
Profiling single-cell histone modifications using indexing chromatin immunocleavage sequencing
- Laboratory of Epigenome Biology, Systems Biology Center, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, Maryland 20892-1674, USA
-
↵1 These authors contributed equally to this work.
Abstract
Recently, multiple single-cell assays were developed for detecting histone marks at the single-cell level. These techniques are either limited by the low cell throughput or sparse reads which limit their applications. To address these problems, we introduce indexing single-cell immunocleavage sequencing (iscChIC-seq), a multiplex indexing method based on TdT terminal transferase and T4 DNA ligase-mediated barcoding strategy and single-cell ChIC-seq, which is capable of readily analyzing histone modifications across tens of thousands of single cells in one experiment. Application of iscChIC-seq to profiling H3K4me3 and H3K27me3 in human white blood cells (WBCs) enabled successful detection of more than 10,000 single cells for each histone modification with 11 K and 45 K nonredundant reads per cell, respectively. Cluster analysis of these data allowed identification of monocytes, T cells, B cells, and NK cells from WBCs. The cell types annotated from H3K4me3 single-cell data are specifically correlated with the cell types annotated from H3K27me3 single-cell data. Our data indicate that iscChIC-seq is a reliable technique for profiling histone modifications in a large number of single cells, which may find broad applications in studying cellular heterogeneity and differentiation status in complex developmental and disease systems.
Histone modifications, which are typically measured by chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing (Barski et al. 2007; Johnson et al. 2007; Mikkelsen et al. 2007; Robertson et al. 2007) at the bulk-cell level, are associated with transcriptional regulation. Chromatin regions enriched in H3K4 methylation and H3K27 acetylation are potentially active promoters or enhancers that activate the transcription of target genes; on the other hand, genes enriched in H3K27me3 signals are usually repressed (Kim et al. 2005; Barski et al. 2007; Mikkelsen et al. 2007; Wei et al. 2009; Creyghton et al. 2010). Whereas the genomic profiles of various histone modifications have been extensively characterized at the bulk-cell level, several single-cell epigenomic techniques for detecting histone modification marks are reported recently (Rotem et al. 2015; Ai et al. 2019; Carter et al. 2019; Grosselin et al. 2019; Hainer et al. 2019; Harada et al. 2019; Kaya-Okur et al. 2019; Ku et al. 2019; Wang et al. 2019).
Although single-cell assays including scChIL-seq (Harada et al. 2019), scChIC-seq (Ku et al. 2019), uliCUT&RUN (Hainer et al. 2019), scCUT&Tag (Kaya-Okur et al. 2019), iACT-seq (Carter et al. 2019), CoBATCH (Wang et al. 2019), itChIP-seq (Ai et al. 2019), and scChIP-seq (Rotem et al. 2015; Grosselin et al. 2019) were developed recently for measuring histone marks (Supplemental Table S1), they have specific limitations. Whereas scChIP-seq combined the droplet barcoding approach with ChIP-seq (Barski et al. 2007; Rotem et al. 2015; Grosselin et al. 2019), all other methods except for itChIP-seq replaced the traditional immunoprecipitation with antibody guided digestion of chromatin either via antibody-directed, transposase-mediated integration of a DNA tag and fragmentation (for scChIL-seq [Harada et al. 2019] and scCUT&Tag [Kaya-Okur et al. 2019], iACT-seq [Carter et al. 2019], CoBATCH [Wang et al. 2019]), or via DNA cleavage specifically around nucleosomes containing the target modification (Schmid et al. 2004) (for uliCUT&RUN [Hainer et al. 2019] and scChIC-seq [Ku et al. 2019]). scChIP-seq (Rotem et al. 2015; Grosselin et al. 2019), with a relatively complicated workflow, could detect ∼2000–4000 cells in one experiment with an average of 4000 reads per cell. Although iACT-seq, scCUT&Tag, uliCUT&RUN, itChIP-seq, and scChIC-seq have simpler workflows and are more cost-effective, iACT-seq and scCUT&Tag could detect an average of 2000–6000 reads per cells and the cell throughput of uliCUT&RUN, itChIP-seq, and scChIC-seq is low. Although scChIL-seq and CoBATCH worked well for detecting active marks, they were not optimal for detecting repressive marks in fixed samples considering the attenuated activity of Tn5 in nonaccessible chromatin regions and its intrinsic bias toward open regions (Harada et al. 2019). Therefore, there is a need to develop a single-cell technique for profiling histone marks with higher cell throughput, wider applications, and detection of more reads per cell.
Results
The simultaneous addition of several dG nucleotides to DNA ends by TdT enzyme and ligation of oligo-dC barcode adaptors by T4 DNA ligase is an efficient strategy to barcode chromatin regions following DNase digestion (Gao et al. 2021). We adapted this barcoding strategy to label the DNA ends generated by antibody-guided MNase cleavage in ChIC-seq assays to profile histone modifications in more than tens of thousands of single cells in one experiment through three levels of barcoding and indexing strategy (Fig. 1A,B). Briefly, following antibody-guided MNase digestion of cells cross-linked with formaldehyde and disuccinimidyl glutarate (DSG), several dGs were added to the DNA ends by the activity of TdT in the presence of T4 DNA ligase and oligo-dC barcode adaptors in a 96-well plate. The cells were then pooled from 96 wells and aliquoted into new 96-well plates with 30 cells per well by flow cytometry sorting, followed by two consecutive rounds of PCR amplification. The samples were then pooled, purified, and sequenced using Illumina HiSeq 3000. The barcodes and PCR indexes (Supplemental Table S2) were identified and resolved to reveal single cells using a previous strategy (Cusanovich et al. 2015).
