROBO1 is significantly up-regulated in Turner syndrome (XO) leukocytes and down-regulated in Klinefelter's syndrome (XXY) (P < 0.0001) (Supplemental Fig. S22; Zhang et al. 2020). Similar patterns are observed for other up-regulated mLATEs, including RBFOX2, DOCK1, and CDC42BPA (P < 0.001), suggesting a sex chromosome ploidy-dependent transcriptional mechanism. Across all tissues, ROBO1 expression is positively correlated with 117 genes that include RBFOX2 and DOCK1 (P < 0.05; Bonferroni) (Miller and Bishop 2021). Further investigation into LOY-associated transcriptional effects (LATEs) is required to determine mechanisms and association with disease.
We acknowledge that AD has an increased prevalence in females, which complicates the AD–LOY relationship (Dumitrescu et al. 2019). Nevertheless, similar to LOY, monosomy X is commonly observed in blood (Liu et al. 2022) and brain tissue (Yurov et al. 2014) of healthy elderly females, with increased rates in AD patients (Yurov et al. 2014; Spremo-Potparevic et al. 2015). Given the dosage-sensitivities of PAR and X-inactivation escape genes, we and others (Bajic et al. 2020) hypothesize that monosomy X and LOY could potentially share mechanisms that contribute to AD development. In agreement, decreased Chromosome X expression is observed in female neurodegenerative brain samples (Swingland et al. 2012) and is associated with age-related cognitive decline and tau pathology (Davis et al. 2021). Future studies on the prevalence and impact of monosomy X in microglia may elucidate additional mechanisms involved in AD development.
Although we provide a valuable exploratory analysis of LOY in the brain, there are limitations with our study. The MSY region used to classify LOY only contains approximately 12 genes that are commonly detected using scRNA-seq, and only 10 that are well expressed (Supplemental Fig. S20). Given the sparsity of 10x data, classifying Chromosome Y status using this small group of genes strongly relies on per cell sequencing depth. Because per cell sequencing depth varies significantly between experiments, batches, chemistries, and cell types, we often observe correlations with technical variables. Although these correlations can be mitigated through strict QC thresholds and MSY sparsity adjustment, these methods can remove a significant proportion of data, which can severely hamper signal. Mattisson et al. (2021) noted similar issues of transcriptional variability when using scRNA-seq and found that cell-surface proteins are significantly more reliable for characterizing LOY. CITE-seq has been applied to CNS immune cells in mice (Golomb et al. 2020), and similar studies in an aging human cohort would be valuable for further characterizing LOY in the CNS. Lastly, our findings linking microglial LOY frequency to AD are based on association and cannot prove a causal relationship. It remains possible that in the brain LOY is a benign passenger mutation that arises in response to increased genomic instability, which is common in AD patients. At the same time, recent evidence suggests hematopoietic LOY can act as a causal pathogenic aberration. Mice given bone marrow grafts with mosaic LOY cells displayed fibrosis, heart dysfunction, and reduced lifespan (Sano et al. 2022). Similar in vivo studies focusing on the brain are required to further clarify the causality of LOY in AD.
In conclusion, using single-cell transcriptomes, we present the first evidence of LOY affecting microglia in the brain, disproportionately affecting elderly donors diagnosed with AD. Our results show microglial LOY affects the expression of hundreds of autosomal genes with diverse functions that require additional experimental investigation. We believe LOY in the microglia could represent an additional, understudied biological process that could alter microglia phenotypes and play a role in male-specific neurodegeneration.
Methods
Single-cell/nuclei data set curation and selection
Single-cell transcriptome data used in this study was acquired from both public and controlled sources (Supplemental Table S1). Publicly available sc/snRNA-seq data sets were primarily downloaded from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/), Human Cell Atlas (HCA), and 10x Genomics data set page (https://www.10xgenomics.com/resources/datasets). Controlled access data were downloaded from Synapse through the AMP-AD Knowledge Portal. We searched for data sets that met the following criteria: (1) were available as raw FASTQ files (single-cell) or unnormalized count matrix (single-nuclei), (2) were generated using the 10x Genomics Chromium platform (3′v2, 3′v3, or 5′v1), and (3) were profiled human samples from primary brain tissue. By using data produced via 10x Chromium library preparation, we attempted to reduce undesired technical variation. In total (without QC), we collected 30 data sets across a diverse set of brain regions that totaled 1,227,300 male cells/nuclei from 310 male individuals (median age: 68; range age: 0–96; nUMI of more than 1500 and nGene of more than 1000). Both male and female cells were included for initial clustering and cell type annotation.
scRNA-seq and snRNA-seq processing
Generating expression matrices (single-cell)
Beginning with FASTQ files, all scRNAseq data sets used in this study (n = 4) were processed through the same bioinformatic pipeline (Supplemental Fig. S2). For each data set, alignment of reads and cell barcode filtering were performed using Cellranger 5.0.0 (10x Genomics). Reads were aligned to the GRCh38 2020-A reference (10x Genomics) using Cellranger count in intron-retaining mode. To include intronic reads, we used the “–include-introns” flag. This flag considers confidently mapped intronic reads as candidates for UMI counting. Although the use of intronic reads is commonly used for samples enriched with pre-mRNA (i.e., snRNA-seq experiments), we observed improved gene detection sensitivity with minimal impact on clustering, which is in agreement with a previous report (10x Genomics 2021, CG000376-Rev A). Our data show when performing LOY estimation using scRNA-seq, increased UMI depth, feature diversity, and cell/nuclei counts improve the power of DE tests. Sample-specific UMI count matrices output from count were aggregated using Cellranger aggr without read count normalization.
