
A new emu genome illuminates the evolution of genome configuration and nuclear architecture of avian chromosomes
- Jing Liu1,2,16,
- Zongji Wang1,2,3,16,
- Jing Li1,
- Luohao Xu2,
- Jiaqi Liu4,
- Shaohong Feng5,
- Chunxue Guo5,
- Shengchan Chen6,
- Zhanjun Ren7,
- Jinpeng Rao8,
- Kai Wei8,
- Yuezhou Chen9,
- Erich D. Jarvis10,11,
- Guojie Zhang12,13,14,15 and
- Qi Zhou1,2,8
- 1MOE Laboratory of Biosystems Homeostasis & Protection and Zhejiang Provincial Key Laboratory for Cancer Molecular Cell Biology, Life Sciences Institute, Zhejiang University, Hangzhou 310058, China;
- 2Department of Neuroscience and Developmental Biology, University of Vienna, Vienna 1090, Austria;
- 3Institute of Animal Sex and Development, Zhejiang Wanli University, Ningbo 315100, China;
- 4Wuhan Gooalgene Technology Company, Wuhan 430070, China;
- 5BGI-Shenzhen, Beishan Industrial Zone, Shenzhen 518083, China;
- 6Longteng Ecological Culture Company, Limited, Zhashui 711400, China;
- 7Key Laboratory of Animal Genetics, Breeding and Reproduction of Shaanxi Province, College of Animal Science and Technology, Northwest A&F University, Yangling 712100, China;
- 8Center for Reproductive Medicine, The 2nd Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310052, China;
- 9Jianzhou Poultry Industry Company, Limited, Yong'an 366000, China;
- 10Laboratory of Neurogenetics of Language, The Rockefeller University, New York, New York 10065, USA;
- 11Howard Hughes Medical Institute, Chevy Chase, Maryland 20815, USA;
- 12China National GeneBank, BGI-Shenzhen, Shenzhen 518120, China;
- 13State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming 650223, China;
- 14Section for Ecology and Evolution, Department of Biology, University of Copenhagen, DK-2100 Copenhagen, Denmark;
- 15Center for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, Kunming 650223, China
-
↵16 These authors contributed equally to this work.
Abstract
Emu and other ratites are more informative than any other birds in reconstructing the evolution of the ancestral avian or vertebrate karyotype because of their much slower rate of genome evolution. Here, we generated a new chromosome-level genome assembly of a female emu, and estimated the tempo of chromosome evolution across major avian phylogenetic branches, by comparing it to chromosome-level genome assemblies of 11 other bird and one turtle species. We found ratites exhibited the lowest numbers of intra- and inter-chromosomal changes among birds since their divergence with turtles. The small-sized and gene-rich emu microchromosomes have frequent inter-chromosomal contacts that are associated with housekeeping genes, which appears to be driven by clustering their centromeres in the nuclear interior, away from the macrochromosomes in the nuclear periphery. Unlike nonratite birds, only less than one-third of the emu W Chromosome regions have lost homologous recombination and diverged between the sexes. The emu W is demarcated into a highly heterochromatic region (WS0) and another recently evolved region (WS1) with only moderate sequence divergence with the Z Chromosome. WS1 has expanded its inactive chromatin compartment, increased chromatin contacts within the region, and decreased contacts with the nearby regions, possibly influenced by the spreading of heterochromatin from WS0. These patterns suggest that alteration of chromatin conformation comprises an important early step of sex chromosome evolution. Overall, our results provide novel insights into the evolution of avian genome structure and sex chromosomes in three-dimensional space.
Most birds have about 10 pairs of relatively large-size macrochromosomes, including one pair of sex chromosomes (male ZZ, female ZW), and about 30 pairs of smaller microchromosomes, some of which can be hardly discerned by light microscopy (Takagi and Sasaki 1974). It was hypothesized that microchromosomes represent archaic linkage groups of ancestral vertebrates (Ohno et al. 1969; Tegelström and Ryttman 1981; Burt 2002; Uno et al. 2012). This was implicated by reconstruction of the ancestral vertebrate karyotype, first using the genomes of a few (Nakatani et al. 2007), and recently many more available species (Sacerdot et al. 2018). Direct supporting evidence for the hypothesis came from genomic comparisons of chicken versus the amphibians Mexican axolotl and Western clawed frog (Voss et al. 2011), the spotted gar fish (Braasch et al. 2016), and the invertebrate amphioxus (Simakov et al. 2020). These studies found one-to-one correspondence between many but not all chicken microchromosomes versus (micro-)chromosomes of amphibians and gar fish or the reconstructed chromosomes of the vertebrate/gnathostome common ancestor using amphioxus (Simakov et al. 2020), sea lamprey (Smith et al. 2018), or elephant shark (Venkatesh et al. 2014) genome, dating the likely existence of microchromosomes at least to the ancestor of jawed vertebrates.
Chicken microchromosomes tend to be gene-rich and have higher recombination rate and GC content than macrochromosomes (International Chicken Genome Sequencing Consortium 2004). Such a distinct genomic composition probably dictates the segregated nuclear architecture in chicken cells that might also have existed in the vertebrate ancestor. Similar to the small-sized human chromosomes (Cremer et al. 2001), the chicken microchromosomes predominantly occupy the nuclear interior (Habermann et al. 2001), which corresponds to the transcriptionally active or A compartments revealed by Hi-C analysis (Lieberman-Aiden et al. 2009); whereas macrochromosome regions are mainly located at the nuclear periphery (Habermann et al. 2001) and correspond to the inactive heterochromatin or B compartments. The scarcity of high-quality chromosome-level genomes of birds, particularly underrepresentation of identified microchromosome sequences except for chicken, has hampered the reconstruction of the evolutionary trajectories of avian and thus vertebrate chromosome architectures.
We previously generated a near chromosome-level genome of ostrich (Zhang et al. 2015) with Illumina reads and optical-mapping, and found that its Z Chromosome (Chr Z) harbors many fewer inversions than chicken and zebra finch, when all three species were compared to the autosomal counterparts of lizard and snake (Zhou et al. 2014). As Chr Z usually exhibits disproportionately more inversions than any other chromosomes in birds (Hooper and Price 2017), this highly conserved intrachromosomal synteny between ratites and reptiles is very likely a genome-wide pattern. This was confirmed by recent refinements of our ostrich genome (O'Connor et al. 2018). These results are consistent with a much lower genome-wide substitution rate in ratites, associated with their larger body size and longer generation time (Bromham 2011; Jarvis et al. 2014; Wang et al. 2019). Particularly, ratites have a pair of homomorphic sex chromosomes with a much lower pairwise substitution rate, in contrast to the heteromorphic sex chromosomes of most other birds and mammals (Cortez et al. 2014; Zhou et al. 2014). We recently determined over two-thirds of the ratite (except for kiwis) sex-linked regions are still recombining with each other as the pseudoautosomal region (PAR) (Wang et al. 2019; Xu et al. 2019b). In the remaining non-recombining sexually differentiated regions (SDRs), ratites have undergone at least one ancestral recombination suppression (RS) shared by all birds and another lineage-specific RS (Wang et al. 2019), which demarcated the SDRs into two regions of ‘evolutionary strata’ (the older stratum is named as stratum S0 and the younger one as S1) by their different levels of Z/W pairwise sequence divergence (Zhou et al. 2014; Wang et al. 2019). Among the studied ratites, emu and cassowary are even less differentiated between sex chromosomes than ostrich and any other Neognathae species, thus best preserving the ancestral status of avian sex chromosomes (Zhou et al. 2014).