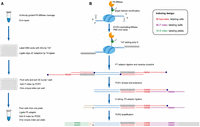
Schematic of iscChIC-seq. (A) Experimental flow. (1) Bulk cells were split into the first 96-well plate after antibody-guided MNase cleavage and end repair. (2) Barcoded cells were pooled together and sorted into the second 96-well plate to introduce the i7 index. (3) Cells were pooled together again from each plate and labeled with the i5 index in PCR2. (B) Illustration of poly(dG) addition to DNA ends by TdT, oligo dC adaptor ligation by T4 DNA ligase, and PCR-mediated barcoding process. Cell barcode (red) is designed into the oligo dC P7 adaptor in which 3′ ends are blocked to prevent nontemplate tailing by TdT. After reverse crosslinking, barcoded DNA fragments could be efficiently labeled with the i7 index (purple) through annealing and PCR extension. The barcoded P5 adaptor is added to the other end of genomic DNA fragments by ligation and PCR2, which is used to amplify the library DNA for NGS sequencing.
We first examined the collision rate by applying iscChIC-seq to a mixture of mouse NIH3T3 and human HEK293T cells. We found that reads from cells were mainly mapped either to the mouse genome (n = 439) or the human genome (n = 2371), whereas 149 barcodes were categorized as human and mouse doublets. The human-mouse cell doublet rate is ∼10% (Supplemental Fig. S1; Methods), which is similar to the collision rate of 12% obtained from the estimation based on the number of cells per well (Rubin et al. 2019). We then applied iscChIC-seq to white blood cells isolated from human blood for profiling the H3K4me3 modification, which is an active histone modification mark, at a single-cell resolution. Using a cutoff to filter cells with less than 1000 reads, we detected 10,000 single cells and about 9000 reads per cell on average in one single experiment (Supplemental Table S3). Using a more stringent filtering criteria (a cell has at least 3000 reads), this resulted in ∼7800 single cells each having about 11,000 reads on average. Note that the number of total reads in some wells of the PCR plate is much lower than other wells (Supplemental Table S3), which could be caused by the irregularities of the PCR machine—for example, temperature control at those few wells. The cell number and unique reads number per cell detected by iscChIC-seq are significantly improved as compared with the previous published single-cell methods (Supplemental Table S3), whereas the precision of reads detected by iscChIC-seq is comparable to other methods (Grosselin et al. 2019; Kaya-Okur et al. 2019). The duplication rate of iscChIC-seq reads is about 73%, whereas it is 87% for scCUT&Tag (Kaya-Okur et al. 2019) and 23% for scChIP-seq (Grosselin et al. 2019). The genomic profiles of the sequencing read from pooled single cells displayed specific peaks around transcription start sites (TSSs) and were highly consistent with that of the bulk-cell H3K4me3 ChIP-seq data from ENCODE (Fig. 2A; Supplemental Fig. S2A,B). Using SICER (Zang et al. 2009; Xu et al. 2014), 36,169 H3K4me3 peaks were detected from the pooled single cells. Using a similar strategy, 52,798 H3K4me3 peaks were detected from the ENCODE ChIP-seq data from different immune cells in human WBCs (Supplemental Table S4). Comparison of the ENCODE data with our single-cell data revealed that 31,432 out of 36,169 (87%) H3K4me3 peaks from the pooled cells overlapped with the peaks from the bulk-cell H3K4me3 ChIP-seq data (Fig. 2B). The read densities of the pooled single cells and the bulk cell ChIP-seq data were highly correlated (r = 0.89) (Fig. 2C). Also, the pooled single-cell data showed high enrichment and nucleosome phasing around the transcription start site (Fig. 2D), as found from ChIP-seq data (Barski et al. 2007). Together, these results indicate that our iscChIC-seq data can effectively detect H3K4me3 marks in single cells.
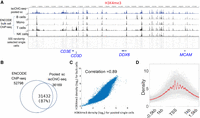
iscChIC-seq robustly detects H3K4me3 profiles in human white blood cells. (A) A genome browser snapshot showing panels of H3K4me3 profiles in human white blood cells. The top blue track shows the pooled single-cell data from iscChIC-seq. The bottom track shows 500 randomly selected single cells. The middle tracks display the ENCODE bulk cell ChIP-seq data from different cells indicated on the left. (B) A Venn diagram showing the overlap of the enriched regions (peaks) of H3K4me3 profiles measured by ChIP-seq using bulk cells and by the pooled single-cell data. (C) A scatterplot of the H3K4me3 read density of ChIP-seq (bulk-cell) versus that of pooled single cells from iscChIC-seq (2000 cells were randomly selected) at the genome-wide divided bins (the size of the bin is 5 kb). The Pearson's correlation is equal to 0.89. (D) A TSS profile plot showing the H3K4me3 profile around TSSs for all single cells (gray) and the pooled single cells (red).
To further study the performance of iscChIC-seq, we compared the sensitivity (i.e., percentage of true peaks recovered) and precision (i.e., percentage of reads located in true peaks) of iscChIC-seq to both scCUT&Tag (Kaya-Okur et al. 2019) and scChIP-seq (Grosselin et al. 2019). Note that H3K4me2 from scCUT&Tag was compared to H3K4me3 from either iscChIC-seq and scChIP-seq due to the lack of published single-cell H3K4me3 data using scCUT&Tag. The results showed that iscChIC-seq has the best performance in sensitivity, whereas its precision is either compatible with or slightly lower than scChIP-seq or scCUT&Tag (Supplemental Fig. S3). To check the reproducibility of iscChIC-seq, we generated two sets of single-cell H3K4me3 data. We pooled the single cells in each set of data and compared the H3K4me3 density between the two pooled sets. The results showed that the two replicates are highly correlated (r = 0.96) (Supplemental Fig. S4).