Generating expression matrices (single-nuclei)
Most published single-nuclei data sets are generated using intronic information, and therefore, processing through our local pipeline did not provide noticeable benefits. Furthermore, because of patient privacy concerns, several single-nuclei data sets had restricted access to raw FASTQ files. To save time and computational resources, we used publicly available, unnormalized expression matrices for most single-nuclei data sets. Processing details for each data set are available in Supplemental Table S1.
QC, filtering, and clustering
Gene expression matrices were imported into R (v4.0.2) (R Core Team 2022) for further analysis using Seurat (v4.0.2) (Hao et al. 2021). Filters were applied to retain cells/nuclei with adequate gene representation (more than 1000 genes), UMI counts (more than 1500 UMI), mitochondrial UMI percentage (<15%), and ribosomal UMI percentage (<45%). These filters were applied to discard empty droplets and apoptotic cells. scDblFinder (v1.6.0) (https://github.com/plger/scDblFinder) was applied using default settings to identify and remove putative doublets. Normalization, dimension reduction, and clustering was performed using Seurat (Supplemental Methods). Clusters were annotated using known cell type markers (Supplemental Fig. S21). If systemic biases were observed, batch correction was applied using Harmony (v1.0.0) (Korsunsky et al. 2019).
In-depth microglia subcluster analysis was completed for nine data sets, including syn12514624, GSE148822, GSE137444 and GSE160936, GSE178265, GSE167494, GSE183068, GSE174367, and GSE174332 (Supplemental Fig. S16). Smith et al. (2022) enriched for microglia and astrocyte nuclei from the ECs and SSCs of AD diagnosed and control donors, whereas Olah et al. (2020) and Mancuso et al. (2019) enriched for microglia from DLPFC and temporal cortex tissue, respectively. Sadick et al. (2022) enriched for astrocytes (prefrontal cortex) and Gerrits et al. (2021) enriched for microglia and astrocytes from the occipital cortex. The remaining microglia data sets consisted of unsorted nuclei from the substantia nigra, DLPFC, prefrontal cortex, and primary motor cortex (Morabito et al. 2021; Pineda et al. 2021; Sayed et al. 2021; Kamath et al. 2022). Each included data set was initially preprocessed, clustered, and annotated (Supplemental Methods). Only microglia and detected brain macrophages (i.e., CNS-associated macrophages, monocytes) were retained for downstream analysis. Individual samples were integrated using canonical correlation analysis (CCA). Seurat CCA integration was used instead of Harmony when analyzing in-depth subsets and clustering patterns. All other filtering and processing were completed as aforementioned. To annotate biologically relevant subclusters within microglia, we complied gene sets from several published studies and calculated module scores via the Seurat AddModuleScore function (default settings). All gene sets used in the study are provided in Supplemental Table S8.
Loss of Y classification
Our method of determining LOY classification from each single-cell transcriptome was adapted from previous studies (Thompson et al. 2019; Dumanski et al. 2021). Most male cells commonly express several genes located in the male-specific region of Chromosome Y (GRCh38:Y: 2,781,480–56,887,902). In each data set, cells were classified as LOY if they lacked expression of all commonly expressed genes residing on the MSY region, which were defined as MSY genes with more than 0.05 normalized expression and/or with expression in >5% of cells. Ultimately, cells were labeled LOY if they lacked detection of all expressed MSY genes and were labeled normal/non-LOY if they did not. Functions and scripts used to classify LOY can be accessed from GitHub (https://github.com/michaelcvermeulen/microglia-loss-of-y) and in Supplemental Code. Raw LOY values for each cell type in each included sample are available in Supplemental Data S2.
Loss of Y percentage
Initial LOY cell percentages were calculated within each broad cell type cluster, within each subject. Additional filters were applied to limit variability and technical biases. For LOY frequency estimation, we used stricter QC threshold (nUMI more than 3000 and nGene more than 1000) to limit false-positive LOY calls. Cell type populations within each subject with fewer than 100 cells per nuclei were removed to limit undesired LOY variability. Additionally, LOY estimates were strongly associated with the sparsity of male-specific Y (MSY) gene expression. To remove cell type populations with insufficient MSY expression, we filtered the data using a MSY sparsity score (Supplemental Methods). To calculate this score, we iteratively found the percentage of cells expressing each detected MSY gene using the Seurat DotPlot function, subsetting for cells with evidence of Chromosome Y presence (MSY UMI ≥ 1). MSY sparsity values were determined by summing the percentage of expressed values for each cell type cluster/population in each donor. Afterward, MSY sparsity values were scaled and centered. When making LOY frequency estimates, cell type populations with a MSY sparsity score of less than −1.75 were removed (Supplemental Fig. S4).