Here, we chose emu, a unique model for studying the evolution of vertebrate chromosome architectures and avian sex chromosomes, for high-quality genome assembly using the cutting-edge third-generation long-read sequencing and Hi-C technologies with the pipeline of the Vertebrate Genomes Project (Rhie et al. 2020). By comparing the new emu genome to other chromosome-level genome assemblies of 11 bird and one turtle species, we estimated the tempo of inter- and intrachromosomal rearrangements in major lineages of birds in order to test the hypothesis that emu and other ratites have the lowest lineage-specific chromosome evolution rate among birds, therefore best representing the ancestral avian genome configuration. We further compared the chromatin architectures between the emu macro- and microchromosomes, and between the Z and W Chromosomes using the liver Hi-C data, in order to gain insights into the nuclear architecture of the avian ancestor and the evolutionary process of avian sex chromosomes in the three-dimensional (3D) nuclear space.
Results
Chromosomal assembly of a female emu genome
To generate a high-quality reference genome of emu, including the repetitive Chr W, we produced 70-fold genomic coverage of Pacific Biosciences (PacBio) reads (subread N50 length 15.5 kb), and long-range linkage data of 124-fold 10x Genomics linked reads, 154-fold Dovetail Chicago data, and 46-fold Hi-C data from three female emu individuals (Fig. 1A). Our preliminary assembly derived from PacBio reads alone produced 1389 gapless contig sequences with an N50 size of 13.3 Mb. Using the three different types of linkage data followed by manual curation, these contigs were oriented and connected into 802 scaffolds with an N50 size of 82.7 Mb. We performed a final gap filling step and assembly polishing using the raw reads of both PacBio and Illumina data that are compensatory to each other and assembled the entire genome into 1.26 Gb (97.9% of the 1.29-Gb estimated genome size by GenomeScope [Vurture et al. 2017]). About 1.20 Gb sequences or 94.9% of the assembled genome have been anchored into 29 autosomes (macrochromosomes Chr 1–Chr 10 and microchromosomes Chr 11–Chr 28 and Chr 33), one Chr Z (82.7 Mb), and one Chr W SDR (7.92 Mb or 29.1% of the homologous Z-linked region, which is 27.2 Mb long) (Supplemental Table S1; Supplemental Fig. S1).
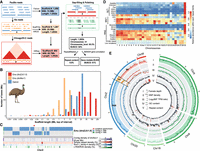
Genome assembly of a female emu. (A) The PacBio long reads were first used to generate contigs, then used various linkage data of 10x linked reads, Chicago, and Hi-C reads to connect the contigs into chromosomal sequences, and finally polished the assembly with corrected PacBio long reads and Illumina reads. (B) Comparison of scaffold length distribution between droZJU1.0 and droNov1 emu assemblies and ostrich assembly, the latter two of which were generated by Illumina reads. (C) An example showing that the new droZJU1.0 assembly is more continuous than droNov1. In almost all the cases, one droZJU1.0 contig corresponds to multiple droNov1 contigs, and regions of high repeat content (e.g., LTR/ERVK) coincide with the breakpoints between contigs. (D) The abundance of each repeat family was normalized to a range from 0 and 1 for each chromosome, respectively. (E) The genomic landscape of emu chromosomes. We showed two macrochromosomes (Chr 5 and Chr 9), two microchromosomes (Chr 18 and Chr 20), and the Z/W sex chromosomes for their genomic compositions. (I) PAR (blue), autosomes (green), S1 (orange) show a twofold higher female read coverage than the S0 and Chr W (red). (II) S1 shows a higher female SNP density than any other genomic regions. (III) S0 shows a male-biased expression pattern. (IV) Microchromosomes have a higher GC content than macrochromosomes. (V) Microchromosomes have a lower repeat content than macrochromosomes.
This version of the emu genome (droZJU1.0) has a 77-fold increase of continuity measured by N50 size relative to a recent Illumina-based assembly (droNov1) (Sackton et al. 2019), including a 7.6-fold increase of the size of assembled Chr W SDR sequences (Wang et al. 2019). The distribution of the scaffold lengths was shifted toward much longer sequences in the droZJU1.0 assembly relative to droNov1 or the most recent ostrich chromosomal assembly (Fig. 1B; Zhang et al. 2015). A major improvement came from the resolution of repetitive sequences that are enriched within or between the previous Illumina scaffolds, by the long reads and long-range linkage information (Fig. 1C). This was reflected by the increased annotated repeat content (9.9% vs. 7.2%) of droZJU1.0 versus droNov1 genomes, which is mainly concentrated at several long terminal repeat (LTR) retrotransposon families (Supplemental Fig. S2). We annotated a total of 20,823 consensus gene models combining homology-based predictions, 14 tissue-specific transcriptomes, and de novo predictions. The structural accuracy of our chromosome assembly was demonstrated by its highly consistent synteny with another emu chromosomal genome independently produced by DNA Zoo from Illumina reads and Hi-C data (Supplemental Fig. S3; Dudchenko et al. 2017). Its completeness was reflected by a high BUSCO gene value (95%) and our annotation of the putative centromeric regions of 26 chromosomes and telomeres of eight chromosomes as well as the interstitial telomeric repeats (ITRs) (Supplemental Table S2; Supplemental Figs. S4, S5), which were corroborated by the published cytogenetic results of the emu (Nanda et al. 2002).
The new emu genome exhibited distinctive genomic features between sex chromosomes and autosomes, and between macro- and microchromosomes. Because of RS, Chr W is expected to accumulate transposable elements (TEs) and diverge from Chr Z in genomic sequences (Charlesworth et al. 2005). This was confirmed by a 3.9-fold increase of TE content, particularly various subfamilies of LTR retrotransposons and DNA transposons on Chr W (10.5%) compared to the rest of the genome (0.88%) (Fig. 1D). We managed to assemble the Z- and W-linked regions of S1 into two separate sequences (ZS1 and WS1) of similar lengths (about 4.8 Mb) that showed the same level of female read depth, but a much higher level of female single-nucleotide polymorphism (SNP) density because of Z/W divergence, compared with autosomes and the juxtaposed long PAR (0 Mb–55.5 Mb, 67.1% of the Chr Z). The ancient stratum S0 (60.3 Mb–82.7 Mb on the Chr Z) that suppressed recombination in the ancestor of birds has become much more repetitive or diverged on the Chr W than S1. Therefore, its W-linked sequence assembly is quite fragmented, and its Z-linked region exhibited half the female read coverage relative to autosomes (Fig. 1E). The two evolutionary strata constituted the SDR of emu sex chromosomes and showed a generally male-biased gene expression pattern of Z-linked genes because of the lack of global dosage compensation (Fig. 1E; Supplemental Table S3; Supplemental Fig. S6; Wang et al. 2014).