Next, we examined if different cell types of the human WBCs, which contain T cells, NK cells, monocytes, and B cells, could be identified from the iscChIC-seq data. For this purpose, a combined reference set of H3K4me3 peaks for human WBCs was first computed using the ENCODE bulk-cell H3K4me3 ChIP-seq data (Methods). By applying the silhouette analysis (Rousseeuw 1987), six was found to be the optimal number of clusters (Supplemental Fig. S5A; Fig. 3A). To annotate the cells in each cluster, we pooled the cells from each cluster and identified the H3K4me3 peaks that are specific to each cluster. Using the ENCODE T cell, B cell, NK cell, and monocyte bulk-cell H3K4me3 ChIP-seq data, we identified the peaks that are specific to each cell type. Next, the statistical significance of the overlap between the two types of specific peaks was calculated using a hypergeometric test, which robustly annotated four of the six clusters to be monocytes, T cells, B cells, and NK cells whereas the other two clusters could not be clearly annotated (Fig. 3A,B; Methods). Subsampling using 33% of single cells from each cluster confirmed the accurate and reproducible annotation of these cells (Supplemental Fig. S5B; Methods). From the four annotated clusters, 1610 monocytes, 1265 T cells, 898 NK cells, and 446 B cells were obtained.
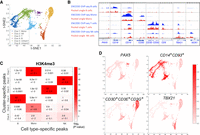
Identification of sub-cell types in white blood cells based on clusters generated from single-cell H3K4me3 profiles. (A) A t-SNE visualization of cells by applying the t-SNE analysis on the matrix Ec. Cell type annotations of clusters were obtained by the analysis in part B. (B) A heat map showing the significance of the overlap between the cluster-specific peaks from the H3K4me3 iscChIC-seq data (Fig. 3A) and cell type–specific peaks from ENCODE H3K4me3 ChIP-seq data. The y-axis refers to the cluster-specific peaks and x-axis refers to the cell type–specific peaks. The values before the +/− sign refer to the average negative logarithm of the P-value for the overlap between the two types of peaks over 100 subsamples. The values behind the +/− sign refer to the standard deviation of the negative logarithm of the P-value over 100 subsamples. (C) Genome browser snapshots showing the H3K4me3 profiles from bulk-cell ChIP-seq data and pooled single-cell iscChIC-seq data. The ChIP-seq data for B cells, monocytes, T cells, and NK cells were downloaded from ENCODE (red). The pooled H3K4me3 iscChIC-seq data for each identified cell type (Fig. 3A) are displayed (blue). For the iscChIC-seq data, 1610 monocytes, 1265 T cells, 898 NK cells, and 446 B cells were used. (D) A t-SNE visualization of cells by applying the t-SNE analysis on the matrix Ec. H3K4me3 density of regions associated with different genes is plotted. The color level indicates the H3K4me3 density level.
Next, we compared the genomic profiles of the annotated pooled single-cell data (from cluster T, B, NK, and monocyte) with the genome profiles of ENCODE bulk-cell ChIP-seq data for the corresponding cell types. The analysis revealed that the annotated cluster of single cells showed a genomic profile highly similar to that of the corresponding bulk cells at the cell type–specific gene loci including PAX5, CD19, CD14, CD93, CD3D, CD5, TBX21, and NCR1 (Fig. 3C). By comparing the cell type–specific peaks identified from the ENCODE data and cluster-specific peaks identified from the pooled single cells, we found that ∼80%–90% of cell type–specific peaks were detected in the pooled single cells from the NK, monocyte, and T clusters, whereas only 26% of cell-specific peaks were detected in the pooled single cells from the B cluster (Supplemental Fig. S6), which may be related to the relatively small number of cells in the B cluster. However, in all cases, much lower fractions of cell type–specific peaks were detected from other cell types than the annotated cell type in the single-cell cluster, indicating that the signals from the pooled single cells are specific. Because H3K4me3 is an active mark, we compared the expression levels of genes associated with the specific peaks identified in the pooled single cells from each annotated cluster. The analysis indicated that the genes associated with cluster-specific peaks are expressed at significantly higher levels in the annotated cell type than the other cell types (Supplemental Fig. S7).
At the single-cell level, the majority of cells annotated as T cells, B cells, NK cells, and monocytes exhibited high H3K4me3 density in regions associated with CD3D+CD3E+CD3G+ (T cell–specific), PAX5 (B cell–specific), TBX21 (NK and T cell–specific), and CD14+CD93+ (monocyte-specific), respectively (Fig. 3D). Overall, these results indicate that iscChIC-seq could reliably identify different cell types from a complex population of cells such as WBCs. To estimate the minimum number of reads that was required for reliable clustering analysis, we have subsampled the reads in single-cell H3K4me3 data using different percentages (90%, 80%, 70%, 60%, and 50%) of reads from the original set of reads for each cell. An original cluster is considered to be successfully recovered if there is only one new cluster that has more than 40% of cells overlap with that of the original cluster. For H3K4me3, about 90% of reads were required to recover the original six clusters (Supplemental Fig. S8A,B). Similarly, to estimate the minimum number of cells that was required for reliable clustering analysis, we have subsampled the cells in single-cell H3K4me3 data using different percentages (90%, 70%, 50%, 30%). We observed that about 50% of cells (3000 cells) were required for H3K4me3 single-cell data to recover the six clusters (Supplemental Fig. S8C,D).