LOY adjustment using MSY sparsity
We further used MSY sparsity scores to adjust LOY percentage estimates. Using a collection of 1.4 million male single-cell transcriptomes (10x chemistry) from multiple tissues, we calculated the nonlinear least-square best fit using an exponential decay model with the formula: LOY_percent ∼ MSY score (Supplemental Fig. S7). The drm function (drc v3.0-1) was used to fit a nonlinear model to the data using the DRC.expoDecay function (aomisc v0.647) (Supplemental Code). The exponential decay model fit our brain data closely (P < 2.2 × 10−16). Positive residuals were taken as adjusted LOY percentage estimates, and negative residuals were set to zero. Simply, our adjusted LOY represents the deviation above the expected LOY of a donor/cell type population given the MSY expression sparsity of the Y-expressing cells in the population. LOY data used for the adjustment process are available in Supplemental Data S1.
Loss of Y differential gene expression
We identified LATEs by performing DE tests between LOY and non-LOY populations. DE tests were performed on pooled microglia and within the microglia of each cohort and subject. To improve statistical power for DE tests, we reduced the strictness of cell information filters. Minimum UMI and gene filters were set to 2000 and 1000, respectively. DE analysis was performed for each cell type using the MAST algorithm via the Seurat FindMarkers function using both full data sets (tissue specific) and individual subjects. When performing DE on a full data set, latent variables, including sample, percentage of mitochondrial UMI, nUMI, and nGenes were added to the hurdle-model. For tests on individual subjects, we used the percentage of mitochondrial UMI, nUMI, and nGenes as latent variables. Genes were considered if they were expressed in >5% of cells/nuclei.
Gene set enrichment analysis
Genomic region overrepresentation of DE genes was performed using Molecular Signatures Database (MSigDB) gene sets (Subramanian et al. 2005) and the hypergeometric test provided by the hypeR package (Federico and Monti 2020). DE genes with FDR < 0.05 were used, and significance for all gene set enrichment analysis (GSEA) tests was declared using FDR < 0.05. To determine PAR enrichment, the c1.all.v7.4.symbols.gmt file was edited to include PAR1 and PAR2 as independent cytogenic bands. Across our data sets, all PAR2 genes lacked adequate expression so only PAR1 was considered. For pathway enrichment, we used Metascape (Zhou et al. 2019). mLATE genes with FDR < 0.05 were entered into the Metascape webservice, which provides a wide range of well-established gene sets. Gene sets with q < 0.05 were considered significant. The results of the Metascape analysis are provided in Supplemental Table S5.
Single nuclei multi-ome ATAC LOY
The multimodal LOY analysis was performed on sample data provided by 10x Genomics (https://www.10xgenomics.com/resources/datasets/fresh-frozen-lymph-node-with-b-cell-lymphoma-14-k-sorted-nuclei-1-standard-2-0-0) and processed using Signac (Stuart et al. 2021) and Seurat (Supplemental Methods). To quantify the relative activity of each gene based on chromatin accessibility, we used the GeneActivity function (Signac). Within each cell, GeneActivity extracts gene coordinates (extended 2 kb upstream to include promoter) and counts fragments in each region. The gene activity matrix was log normalized and scaled. For each cell, MSY gene activities were calculated by taking the mean normalized gene activity of all detected MSY genes (RPS4Y1, ZFY, TBL1Y, USP9Y, DDX3Y, UTY, TMSB4Y, NLGN4Y, KDM5D, EIF1AY). Cells with null MSY gene activity were labeled LOY.
Other statistical analyses
All statistical analyses were performed using R (v4.0.4). Fisher's exact tests used to determine overlap significance between gene sets were performed using the GeneOverlap package (http://shenlab-sinai.github.io/shenlab-sinai/). Wilcoxon tests were performed using the wilcoxon.test function from the R stats package. Global gene coexpression analysis was performed using correlationAnalyzeR (Bonferroni; P < 0.05) (Miller and Bishop 2021). Plotting was performed using a combination of the Seurat, Signac, ggpubr (https://rpkgs.datanovia.com/ggpubr/), ggplot2 (Wickham 2016), and dittoSeq (Bunis et al. 2020) packages.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We acknowledge the support from the Canadian Institute for Advanced Research (CIFAR) and a Natural Sciences and Engineering Research Council of Canada (NSERC) grant to S.M. This work was supported by the National Institutes of Health (U01AG072572, U01AG061356, RF1NS117446, R01AG055909).
Author contributions: M.C.V. and S.M. conceived the project. M.C.V. performed all analyses and wrote the first draft of the paper. S.M., T.Y.-P., and R.P. provided feedback. All authors contributed to writing and finalizing the draft.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276409.121.
-
Freely available online through the Genome Research Open Access option.
- Received November 19, 2021.
- Accepted August 19, 2022.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