The emu macrochromosomes have a significantly higher content of LINE and DNA transposon families (P = 1.16 × 10−5, Wilcoxon test) but a lower content of simple repeats (P = 0.036, Wilcoxon test) than the microchromosomes, with an apparent gradient of change in some TE subfamilies (e.g., L2) (Fig. 1D; Supplemental Fig. S7). Meanwhile, the emu macrochromosomes exhibited lower GC content (P = 9.89 × 10−6, Wilcoxon test) and gene density (P = 3.85 × 10−4, Wilcoxon test) than the microchromosomes (Fig. 1E; Supplemental Figs. S7–S9). These findings indicated that the autosome organization of and relative genomic features in macro- and microchromosomes of the emu are similar to other birds (Burt 2002; International Chicken Genome Sequencing Consortium 2004; Zhou et al. 2014) but are revealed here at a higher and more complete resolution due to the more complete assembly.
Emu and other ratites have best preserved the ancestral avian chromosome configuration
Avian chromosome evolution has been proposed to be dominated by intrachromosomal changes, but this was based on cytogenetic methods of low resolution, with a limited number of species (Griffin et al. 2007), and with major underrepresentation on the microchromosomes. A recent genomic investigation covered nearly 30 bird species but for the Palaeognathae lineage, with 22 of the 31 studied species having only a scaffold-level Illumina-based genome assembly (Damas et al. 2018). With the newly produced more complete emu genome, we were motivated to compare its chromosome evolution rate versus those of other Neognathous birds to quantitatively delineate a finer picture of the tempo of avian chromosome evolution. Using the sea turtle (Chelonia mydas) (Wang et al. 2013; Dudchenko et al. 2017) as an outgroup, we identified the inter- and intrachromosomal rearrangements of 12 birds with all chromosome-level genomes (Supplemental Tables S4, S5).
All but eight of the assembled emu chromosomes together accounted for 87.3% of the genome that was each mapped to one single homologous chromosome in the turtle (Fig. 2A). The eight outlier emu chromosomes were still aligned to four turtle chromosomes, which can be the result of either four chromosome fusions in the turtle lineage or four fissions in the avian lineage. To discriminate between the two scenarios, we inspected other reptile species and found five emu outlier chromosomes (Chr 4, Chr 10, Chr 14, Chr 23, and Chr 25) were mapped to one single chromosome in rattlesnake (Fig. 2B; Schield et al. 2019), whereas among others, the same combination of two emu chromosomes that were mapped to one turtle chromosome could not be found in rattlesnake or alligator (Fig. 2C; Dudchenko et al. 2017; Rice et al. 2017). For example, emu Chr 6 and Chr 12 were homologous to turtle Chr 7 but were homologous to parts of snake Chr 5 and Chr 2, respectively. Therefore, it is likely that the eight outlier emu chromosomes were also ancestrally single chromosomes that have undergone independent fusions or translocations in other reptile species. Similar to emu, all the other investigated birds showed few fusions or fissions of chromosomes compared to the turtle (Supplemental Fig. S10), except for the golden eagle (Aquila chrysaetos) (Dudchenko et al. 2017; Van Den Bussche et al. 2017), where we identified four fissions and 10 fusions/translocations (Fig. 2D) after it diverged from the California condor (Gymnogyps californianus) (Dudchenko et al. 2017). A lack of any identified fusions or fissions in the condor indicated that the extensive interchromosomal changes are not a universal feature of birds-of-prey. Overall, these results provided evidence that the common ancestor (Archelosauria) of birds and turtles had almost the same karyotype as that of emu, where certain other bird lineages went on to evolve more chromosomal rearrangements.
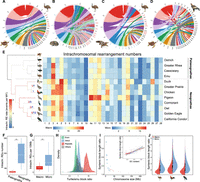
Tempo of avian chromosome evolution. (A–D) Genomic synteny between green sea turtle, rattlesnake, American alligator versus emu and green sea turtle versus golden eagle, respectively. Chromosome names with blue/red color denote the fused chromosomes in reptiles and the homologous chromosomes of emu, respectively. Each chromosome is indicated by the first letter of species name and chromosome number. (E) Chromosomal rearrangements across all major phylogenetic branches of birds. The phylogenetic branches (Jarvis et al. 2014; Claramunt and Cracraft 2015) are color-coded according to the respective average rate of intrachromosomal changes, and the numbers with different colors indicate those of detected chromosomal fissions (red) and fusions (blue). The intrachromosomal rearrangement number per chromosome of birds compared to sea turtle is shown in the heat map with a different color scale for macro- and microchromosomes, given their drastically different size and rearrangement numbers. (F) Paleognaths show more rearrangements per chromosome than neognaths. (***) P < 0.0005. (G) Microchromosomes have a higher rearrangement number per 10 Mb length than macrochromosomes. (***) P < 0.0005. (H) The distributions of the length ratios of syntenic blocks comparing turtle versus emu across different types of genomic regions, which indicate that the major source of sequence loss in birds is from repeat regions. (I) The outer dot plot shows the correlation between the overall turtle versus emu syntenic length ratio per chromosome versus the size of the chromosome (blue for microchromosomes, red for macrochromosomes). The size of the dots is scaled to the average GC content of each chromosome. The inner plot shows the positive correlation between GC content and turtle versus emu syntenic length ratio. Each dot represents one syntenic block, with the red ones for the macrochromosome blocks and the blue ones for the microchromosome blocks. (J) Microchromosomes have higher turtle versus birds syntenic length ratios than macrochromosomes, suggesting that microchromosomes experienced more severe sequence loss in birds.
We found much greater variations of intrachromosomal rearrangements (inversions and translocations) among the studied birds using the turtle as an outgroup (Fig. 2E; Supplemental Table S6). By determining the presence or absence of orthologous rearrangement regions of the focal species in its related species, we inferred the rate of intrachromosomal rearrangements on each phylogenetic branch based on parsimony (Methods). Overall, ratites have undergone significantly fewer genomic rearrangements (P < 2.20 × 10−16, Wilcoxon test) including both intra- (Fig. 2F) and interchromosomal changes than Neognathae species after they diverged from the avian ancestor, supporting that the ratites have better preserved the ancestral avian genomic configuration than any other birds. The intrachromosomal evolution rates at the external lineages are significantly lower (P = 0.022, t-test) than those at the internal lineages of ratites. This reflects the impacts of independently evolved gigantism and elongated generation time among ratites (Sackton et al. 2019). In contrast, the intrachromosomal evolution rate, greatly accelerated at the ancestor of Neognathae, accompanied by their lineage species radiation (Jarvis et al. 2014), was maintained at a high level along the internal branches and then became decelerated in most of the external branches (Fig. 2E; Supplemental Figs. S11, S12).