To test if iscChIC-seq works for detecting repressive histone marks, we applied it to profiling H3K27me3 in WBCs. Using a filtering approach similar to that used for H3K4me3 iscChIC-seq libraries, we detected 10,000 single cells each having about 40,000 unique reads on average. Using more stringent filtering criteria such that a cell has at least 4000 unique reads, it resulted in ∼9000 single cells each having about 45,000 reads on average. The genomic profiles of the pooled single cells were highly consistent with the profiles of the bulk-cell H3K27me3 ChIP-seq data from ENCODE (Fig. 4A; Supplemental Fig. S9). We detected a total of 79,110 and 35,246 enriched regions from the ENCODE bulk cell ChIP-seq data and the pooled single-cell data, respectively. Comparison of the ENCODE data with our single-cell data revealed that 31,726 of 35,246 (90%) H3K27me3 peaks from the pooled single cells overlapped with the peaks from the ENCODE H3K27me3 ChIP-seq data (Fig. 4B). The read densities of the pooled single-cell and the bulk-cell ChIP-seq data were highly correlated (r = 0.92) (Fig. 4C). Applying the silhouette analysis to H3K27me3 iscChIC-seq data, an optimal number of clusters equal to six was found (Supplemental Fig. S5C), which was the same as the H3K4me3 iscChIC-seq data. Similar to the H3K4me3 data, the clustering analysis of the H3K27me3 iscChIC-seq data revealed six clusters of cells (Fig. 4D; Methods). After pooling the cells from each cluster, the cluster-specific peaks were identified and compared to the T cell-, B cell-, NK cell-, and monocyte-specific peaks identified from the ENCODE bulk cell ChIP-seq data. Four cell clusters, including 1146 T cells, 432 B cells, 749 NK cells, and 2192 monocytes, were annotated by the significant overlap between the two types of peaks (Fig. 4E). Similar to H3K4me3, we estimated the minimum of number of reads and cells that was required for reliable clustering analysis. We found that about 80% of reads for H3K27me3 were required to recover the original six clusters (Supplemental Fig. S10A,B), whereas about 70% of cells (4900 cells) were required for H3K27me3 single-cell data to recover the six clusters (Supplemental Fig. S10C,D). Overall, these results indicate that iscChIC-seq could also reliably profile repressive histone marks in a mixed population of cells.

iscChIC-seq robustly detects H3K27me3 profiles in human white blood cells. (A) A genome browser snapshot showing H3K27me3 profiles in human white blood cells. The top blue track shows the pooled single-cell data from iscChIC-seq. The bottom track shows 500 randomly selected single cells. The middle tracks display the ENCODE bulk-cell ChIP-seq data from different cells indicated on the left. (B) A Venn diagram showing the overlap of the enriched regions (peaks) of H3K27me3 profiles measured by ChIP-seq using bulk cells and by the pooled single-cell data. (C) A scatterplot of the H3K27me3 read density of ChIP-seq (bulk-cell) versus that of pooled single cells from iscChIC-seq (2000 cells were randomly selected) at the genome-wide divided bins (the size of bin is 50 kb). The Pearson's correlation is equal to 0.92. (D) A t-SNE visualization of cells by applying the t-SNE analysis on the matrix Ec. Cell type annotations of clusters were obtained by the analysis in part E. (E) A heat map showing the significance of the overlap between the cluster-specific peaks from the H3K27me3 iscChIC-seq data (Fig. 4D) and cell type–specific peaks from ENCODE H3K27me3 ChIP-seq data. The y-axis refers to the cluster-specific peaks and x-axis refers to the cell type–specific peaks. The values before the +/− sign refer to the average negative logarithm of the P-value for the overlap between the two types of peaks over 100 subsamples. The values behind the +/− sign refer to the standard deviation of the negative logarithm of the P-value over 100 subsamples.
Different from ChIP-seq, ChIC-seq depends on antibody-guided cleavage of chromatin by MNase and thus may have bias toward open chromatin regions. To address this question, we identified all the DHSs from the ENCODE DNase-seq data sets from T, B, NK, and monocyte cells and analyzed the fraction of the ENCODE bulk-cell H3K4me3 ChIP-seq reads that overlapped with DHSs in each cell type. The analysis revealed that ∼60%–67% of H3K4me3 ChIP-seq reads from the ENCODE bulk-cell H3K4me3 ChIP-seq libraries fell into the DHS regions (Supplemental Table S5A). In contrast, ∼52%–56% of the H3K4me3 reads from the pooled single cells fell into the DHS regions (Supplemental Table S5A), suggesting that the specificity of the H3K4me3 reads from the iscChIC-seq libraries is slightly lower than that of the bulk-cell ChIP-seq libraries, which may be caused by differences in washing conditions and/or differences in cell numbers used for the experiments. We also similarly analyzed the H3K27me3 data. Our results indicate that, whereas ∼38%–53% of H3K27me3 reads from the ENCODE bulk-cell H3K27me3 ChIP-seq libraries fell into the DHS regions (Supplemental Table S5B), ∼33%–41% of the H3K27me3 reads from the pooled single cells fell into the DHS regions. Thus, the percentage of the H3K27me3 reads from the iscChIC-seq libraries in DHS regions is slightly lower than that from the bulk-cell libraries, indicating that the H3K27me3 reads detected by iscChIC-seq are not substantially biased toward open chromatin regions. To test if the iscChIC-seq reads are depleted from heterochromatic regions, we compared the overlap of the H3K27me3 iscChIC-seq reads or ENCODE bulk-cell H3K27me3 ChIP-seq reads with the ENCODE H3K9me3 ChIP-seq peaks. We found that a quarter of them are overlapping with H3K9me3 peaks. (Supplemental Fig. S11). The similar percentage between H3K27me3 iscChIC-seq data and ENCODE H3K27me3 bulk-cell ChIP-seq data suggested that the iscChIC-seq reads are depleted from heterochromatic regions. To further estimate the true positive and false positive rates of the iscChIC-seq reads, we assumed that the peaks from pooled single cells that overlap with those from ENCODE data are true positives and the peaks not overlapping with the ENCODE peaks are false positives. The analysis revealed that, whereas the false positive rate ranges from 1.6% to 2.0%, the true positive rate is ∼27%–22% for H3K4me3 and H3K27me3, respectively (Supplemental Table S5C). For scCUT&Tag (Kaya-Okur et al. 2019), the false positive rate ranges from 11% to 7%, and the true positive rate is ∼44%–51% for H3K4me2 and H3K27me3, respectively. For scChIP-seq (Grosselin et al. 2019), the false positive rate ranges from 14% to 28%, and the true positive rate is ∼77%–57% for H3K4me3 and H3K27me3, respectively. Although iscChIC-seq has a lower true positive rate compared to other methods, it also has a much lower false positive rate.