Some chromosomes (e.g., Chr 3) seem to be a hotspot for rearrangements across all the investigated birds, whereas some (e.g., Chr 10) seem to be highly conserved for their gene synteny between all species (Fig. 2E). The cause for such variations of the numbers of rearrangements between chromosomes remains unclear: The variations do not seem to correlate with those of chromosome-wide expression levels, gene density, GC content, or repeat content (Supplemental Table S7). The chromosome evolution rate of Chr 10 seems to have slowed down in the ancestor of alligator and emu, that is, archosaurs, whereas that of Chr 3 has accelerated in the ancestor of reptiles and independently slowed down in the alligator lineage (Supplemental Fig. S13). Chr Z of Neognathae, but not Palaeognathae, have fixed a significantly higher number of rearrangements (P = 1.21 × 10−5, Wilcoxon test) than other macrochromosomes compared to the homologous turtle autosome. This is consistent with the recent cytogenetic examination of over 400 passerine species which characterized the Chr Z with more fixed inversions than any other macrochromosomes (Hooper and Price 2017; Damas et al. 2018). The faster evolution of Chr Z genomic structure (the ‘fast-Z’ effect) (Meisel and Connallon 2013) can be explained by a hemizygous Chr Z that is more likely to fix genomic rearrangements by genetic drift, due to a reduced effective population size, or it is driven by selection for incompatible inversion alleles between species. As expected, ratites do not exhibit such a fast-Z pattern because most parts of their Chr Zs are evolving predominantly like autosomes (Fig. 1E; Xu et al. 2019b). Microchromosomes generally have a higher rate of intrachromosomal rearrangements than the macrochromosomes after scaling for chromosome size or removing the outlier species (e.g., pigeon; P = 0.004, Wilcoxon test) (Fig. 2G; Supplemental Fig. S14). This may be influenced by the higher recombination rate and GC content of the microchromosomes leading to more frequent DNA double-strand breaks (DSBs). To test this hypothesis, we examined the rearrangement breakpoint regions, that is, evolutionary breakpoints in three species representing each major avian group, and found that the breakpoints indeed had significantly higher GC content than the genome average (P = 1.17 × 10−10, Wilcoxon test) (Supplemental Fig. S15), probably driven by the GC-biased gene conversion caused by a high local recombination rate (Weber et al. 2014).
We hypothesized that the different GC content of macro- versus microchromosomes (Supplemental Fig. S7) is also associated with their different contributions to the genome size reduction of birds, relative to their reptile relatives. To quantitatively measure this chromosomal difference, we calculated the length difference between turtle and emu in their syntenic blocks, whose aligned sizes together accounted for 95.9% of all the investigated emu chromosomes (Chr 1–Chr 28, Chr Z) (Supplemental Table S6). Repetitive regions within the syntenic blocks exhibited a much larger length difference between the two species (P < 2.20 × 10−16, Wilcoxon test) than any other genomic regions, whereas the exonic regions have maintained approximately the same lengths, with slightly larger exons in emu (Fig. 2H; Supplemental Fig. S16). These findings confirm that the reduction of avian genome size is mainly attributed to the genome-wide contraction of TE content (Zhang et al. 2014), possibly related to the evolution of flight (Kapusta et al. 2017) and associated high metabolic requirements (Organ et al. 2007; Zhang and Edwards 2012). We found that the smaller an emu chromosome is, the larger the GC content (P = 7.45 × 10−5, Pearson's correlation r = −0.67) and the more sequence loss, particularly repeat sequence loss (P < 2.20 × 10−16, Pearson's correlation r = 0.71), relative to the turtle (Fig. 2I; Supplemental Fig. S17). This pattern is consistent among all the studied birds where microchromosomes exhibit significantly more extensive sequence loss (P < 2.20 × 10−16, Wilcoxon test) than macrochromosomes, when compared to turtle or crocodile genomes (Fig. 2J; Supplemental Figs. S18, S19).
These results suggest that, due to the higher GC content on microchromosomes resulting from the higher recombination rate than macrochromosomes, their noncoding sequences are more prone to deletions, triggered by, for example, replication slippage (Kiktev et al. 2018). Using the available population genomic data of duck (Zhou et al. 2018), we found a significant positive correlation (P = 4.73 × 10−8, Pearson's correlation r = 0.31) between the recombination rate and the extent of sequence loss within its syntenic region with turtle (Supplemental Fig. S20). This is consistent with the reported negative correlation between the GC content and the genome size among mammals (Romiguier et al. 2010). Birds have maintained a higher number of microchromosomes since the divergence with other related reptile species (Uno et al. 2012). This may also have contributed to their genome size reduction.
Microchromosomes have an excess of interchromosomal contacts associated with housekeeping genes
The nuclear arrangement of chicken microchromosomes in the interior and macrochromosomes at the periphery (Habermann et al. 2001; Maslova et al. 2015) should give rise to more frequent interchromosomal (trans-) contacts between microchromosomes. To test this hypothesis, we measured the trans- and cis-chromosomal contacts by the emu normalized Hi-C read pairs that are derived from the different and the same chromosomal regions, respectively. These contacts quantify the frequency of spatial proximity between two distant genomic regions captured by the Hi-C technique that may be related to but not necessarily demonstrate functional association between these regions (Lieberman-Aiden et al. 2009). Similar to the reported patterns of mammals and Drosophila (Szabo et al. 2019), cis-contacts were the dominant type of chromatin interactions (Fig. 3A). There are much more abundant and stronger trans-contacts between microchromosomes than between macrochromosomes, and the overall trans-contact frequencies are negatively correlated with the chromosome size (P = 9.13 × 10−11, Pearson's correlation r = −0.88) (Fig. 3B). A previous study showed that human Chr 18 and Chr 19, albeit having a similar size, occupy distinct nuclear territories with the gene-poor Chr 18 located at the nuclear periphery, whereas the gene-rich Chr 19 is at the interior (Croft et al. 1999; Cremer and Cremer 2001). This suggested that the gene content rather than the chromosome size is underlying the segregated nuclear territories. Indeed, we found that the gene density and GC content was positively correlated with the trans-contact numbers per chromosome (Supplemental Figs. S21–S23). Similar to humans, we noted that some emu macrochromosomal regions with high gene densities also showed robust trans-contacts with other chromosomes (P < 2.20 × 10−16, Wilcoxon test) (Supplemental Fig. S24). Such high trans-contacts between microchromosomes were also found in our companion study on the duck genome (Li et al. 2021) and were reported previously for the rattlesnake (Schield et al. 2019), as well as for small-sized chromosomes in human cells (Lieberman-Aiden et al. 2009), and therefore it is probably a conserved chromosome territorial feature of vertebrates (Perry et al. 2020).
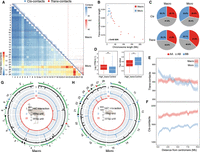
3D chromatin contacts of macro- and microchromosomes. (A) Upper right panel: each blue triangle shows the cis-contacts of each chromosome measured by the numbers of Hi-C reads connecting any of the two 40-kb regions of the same chromosome. Microchromosomes exhibit more frequent trans-contacts measured by the number of Hi-C reads connecting any of the two 40-kb regions of two different chromosomes. The color is scaled to the contact strength, that is, the Hi-C read numbers. The lower left heat map shows the chromosome-wide average strength of trans-contacts between any of the two chromosomes. The dashed lines demarcate macro- and microchromosomes. (B) Microchromosomes show more trans-contacts than macrochromosomes, after being scaled by chromosome size, and the SDR of Chr W (black dot) shows very few trans-contacts. Each dot represents one chromosome (blue for microchromosomes, red for macrochromosomes). (C) Comparing different types of contacts connecting two active compartments (AA), two inactive compartments (BB), or active and inactive compartments (AB) between macro- and microchromosomes. (D) Genes that are overlapped with any of the 40-kb windows exhibiting the top 10% high levels of trans-contacts (‘High_trans’) have significantly higher expression levels and lower tau values (the lower the tau value is, the broader tissue expression the gene has) than the other genes, suggesting these are likely housekeeping genes. (***) P < 0.0005. (E,F) The average distributions of trans- or cis-contacts along the chromosomes with the distance away from the centromeres, suggesting centromeres have more impacts on the trans-contacts of microchromosomes than those of macrochromosomes. (G,H) From the outer to inner circles: (I) Hi-C contacts where the black lines indicate the punctuation of such contacts; (II) genomic distribution of 65-bp putative centromeric repeats (blue); (III) genomic distribution of 81-bp (red) centromeric repeats. The putative centromeres were annotated by one or two of these three sources of information, corroborated with karyotype information, and then color-coded accordingly on the chromosomes.
To explore the functional significance of these trans-contacts, we divided the entire emu genome according to their Hi-C interaction profiles into the active (A) and inactive (B) compartments (Lieberman-Aiden et al. 2009) and then compared the trans-contact frequencies within or between the two types of compartments. We found that the trans-contacts more frequently involved two regions that were both from active compartments (AA contacts) than the cis-contacts (P < 2.20 × 10−16, Fisher's exact test), and microchromosomes had more frequent AA trans-contacts than the macrochromosomes (P < 2.20 × 10−16, Fisher's exact test) (Fig. 3C). Further, genes exhibiting high frequencies (ranked top 10%) of trans-contacts detected by our liver Hi-C data have significantly higher expression levels (P < 2.20 × 10−16, Wilcoxon test), particularly in the liver (Supplemental Fig. S25), and broader tissue expression patterns (P < 2.20 × 10−16, Wilcoxon test) than the other genes (Fig. 3D). These trans-contacting genes defined are enriched in cell regulatory and metabolic functional categories (Supplemental Fig. S26). Therefore, trans-contacts between active compartments of different chromosomes probably play an important role in regulating housekeeping gene expression. One caveat about this conclusion is that our cis- or trans-contacts were calculated from Hi-C data of only the liver tissue, and the conclusion needs to be verified in other tissues in the future.
The frequency of trans-contacts, but not cis-contacts, is on average the highest at the centromeric regions of micro- but not macrochromosomes and decays by the distance away from the centromeres (Fig. 3E,F). The chromosomal distribution of trans-contacts is consistent with the radial 3D conformation of the predominantly acrocentric microchromosomes of birds, whose pericentromeric heterochromatin associates with the interior nucleolus (Habermann et al. 2001; Maslova et al. 2015). To search for the candidate genomic determinants of such nuclear conformation, we compared the centromeric sequences between the emu macro- and microchromosomes. We identified two GC-rich (GC content > 55%) repeat monomers of 65 bp and 81 bp long enriched at the putative centromeres, whose copy numbers are among the most abundant throughout the entire emu genome. The 65-bp repeats are more enriched in microchromosomes than macrochromosomes (Fig. 3G,H; Supplemental Fig. S4). Similar microchromosome centromere enriched repeats, but with different sequences, have also been reported in chicken (chicken nuclear membrane associated repeats, CNM) (Matzke et al. 1990; Shang et al. 2010) and other bird and turtle species (Yamada et al. 2002; Yamada et al. 2005; Nishida et al. 2013). Thus, we named this repeat emu nuclear interior associated (ENI) repeat. Whether the differential chromosomal distribution of ENI repeats drives the segregated nuclear architecture of emu macro- versus microchromosomes remains a question for future functional investigation.
Emu sex chromosome evolution involves alteration of chromatin conformation
The newly assembled Chr W of emu comprises a model for studying the stepwise evolution of genome and chromatin conformation under a non-recombining environment. The long PAR is shared between sex chromosomes and represents the ancestral autosome state before recombination was suppressed, whereas S1 and S0, respectively, represent the early and late phases of sex chromosome differentiation. This was demonstrated by the gradient of accumulated TEs and functional gene loss formed by the W-linked S0 (WS0), S1 (WS1), and PAR. The greatly different TE content between WS0 (43.5%), WS1 (9.3%), and PAR (7.3%) has demarcated the three regions (Supplemental Fig. S27), with a small part of WS0 having been reshuffled between WS1 and PAR (Fig. 4A). The highly heterochromatic WS0 had RS over 150 million years ago (MYA) in the avian ancestor and has only about 3.6 Mb sequences assembled, compared to the 22.4-Mb-long Z-linked S0 (ZS0) region. The ZS0 also seems to have undergone much more intrachromosomal rearrangements than other Z-linked regions when being compared to the homologous autosomal region of the sea turtle (Supplemental Fig. S28), presumably due to the reduction of recombination rate. The few alignable sequences between ZS0 and WS0 hampered our inference of how RS occurred within S0. In contrast, the younger S1 emerged 23 MYA (Wang et al. 2019) and exhibited a low level of average sequence divergence (5%) between the Z/W, confirming the low rate of emu sex chromosome evolution. The entire WS1 formed a large inversion compared to its Z-linked homolog, as well as the ostrich Chr Z and the homologous turtle autosome (Fig. 4B). This suggests that the emu S1 likely evolved RS through a W-linked chromosome inversion.
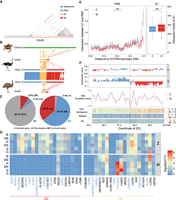
Emu sex chromosome evolution. (A) Dot plot of SDR of Chr W, which is segregated into a highly repetitive S0 (red) and only moderately repetitive S1 (orange). Part of the S0 region was reshuffled between the PAR (blue) and the S1, which was inferred based on its homology with the Z-linked S0 region (Fig. 1E). (B) Syntenic plot between the ZW Chromosomes of emu and ostrich and the homologous autosome of green sea turtle, which suggests a W-linked inversion created the emu S1. (C) There are more genes that have become deleted (gray) or pseudogenes (blue, those containing premature stop codons or frameshift mutations) in S0 than in S1 on the W Chromosome. (D) The log expression levels (TPM) of single-copy homologous genes in the S0 and S1 regions of Z and W Chromosomes. (B) Brain, (EB) embryonic brain, (K) kidney, (EK) embryonic kidney, (S) spleen, (O) ovary. The genes with blue color indicate the pseudogenes. (E) The left panel plot shows the cis-contacts between the PAR and the Z- (blue) and W- (red) linked S1 regions, which exhibit reduced W-linked contacts compared to the Z-linked contacts. The x-axis shows the distance to the S1/PAR boundary. The right panel plot indicates higher cis-contacts within the W-linked S1 region compared to its homologous Z-linked region. (***) P < 0.0005. (F) The W-linked S1 is segregated into two compartments WS1A (left, blue) and WS1B (right, red). From upper to lower: A(blue)/B(red) compartment division based on the Hi-C contact profiles; TAD insulation scores (the lower values correspond to the TAD boundaries); ZW pairwise sequence divergence level, repeat, and GC content. The black line indicates the boundary (58.22 Mb on Chr Z and 5.28 Mb on Chr W) between WS1A and WS1B. Note: The WS1 has been reversed for the convenience of ZW comparison.
Out of 273 WS0 genes, 247 (90.5%) have become deleted or accumulated nonsense mutations, compared to 27 out of 42 (64.3%) WS1 genes (Fig. 4C). Among the retained W-linked genes with intact open reading frames, they are expressed at a significantly lower level (P = 0.020, Wilcoxon test) than their Z-linked homologs, with the WS0 region showing more severe down-regulation of gene expression (P = 8.16 × 10−9, Wilcoxon test) than the WS1 (Fig. 4D). Despite the low level of sequence divergence, WS1 nevertheless shows clear signatures of down-regulation of gene expression, suggesting regulatory changes simultaneously occurred with or even preceded the amino acid changes during W Chromosome evolution.