Because the same WBC populations were used in profiling single-cell H3K4me3 and single-cell H3K27me3, it would be important to examine if a cluster annotated with a cell type from H3K4me3 iscChIC-seq data is specifically correlated with the cluster annotated with the same cell type from H3K27me3 iscChIC-seq data. H3K4me3, an active modification, and H3K27me3, a repressive modification, are colocalized at some key regulatory genomic regions due to either bivalent modifications or cellular heterogeneity (Bernstein et al. 2006; Roh et al. 2006; Wang et al. 2009; Wei et al. 2009). The relative levels of these two modifications at these regions are related to each other and influence the expression of underlying genes (Roh et al. 2006). To test this possibility, we first identified 7873 TSS regions (±2.5 kb) which exhibited overlapping H3K4me3 and H3K27me3 peaks from the bulk-cell H3K4me3 and H3K27me3 ChIP-seq data in monocytes, T cells, B cells, and NK cells. Next, we identified cluster-specific H3K4me3 peaks among the 7873 bivalent genes from the H3K4me3 iscChIC-seq data, which are peaks that have a higher H3K4me3 methylation level in one cell cluster compared to all other clusters. To relate the H3K4me3 modification with the H3K27me3 modification in the iscChIC-seq data sets, we reasoned that, when the H3K4me3 level becomes higher, the H3K27me3 level should become lower. Thus, from the four cell clusters based on the H3K27me3 iscChIC-seq data, we identified the cluster-specific peaks among the 7873 bivalent genes, which are peaks that have a lower H3K27me3 methylation level in one cluster compared to all other clusters. Comparison between these two kinds of cluster-specific peaks revealed that the specific peaks of an H3K4me3 cluster is significantly overlapped with the specific peaks of the H3K27me3 cluster if they are annotated as the same cell type (Fig. 5A). These results indicate that the H3K4me3 level is negatively correlated to the H3K27me3 level in the bivalent genes. Further, we observed that cell-to-cell variation in H3K4me3 and H3K27me3 was positively correlated at bivalent domains in monocytes (Fig. 5B). To match the clusters from single-cell H3K4me3 and H3K27me3 data, we repeated the correlation analysis for B cells, NK cells, and T cells. Therefore, clusters annotated as B, T, monocyte, and NK from H3K4me3 data were compared with the clusters annotated as B, T, monocyte, and NK from H3K27me3 data. By computing the correlation between the cell-to-cell variation in these clusters (Methods), we found that B, T, monocyte, and NK clusters from H3K4me3 data have the highest correlation with B, T, monocyte, and NK clusters from H3K27me3 data, respectively (Fig. 5C). The P-value of this observation is 4 × 10−4 (Methods). This result suggests that cell-to-cell variations in H3K4me3 and H3K27me3 are potentially coregulated in the bivalent domains, which can be used to correlate the cell clusters identified from H3K4me3 and H3K27me3 single-cell data.
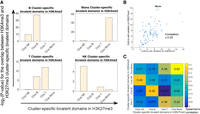
Correlation of cell clusters revealed from the single-cell H3K4me3 and H3K27me3 data by bivalent domains. (A) The cluster-specific peaks identified from the single-cell H3K4me3 and H3K27me3 data exhibit the highest overlap if they are from the same cell type. For each subplot, the cluster-specific peaks of H3K4me3 from one annotated cluster (as indicated on the top) were compared with the cluster-specific peaks of H3K27me3 from different clusters (as indicated below the plot). The y-axis in each subplot indicates the −log2 of P-value for the overlap between the cluster-specific peaks of H3K4me3 and cluster-specific peaks of H3K27me3. (B) A scatterplot between the cell-to-cell variation of H3K4me3 and H3K27me3 for clusters annotated as monocytes in bivalent domains (Methods). (C) Cluster-specific bivalent domains associated with H3K4me3 and H3K27me3 were computed for the purpose of finding the relationship between cell-to-cell variation in H3K4me3 and H3K27me3. For each comparison between the H3K4me3 and H3K27me3 clusters, the overlap between cluster-specific bivalent domains was considered; the Spearman's correlation between the coefficient of variation in H3K4me3 and H3K27me3 for these selected bivalent domains was calculated.
In this study, we developed iscChIC-seq to profile histone modification marks in single cells. This technique employs the highly efficient TdT enzyme combined with T4 DNA ligase to add a unique barcode to the DNA ends generated by antibody-guided MNase cleavage in each cell. Overall, we conclude that iscChIC-seq is a reliable method for studying histone modifications at the single-cell level, which provide important information for the differentiation status of cells.
Discussion
H3K4me3 is usually associated with gene activation, whereas H3K27me3 is associated with gene repression. Our previous single-cell H3K4me3 data indicated that the cell-to-cell variation in H3K4me3 is correlated with the cell-to-cell variation in gene expression (Ku et al. 2019), suggesting that single-cell histone modification data is useful in understanding the cellular heterogeneity in gene expression. However, due to the relatively small number of single-cells (scChIC-seq assay) or relatively sparse unique reads (iACT-seq and scCUT&Tag), the application of current techniques is limited. In this study, we combined the TdT + T4 DNA ligase-mediated barcoding strategy with the scChIC-seq protocol for iscChIC-seq, which enabled the analysis of either active or repressive histone modification profiles in more than 10,000 single cells in one experiment. The assay captured 11,000 unique reads for H3K4me3 or 45,000 reads for H3K27me3 per single cell, which is better than other high-throughput techniques for histone modifications. Different from PA-TN5-based techniques, iscChIC-seq works well for both active and repressive marks. Comparison with the bulk cell ChIP-seq data indicated that iscChIC-seq does not have substantial bias toward open chromatin regions for either active or repressive histone modification marks. In addition, iscChIC-seq does not require expensive equipment or special reagents and is thus easily accessible to most laboratories with molecular biology capabilities. Because PA-MNase effectively cleaves chromatin even in the presence of formaldehyde crosslinking, which stabilizes chromatin binding proteins, bulk-cell ChIC-seq is capable of detecting genome-wide binding sites of transcription factors and other chromatin proteins. Thus, although the specificity of iscChIC-seq appears to be comparable or slightly lower than PA-TN5-based methods for detecting histone modifications, it is potentially applicable to profiling transcription factors and chromatin-modifying enzymes at a single-cell level.
Our analysis in this study indicated that both the active H3K4me3 and repressive H3K27me3 iscChIC-seq data are effective in clustering the complex WBCs and sorting out different cell types. H3K4me3 and H3K27me3 are colocalized to a subset of genomic regions, which are termed “bivalent domains” (Bernstein et al. 2006; Roh et al. 2006). Bivalent modifications are usually associated with key differentiation regulator genes and thus show substantial changes during cell development or differentiation (Bernstein et al. 2006; Wei et al. 2009), and the expression of a bivalent gene is correlated with the relative level of H3K4me3 and H3K27me3 signals at the gene locus (Roh et al. 2006). Although the overlap of H3K4me3 and H3K27me3 peaks at these genomic regions may be caused by different mechanisms, including true bivalent modifications and cellular heterogeneity, the dynamic equilibrium of the two opposing modifications at these regions results from the competition of the corresponding enzymes to these regions. Hence, the two functionally opposite modifications may be coregulated but demonstrate opposite directions. Indeed, our data showed that the increased H3K4me3 levels in bivalent genes in one type of cell cluster are positively correlated with the decreased H3K27me3 levels in the same bivalent genes in the same type of cell cluster. The cell-to-cell variations in H3K4me3 and H3K27me3 are positively correlated and exhibit the highest correlation when the cell cluster annotated from the H3K4me3 iscChIC-seq data matches with the same type of cell cluster annotated from the H3K27me3 iscChIC-seq data. Thus, these properties of bivalent modifications can be used to specifically correlate the cell clusters annotated from different single-cell H3K4me3 and H3K27me3 data.
Overall, our data show that iscChIC-seq is a reliable single-cell technique for measuring histone modifications and potentially for chromatin binding proteins, which may find broad applications in studying cellular heterogeneity and differentiation status in complex developmental and disease systems.
Methods
iscChIC-seq method
Reagents
Histone H3 trimethyl Lys4 antibody was purchased from Sigma-Aldrich (cat. no. 07-473), and histone H3 trimethyl Lys27 antibody was purchased from Diagenode (cat. no. pAb-069-050). Methanol-free formaldehyde solution and DSG (disuccinimidyl glutarate) were purchased from Thermo Fisher Scientific (cat. no. 28906, 20593). Terminal transferase was purchased from New England BioLabs (cat. no. M0315L).
PA-MNase induction and purification
PET15b-PA-MNase plasmid (Addgene #124883) was transformed into BL21-Gold(DE3) competent cells following the standard protocol and grown in 40 mL LB medium (containing ampicillin) overnight. The culture was diluted (1:50) into prewarmed LB medium (containing ampicillin) and shaken for 2 h at 37°C until an OD600 reached ∼0.6. Fresh IPTG was added to the culture to a final concentration of 1 mM and the culture was shaken for another 2.5 h. For PA-MNase purification, the cell pellet was collected, resuspended in 30 mL lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 1× EDTA-free protease inhibitor cocktail, 0.5 mM PMSF) supplemented with 30 mg lysozyme (Thermo Fisher Scientific), and incubated for 30 min on ice. The cell lysate was sonicated for 10 cycles (10 sec on, 10 sec off) and centrifuged at 10,000g for 20 min. A Sonicator Misonix 4000 was used for sonication. In the meantime, 2 mL of the 50% bead slurry were washed with lysis buffer. Then, the supernatant was collected, mixed with the bead slurry, and rotated for 1 h at 4°C. After spinning down, the beads were washed four times with 8 mL wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, 1× EDTA-free protease inhibitor cocktail, 0.5 mM PMSF), followed by three times elution with elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, 1× EDTA-free protease inhibitor cocktail, 0.5 mM PMSF). The purified fraction was mixed with glycerol and finally aliquoted into small tubes and stored at −80°C.