We hypothesized that the global change of chromatin conformation induced by accumulation of TEs may have important contributions to such broad down-regulation of W-linked genes. This was previously shown on the young Y Chromosome of Drosophila miranda, where TE accumulation has increased the level of silencing histone modifications (H3K9me3) and thus induced gene down-regulation (Zhou et al. 2013). Although the TE content of emu WS1 is comparable to that of the PAR and autosomes (9.3% vs. 7.3% and 9.8%), it has become 1.6-fold higher than that of Z-linked S1 (ZS1, 5.8%), suggesting an ongoing process of heterochromatinization. To test this hypothesis, we compared between sex chromosomes for their frequencies of cis-contacts within S1 and between S1 and the neighboring PAR. We expected that, if the WS1 was becoming globally heterochromatic, its chromatin configuration would become more compact and incur more cis-contacts, suggested by the significantly greater number of cis-contacts within the B compartments than the A compartments at the genome-wide level (P < 2.20 × 10−16, Wilcoxon test) (Supplemental Figs. S29, S30). Indeed, Chr W has significantly higher numbers of cis-contacts (P = 0.035, Wilcoxon test) (Fig. 4E) and a moderately increased inactive compartment strength (P = 0.051, Fisher's exact test) than Chr Z within the S1 region but, on average, decreased cis-contacts between WS1 and PAR (P = 7.99 × 10−13, Wilcoxon test) (Fig. 4E).
The chromatin compartments of WS1 also have become more segregated: Compared to the randomly distributed A and B compartments of the ZS1, the WS1 is divided into one large B compartment (WS1B, 3.24 Mb–5.28 Mb) bordering the S0 and one large A compartment (WS1A, 5.28 Mb–7.68 Mb), with expectedly more cis-contacts of WS1B (P < 2.20 × 10−16, Wilcoxon test) than that of WS1A (Supplemental Fig. S31). There are, however, few significant differences in the structure and strength of the finer chromatin units enclosed in the A or B compartments, that is, in the topologically associated domains (TADs) (Fig. 4F; Supplemental Fig. S32). We found WS1B exhibited a significantly higher level of Z/W pairwise sequence divergence (P < 2.20 × 10−16, Wilcoxon test) and repeat content (P = 0.027, Wilcoxon test) than WS1A, a pattern similar to the evolutionary strata. Such a ‘strata within strata’ pattern clearly formed without secondary inversions within S1 (Fig. 4B). We suggest that such pronounced changes of chromatin conformation may be caused by the spreading of heterochromatin from the highly heterochromatic WS0 region to the nearby WS1B, similar to the mechanism of positional effect variegation (PEV) (Elgin and Reuter 2013). As the homologous regions of emu Chr Z and the turtle autosomes of WS1B have a lower gene density (32% vs. 54% genic region) and a higher proportion of the B compartment (60% vs. 51%) than those of WS1A, the ancestrally more inactive WS1B probably has undergone a weaker natural selection against the spreading of S0 heterochromatin after its recombination was suppressed. In addition, the boundary between WS1A/B is overlapped with a TAD boundary with the lowest insulation score, that is, the highest boundary strength within the entire S1 region (Fig. 4F). Selection on such a strong TAD boundary may prevent the further invasion of heterochromatin into WS1A.
Finally, some WS1A genes like ALDH1A1 and ANXA1 have unexpectedly higher expression levels than their Z-linked homologs (Fig. 4D). We recently showed that ANXA1 has a conserved ovary-biased gene expression pattern in various birds and the green anole lizard and was even restored on the W Chromosomes of some songbirds through transposition after the loss of its original copy (Xu and Zhou 2020). In addition, ALDH1A1 has been shown to function at the onset of female meiosis in mice (Bowles et al. 2016). We propose that, once the WS1 became specifically transmitted in females after recombination suppression, the female-specific selection targeting these genes with pre-existing female-related functions may account for their up-regulation and curb the further degeneration or the spreading of heterochromatin from WS1B into WS1A.
Discussion
Among vertebrates, birds have one of the most conserved karyotypes with many of the microchromosomes that have been recently suggested to exist before the divergence of the vertebrate/gnathostome ancestor about 500 MYA (Simakov et al. 2020). Our study here showed that among birds, emu and other ratites are the most informative for the evolution of the ancestral avian or vertebrate karyotype, because they have experienced much less lineage-specific chromosomal changes. It is possible that the vertebrate ancestor had a radial nuclear conformation of chromosomes similar to that of birds (Fig. 5A), with the gene-rich microchromosomes and gene-poor macrochromosomes segregated, respectively, to the transcriptionally active nuclear interiors and more silent peripheries (Habermann et al. 2001). This has been supported by cytogenetic studies of primates, birds, and frogs, which showed that their chromosomes or partial chromosomal regions are all compartmentalized by their gene or GC content in the interphase nuclei (Cremer and Cremer 2001; Federico et al. 2005; Federico et al. 2006). There are some variations between different cell types (Stadler et al. 2004) and an exception of inverted nuclei in rod photoreceptors of nocturnal mammals (Feodorova et al. 2020). The functional significance of such higher-order nuclear architecture (Cremer et al. 2006) is suggested by the interspecific conservation of chromosome territories, regardless of the chromosome rearrangements. For example, in both birds and primates, fusions of microchromosomes or gene-dense chromosomes with other chromosomes do not seem to alter their nuclear positions (Tanabe et al. 2002; Tanabe et al. 2005; O'Connor et al. 2019). Our finding that emu microchromosomes have more trans-contacts between the active compartments of housekeeping genes, and presumably cluster in the nuclear center like other birds, is consistent with the colocalization of ‘splicing speckle’ nuclear structure enriched for splicing factors with microchromosomes in chicken neuronal cells (Berchtold et al. 2011). It remains to be examined in the future with more complete genome assemblies and Hi-C contact maps of more vertebrates whether such an association between trans-contacts and housekeeping genes in the nuclear center is conserved and represents an ancestral gene regulation mechanism in vertebrates.
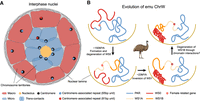
Nuclear architectures of avian chromosomes. (A) Microchromosomes of birds, possibly those of vertebrate ancestors, are clustered around the nucleolus in the nuclear center, which might be associated with specific centromeric repeats. Such a radial chromosome conformation can promote trans-contacts between microchromosomes. In contrast, macrochromosomes are distributed in nuclear peripheries with few trans-contacts. (B) 3D evolution of emu sex chromosomes. About 150 MYA, S0 formed in the ancestor of birds, and WS0 started to degenerate. During the heterochromatinization process, WS0 became anchored to the nuclear lamina like any other heterochromatic regions. About 23 MYA, one W-linked inversion has produced S1. Possibly due to the spreading of heterochromatin of S0 (red), the S1 region (WS1B, orange) adjacent to the WS0 underwent heterochromatinization earlier than the other region (WS1A), and evolved larger inactive/B compartments. This increased the cis-contacts within S1 but decreased cis-contacts between the S1 and PAR. The further spreading of heterochromatin into WS1A may be also halted by the selection on the female-related genes (red dots) located in the WS1A or the natural selection acting to preserve the TAD boundary between the WS1A and WS1B.
On the other hand, the nuclear peripheries are preferentially occupied by the macrochromosomes and possibly the W chromosome, through tethering their heterochromatin to the nuclear lamina (Falk et al. 2019). The conventional model of sex chromosome evolution of birds and mammals involves suppression of recombination through chromosomal inversions, forming the pattern of evolutionary strata (Lahn and Page 1999; Cortez et al. 2014; Zhou et al. 2014). Our recent study of the birds-of-paradise (Xu et al. 2019a), however, suggested an alternative PEV-like model not dependent on inversions, which is demonstrated here by the ‘strata within strata’ pattern of S1. The stratified Z/W sequence divergence levels and repeat content between WS1A and WS1B were likely not caused by inversions indicated by their sequence alignments with the Z Chromosome. It possibly involved the spreading of heterochromatin from the highly heterochromatic S0 to the nearby WS1B region, attracting it closer to the heterochromatin domains of the nuclear periphery (Fig. 5B). Under this scenario, the chromatin conformation change would have initiated the heterochromatinization process first in the WS1B region. The complex suit of selective forces still acting on the W Chromosome, including selection to maintain or even up-regulate the gene expression levels of female-related genes (e.g., ANXA1), and that on the TAD boundaries, may have blocked the further spreading of heterochromatin.
Overall, we demonstrated that emu and other ratite species have better preserved the ancestral avian chromosome composition and nuclear architecture than other birds, probably due to their slower evolutionary rate. We speculated that the ancestor of birds or even vertebrates probably had a segregated nuclear architecture like that of most extant birds, with microchromosomes concentrated at the nuclear center and the macrochromosomes mainly at the nuclear periphery. We showed emu sex chromosome evolution involved alteration of chromatin architecture in the absence of large genomic rearrangements, which may also comprise a critical step at the early stage of sex chromosome evolution of many other species.
Methods
Sample collection and sequencing
All female samples were derived from the Copenhagen Zoo or emu farms in the Fujian and Shaanxi provinces of China. We extracted the high molecular weight (HMW) DNA from the blood of a female emu (Dromaius novaehollandiae) and constructed the libraries for SMRT sequencing. In total, 89 SMRT cells were generated on a PacBio RS II platform (Pacific Biosciences), and 88 Gb subreads with an N50 read length of 15.5 kb were produced. HMW DNA of another female emu individual was used to generate a linked-reads library following the protocol on the 10x Genomics Chromium platform. This 10x library was subjected to MGISEQ-2000 sequencing and 156 Gb PE150 reads were collected. HMW DNA of a third female individual was used for constructing Chicago and Hi-C libraries at Dovetail as described previously (Putnam et al. 2016). Briefly, ∼500 ng of HMW emu gDNA (mean fragment length = 48 kb) was reconstituted into chromatin and digested with DpnII. The DNA was then sheared to ∼350-bp mean fragment size, and sequencing libraries were generated using NEBNext Ultra enzymes and Illumina-compatible adapters. The libraries were sequenced on an Illumina HiSeq X to produce 492 million 2 × 151-bp paired-end reads, which provided 297.50 × physical coverage of the genome (1–100 kb pairs). The libraries were sequenced on an Illumina HiSeq X to produce 211 million 2 × 151-bp paired-end reads, which provided 13,633.10× physical coverage of the genome (10–10,000 kb pairs).
Genome assembly and annotation
The chromosome-length genome assemblies for the ostrich, greater rhea, southern cassowary, greater prairie chicken, double-crested cormorant, spotted owl, golden eagle, California condor, and the green sea turtle as well as the associated Hi-C data sets were downloaded from the DNA Zoo Consortium website (https://www.dnazoo.org), where they were shared ahead of publication. The assemblies incorporated data from Wang et al. (2013), Zhang et al. (2014), Burga et al. (2017), Van Den Bussche et al. (2017), and Sackton et al. (2019) as well as unpublished data sets. More information is available on the corresponding assembly pages at DNA Zoo (https://www.dnazoo.org). The genomes were assembled using methods described in Dudchenko et al. (2017).
For the genome assembly of emu, we produced the contig sequences derived from the PacBio subreads with FALCON (20171207) (Chin et al. 2016), using the option ‘-k24 -e.96 -l2500’ for ovlp_daligner, ‘-e0.75 -l3200’ for pa_daligner. We used Purge Haplotigs (20180325) (Roach et al. 2018) to remove the haplotigs after mapping the raw reads with minimap2 (2.10) (Li 2018) against the contigs to estimate the coverage distribution. The contigs were then polished by Racon (1.3.0) (Vaser et al. 2017) with default parameters. Because the S1 of the sex chromosome evolved recently with sequence similarity between the ZW as high as 95%, we resolved the ZW haploid assembly by partitioning the Z- and W-derived long-reads for separate assemblies. To do so, first we identified the sex-linked contigs by aligning the contigs to the previous reference Z Chromosome of emu (Xu et al. 2019b) and extracted the associated reads. Then, we used FALCON to assemble the ZW-linked reads with more stringent overlapping (-k18 -e0.81 -l3000 for pa_daligner and -k24 -e.96 -l4000 for ovlp_daligner) to avoid haploid collapse, where therefore both Z- and W-linked haploid sequences could be assembled. We distinguished the Z- and W-linked contigs according to their sequence similarity with the reference Z and whether they could be mapped with male reads. Then, we extracted the reads derived from the Z- and W-linked contigs and assembled them with Canu (1.8) (Koren et al. 2017), respectively. The parameters corOutCoverage = 200 correctedErrorRate = 0.15 were used for Canu haploid assembly.
The contigs were then scaffolded first with 10x linked reads using Scaff10X (https://github.com/wtsi-hpag/Scaff10X), then with ARCS + LINKS (Warren et al. 2015; Coombe et al. 2018). Finally, the input de novo assembly, Chicago library reads, and Dovetail Hi-C library reads were used as input data for HiRise, a software pipeline designed specifically for using proximity ligation data to scaffold genome assemblies (Putnam et al. 2016). The Dovetail Chicago scaffolding was performed with HiRise (version 2.1.2-4e9d295dd196), and the Hi-C scaffolding was performed with HiRise (version v2.1.6-072ca03871cc). A previous version of the Dovetail Genomics HiRise assembler (Putnam et al. 2016) is available as an open-source distribution at GitHub (https://github.com/DovetailGenomics/HiRise_July2015_GR); however, Dovetail Genomics has not made the HiRise versions used on this assembly available as open-source software at this time. The intermediate alignment AGP file that relays mapping evidence of Chicago scaffolding is also not available from Dovetail. However, we compared our assembled chromosomal sequences to an independently generated emu assembly from DNA Zoo (Supplemental Fig. S3), which supported the accuracy of our assembly. An iterative analysis was conducted: first, PacBio and Chicago library sequences were aligned to the draft input assembly using a modified SNAP read mapper (http://snap.cs.berkeley.edu). The Chicago read pairs spanning different scaffolds were analyzed by HiRise to produce a likelihood model for estimating the genomic distances between read pairs, and the model was also used to identify and break putative misjoins, to score prospective joins, and to make joins above a threshold. After aligning and scaffolding steps with the Chicago data, Dovetail Hi-C library sequences were aligned for scaffolding the assembly following the similar method. To curate and correct putative assembly errors, we remapped the Hi-C reads and used Juicer tools (Durand et al. 2016) to trim the draft assembly manually. After scaffolding, PacBio long reads were used to close gaps between contigs using the Arrow-corrected PacBio subreads and PBJelly software. The assembly was polished with Illumina reads by Pilon (Walker et al. 2014). Genome completeness was evaluated by BUSCO v3.0.2 (Simão et al. 2015).