WBC preparation
Human blood samples were obtained from healthy donors from the NIH Blood Bank. The WBCs were isolated as previously described (Ku et al. 2019). Two-step fixation was modified from Tian et al. (2012) and performed at room temperature. First, 50 M cells were suspended in 50 mL PBS/MgCl2 containing 2 mM DSG and rotated for 45 min. After washing with PBS, the cells were resuspended in 45 mL culture medium DMEM containing 10% FBS; 3 mL 16% formaldehyde was added to a 1% final concentration and rotated for 5 min, then the reaction was stopped by adding glycine, followed by two washes with PBS. The cells were aliquoted into 2 × 106 cells per tube, frozen on dry ice, and stored at −80°C until use.
MNase digestion
To prepare the ProteinA-MNase and antibody complex, 10 μL antibody and 25 μL PA-MNase were pre-incubated on ice in 40 μL antibody binding buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 150 mM NaCl, 0.1% Triton X-100) for 30 min. Meanwhile, the fixed cells (0.25 million) were thawed on ice and resuspended in 200 μL antibody binding buffer. For H3K27me3 analysis, chromatin need to be first decondensed by suspending the fixed cells in 0.5 mL RIPA buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 150 mM NaCl, 0.2% SDS, 0.1% sodium deoxycholate, 1% Triton X-100) and incubated at room temperature for 10 min, followed by one wash in 0.5 mL antibody binding buffer. Then, the cells were mixed with PA-MNase and antibody complex, incubated on ice for 60 min, followed by three washes with 500 μL high-salt buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 400 mM NaCl, and 1% [v/v] Triton X-100). After washing in 200 μL rinsing buffer (10 mM Tris-HCl [pH 7.5], 10 mM NaCl, and 0.1% [v/v] Triton X-100), the cells were resuspended in 40 μL reaction solution buffer (10 mM Tris-HCl [pH 7.4], 10 mM NaCl, 0.1% [v/v] Triton X-100, 2 mM CaCl2) to activate MNase digestion and incubated for 3 min at 37°C in a water bath. The reaction was stopped by adding 4.4 μL 100 mM EGTA. The cells were pelleted by centrifugation at 500g for 5 min.
TdT + T4 ligation
The MNase cleavage sites were end-repaired by T4 Polynucleotide Kinase (PNK) for removal of 3′-phosphoryl groups and addition of 5′-phosphates to allow subsequent poly(G) tailing and ligation. After digestion, the cells were washed twice with 1 mL 1× T4 ligase buffer containing 0.1% NP-40, then suspended in 300 μL mixed T4 PNK buffer (1× T4 PNK buffer, 1 mM ATP, 30 μL T4 PNK enzyme) and incubated for 30 min at 37°C. Meanwhile, 96 barcode-P7 adaptors (Supplemental Table S2) were thawed, and 2.5 μL 10 μM barcode-P7 adaptors were added to a new 96-well PCR plate with multichannel pipette (one barcode per well). After incubation, the cells were washed once with 1 mL rinsing buffer, suspended with 516 μL nuclei resuspension buffer (1.27× T4 ligase buffer, 2.5 mM dGTP, 0.05% NP-40), and mixed with 526 μL enzyme dilution buffer (1.25× T4 ligase buffer, 52.5 μL terminal transferase, 78 μL T4 ligase). Then, 10 μL cell suspension was aliquoted, mixed with the 2.5 μL barcode-P7 adaptor in each well. Finally, the 12.5-μL reaction mixture (1× T4 ligase buffer, 1 mM dGTP, 0.02% NP-40, 0.5 μL terminal transferase, 0.75 μL T4 ligase) in the 96-well PCR plate was sealed completely and incubated for 60 min at 37°C.
Pool and split
After barcoding the MNase cleavage sites, the reaction system in the 96 wells were pooled together in a solution trough containing 500 μL stop buffer (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 10 mM EDTA, 0.1% [v/v] Triton X-100), the cells were pelleted, resuspended in 800 μL PBS, and sent to the flow cytometry core. Thirty cells were sorted in each well of a new 96-well plate using a BD FACSAria III cell sorter (BD Biosciences) and collected in 10 μL PBS containing 0.1% NP-40. In total, five plates were collected. After adding 3 μL reverse-crosslink buffer (50 mM Tris-HCl [pH 8.0], 25 ng/mL Proteinase K, and 0.1%NP-40) into each well by multichannel pipette, the plates were sealed completely, incubated in a PCR machine overnight at 65°C and for 10 min at 80°C to inactivate Proteinase K.
Library preparation and sequencing
After reverse-crosslinking, the DNA fragments with barcode adaptors were captured and labeled with second library indexes through 12 cycles of annealing and extension with 96 PCR1 index primers (Supplemental Table S2). The reaction was carried out by adding 15 μL 2× Phusion High-Fidelity PCR Master Mix with HF Buffer (New England BioLabs) and 2.5 μL 2 μM index primer (one index per well) into the reverse-crosslinked solution in 96 wells. Then, all the libraries were pooled together as described above and digested with 96 μL Exonuclease I (Thermo Fisher Scientific) for 30 min at 37°C to degrade the excess index primers. The DNAs were purified by a MinElute Reaction Cleanup kit (Qiagen) and eluted with 64 μL EB buffer (Qiagen). The A-tailing was performed in 1× NEBuffer 2 (New England BioLabs) by adding the Klenow fragment (3′ → 5′ exo-) (New England Biolabs) and 1 mM deoxyATP (New England Biolabs). After incubation for 30 min at 37°C, the DNAs were purified and eluted in 23 μL EB buffer. Then, the Illumina P5 adaptor was ligated to the A-tailing fragments using the T4 DNA ligase (New England BioLabs) by incubation overnight at 16°C. The DNAs were purified again and eluted in 15 μL EB buffer. PCR2 amplification was performed by adding the Phusion High-Fidelity PCR Master Mix with HF Buffer, i5 index primer (Supplemental Table S2), and P7-cs2 primer (Supplemental Table S2) in the following conditions: 3 min at 98°C, 3 min at 57°C, 1 min at 72°C, 15 cycles of 10 sec at 98°C, 15 sec at 65°C, 30 sec at 72°C, followed by 5 min at 72°C. Then, the PCR products were run on a 2% E-Gel EX Agarose Gel (Invitrogen), and the 250–600-base-pair (bp) fragments were isolated and purified using a MinElute Gel Extraction kit (Qiagen). The concentration of the library was measured by a Qubit dsDNA HS kit (Thermo Fisher Scientific). The paired-end sequencing was performed on Illumina HiSeq 3000.