For repeat annotation, we first used RepeatModeler (open-1.0.10) to construct the consensus repeat sequence library of the emu. Then, the de novo library and the repeat consensus library in Repbase (Bao et al. 2015) were merged to annotate all repetitive elements in the emu genome using RepeatMasker (open-4.0.9). We integrated evidence of protein homology, transcriptome, and de novo prediction to annotate the protein-coding genes with the MAKER v2.31.10 (Cantarel et al. 2008) pipeline to obtain complete gene models. For the protein homology-based evidence, protein sequences of Gallus gallus, Struthio camelus, and Alligator mississippiensis were downloaded from NCBI. For the transcriptome-based evidence, a genome-guided method was applied to transcriptome assembly. To do so, RNA-seq reads were mapped to the genome with HISAT2 v2.1.0 (Kim et al. 2015) and assembled with StringTie v1.3.4 (Pertea et al. 2015). For the ab initio gene predictions, we used AUGUSTUS v3.3 (Stanke et al. 2006) and SNAP (Korf 2004) to predict gene models using the parameters that were trained based on the results of protein homology and transcriptome predictions. Gene functions were assigned using DIAMOND v0.9.24 (Buchfink et al. 2015) against the UniProtKB (SWISS-PROT + TrEMBL) database with a sensitive mode and an e-value threshold of 1 × 10−5 (‐‐more-sensitive -e 1 × 10−5).
To annotate the putative centromeres, we searched for tandem repeats across the genome using TRFinder v4.09 (Benson 1999) with the parameters: 2 5 7 80 10 50 2000. We overlapped these findings with the prediction that centromeric regions tend to have lower Hi-C contacts (Muller et al. 2019; Tao et al. 2020), where two putative centromeric units (65 bp and 81 bp) were identified. The centromere positions for all chromosomes were further manually checked with the reported karyotype of emu chromosomes (Takagi and Sasaki 1974; Kabir 2012). For telomeres, we used the known vertebrate consensus sequence ‘TTAGGG/CCCTAA’ to search for the clusters of consensus sequences on both strands. Consensus sequence enriched genomic blocks in a 50-kb window were then defined as the putative telomere regions.
Comparative genomic analyses
Twelve chromosome-level genomes of bird species including common ostrich (Struthio camelus) (Zhang et al. 2014), greater rhea (Rhea americana) (Sackton et al. 2019), Southern cassowary (Casuarius casuarius) (Sackton et al. 2019), emu (Dromaius novaehollandiae), Peking duck (Anas platyrhynchos), greater prairie chicken (Tympanuchus cupido) (Johnson et al. 2015), chicken (Gallus gallus) (Warren et al. 2017), band-tailed pigeon (Patagioenas fasciata) (Murray et al. 2017), double-crested cormorant (Phalacrocorax auritus) (Burga et al. 2017), spotted owl (Strix occidentalis) (Dudchenko et al. 2017), golden eagle (Aquila chrysaetos) (Van Den Bussche et al. 2017), and California condor (Gymnogyps californianus) (Dudchenko et al. 2017) were aligned to the chromosome-level genome of green sea turtle (Chelonia mydas) using LAST (http://last.cbrc.jp/). For each investigated species, small syntenic blocks were first merged into larger blocks using the custom Perl script. After two rounds of the merging process, blocks whose lengths were shorter than 50 kb were discarded. Genomic rearrangements including inversions, translocations, and inverted translocations were then detected based on the orientation and position of retained blocks, using the custom Perl scripts. To infer the rearrangement rate for each node of the phylogenetic tree (Claramunt and Cracraft 2015), we followed the principle that, if any rearrangement was shared by two closely related species, that is, the overlap length ratio was larger than 80%, it was present in their common ancestral node. This tracing process was performed iteratively until we reached the ancestral node of all birds. Regarding tolerance for assembly errors, a maximum ratio of one rearrangement missing out of five species was allowed for tracing back to the ancestral node. The rearrangement rate was calculated with the published divergence times for each node (Claramunt and Cracraft 2015).
Hi-C contact analyses
Hi-C read mapping, filtering, correction, binning, and normalization were performed by HiC-Pro v2.10.0 (Servant et al. 2015) with the default parameters. In brief, Hi-C reads from the liver of the female were mapped to the respective reference genome, and only uniquely mapped reads were kept. Then, each uniquely mapped read was assigned to a restriction enzyme fragment, and invalid ligation products were discarded. The interaction contacts were then binned to generate the genome-wide interaction matrix at 5 kb-, 10 kb-, 20 kb-, 40 kb-, 100 kb-, 500 kb-, 1 Mb-, and 10 Mb-resolution. The ICE (iterative correction and eigenvector decomposition) normalization was then applied to the interaction matrix (Imakaev et al. 2012). Then, the cis-contacts and trans-contacts for each 40-kb window were calculated using 40 kb normalized interaction matrix. We identified the A/B compartments using the pca.hic function from the HiTC package (Servant et al. 2012) with default parameters. TADs were identified by HiCExplorer v3.0 (Wolff et al. 2018) with the application hicFindTADs.
Sex chromosomes analyses
The pseudoautosomal region was identified based on an equal ratio of male versus female genomic read coverage. S0 was identified based on the half-ratio of female versus male genomic read coverage. S1 was identified using the different levels of nucleotide diversity between male and female after both reads of both sexes were mapped to the Z Chromosome. To identify the gene repertoire of Chromosome W, we performed a TBLASTN search (Altschul et al. 1990) with the cutoff E-value of 1 × 10−5 against the Chromosome W sequences using the Z-linked genes as query sequences. Aligned sequence fragments were combined into one predicted gene if they belonged to the same query protein. Then, each candidate gene region was extended for 5 kb from both ends to predict its open reading frame by GeneWise v2.4.1 (Birney et al. 2004). We annotated the open reading frames as disrupted when GeneWise reported at least one premature stop codon or frameshift mutation.
Software availability
The software and pipeline for genome de novo assembly, annotation, and Hi-C analysis, and the custom code and scripts for comparative genomics and sex chromosome evolutionary analysis in this work have been deposited at GitHub (https://github.com/JhinAir/Emu) and in the Supplemental Code. All figures were plotted in R (R Core Team 2020).
Data access
The emu assembly from this study has been submitted to the NCBI Nucleotide database (https://www.ncbi.nlm.nih.gov/nuccore/) under the accession number JABVCD000000000.1. The raw PacBio long reads, 10x linked reads, Chicago, and Hi-C linked reads generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under the accession number PRJNA638233.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Dr. Irina Solovei from Ludwig Maximilian University of Munich for her inspiring discussion and constructive comments on the manuscript, and Valentina Peona for sharing the curated repeat library of the emu genome. We thank Erez Lieberman-Aiden and Olga Dudchenko and other staff members from DNA Zoo for sharing the chromosomal genomes. We thank BGI-Shenzhen for providing the 10x linked reads data of emu. Q.Z. is supported by the National Natural Science Foundation of China (31722050, 31671319, 32061130208), the Natural Science Foundation of Zhejiang Province (LD19C190001), and the European Research Council Starting Grant (grant agreement 677696).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.271569.120.
- Received September 9, 2020.
- Accepted December 30, 2020.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