Data analysis
Demultiplexing and data analysis of iscChIC-seq libraries
The scripts for demultiplexing and genome-wide mapping are available at GitHub (https://github.com/wailimku/iscChIC-seq.git). For profiling each type of histone mark, 30 single cells were sorted into each of the 480 wells by FACS and sent to sequencing after the library's preparation steps. All sequencing data was paired-end. The R2 reads contained the information of cell barcodes (Supplemental Table S2), in which the cell barcode sequences followed the common sequence AGAACCATGTCGTCAGTGTCCCCCCCCC. For each well, R1 reads were mapped to the human reference genome (UCSC hg18) using Bowtie 2 (Langmead and Salzberg 2012). Using the cell barcode information from R2 reads, we separated the mapped R1 reads into 96 sets corresponding to the 96 cell barcodes. Reads with mapping quality less than 10 were removed. To remove duplicated reads, for each barcode, all the identical reads were set as one read. Note that hg18 was used in this study because the current study is a follow-up study of scChIC-seq (where hg18 was used). Thus, a direct comparison between the data sets could be performed. We have mapped the reads of some samples to both hg18 and hg19 and observed that their mappability and genome-wide profiles are highly similar. Thus, we believe that the use of hg18 in this study would not affect the conclusions.
The estimated number of single cells in each well is based on the calculation strategy in the previous study (Rubin et al. 2019). In our iscChIC-seq experiment, the cells were distributed into 96 wells such that each well received 30 cells. Therefore, the number of barcodes predicted to not represent any cells = 96(1−1/96)30 = 70.12. There are at least 25 ( = 96 − 70.12) barcodes representing cells. We assumed that the top-ranked barcodes based on the number of reads correspond to cells. Similar to the previous study (Rubin et al. 2019), we used a conservative cutoff of 1000 reads per cell. As a result, combining all single-cell data from the 480 wells, we identified about 10,000 single cells for both H3K4me3 and H3K27me3. The mapping statistics for each of the single cells were included in Supplemental Table S3.
Quality analysis of the single-cell data
Visualization in genome browser
For H3K4me3 and H3K27me3, 2000 single cells were randomly selected (MATLAB command: randperm [N, 2000]) and pooled together as the pseudo-bulk-cell data. This pseudo-bulk-cell data was visualized using the WashU Epigenome Browser (Figs. 2A, 4A; Zhou et al. 2011). For H3K4me3, to compare with a benchmark, the H3K4me3 ChIP-seq data of different human white blood cell types (see Supplemental Table S4) were downloaded from the ENCODE Project (Kazachenka et al. 2018) shown in the genome browser (Fig. 2A). For H3K27me3, to compare with a benchmark, we also downloaded the H3K27me3 ChIP-seq data of different human white blood cell types (see Supplemental Table S4) from the ENCODE Project and visualized in the genome browser (Fig. 4A).
Peak calling
To examine the quality of the single-cell data, we compared the pooled single-cell data to the bulk-cell ChIP-seq data downloaded from ENCODE (Kazachenka et al. 2018). For both H3K4me3 and H3K27me3 marks, the information of the ENCODE data used in comparison could be found in Supplemental Table S4. Peaks of this ENCODE data were called using SICER (Zang et al. 2009; Xu et al. 2014). A final set of peaks for each histone mark was obtained by combining the peaks from different immune cell types. In total, the final combined sets of peaks obtained from ENCODE data contained 52,798 and 79,100 peaks for H3K4me3 and H3K27me3, respectively. Peaks from the pooled single cells were identified using SICER and their widths were fixed to be 3000 and 10,000 for H3K4me3 and H3K27me3, respectively. The overlap between peaks from the pooled single cells and the bulk-cell data were computed using the BEDTools (Quinlan and Hall 2010) intersect commands “BEDTools intersect -wa -a the pooled peak file -b the bulk cell peak file” (Supplemental Fig. S12). Therefore, the criterion of overlapped peaks from pooled cell and bulk-cell data is that there is at least 1 bp overlap.
Scatterplots
The human genome was equally divided into bins (bin size = 5 kb for H3K4me3; bin size = 50 kb for H3K27me3). The 50-kb bin size was selected as in a previous study (Grosselin et al. 2019), because H3K27me3 peaks spanned broad regions. For both bulk-cell and pooled single-cell libraries, the read density (counts per million, CPM) at each bin was calculated. The correlation between the logarithm of the read densities of two libraries was quantified using the Pearson's correlation coefficient (Figs. 2C, 4C).
TSS profile plots
For H3K4me3, the software HOMER (Heinz et al. 2010) was used to calculate the TSS density profile (annotatePeaks.pl tss hg18 -size 3000 -hist 20 –len 1) for each single-cell. In particular, a region of 3 kb around each TSS was considered. This region was then divided into 150 bins. The density profile was generated using the number of reads mapped onto the bin divided by the total number of mapped reads and averaged over all promoters.
Estimate of the collision rate in human and mouse mixing experiments
The same procedure of identification of cells was used for the species mixing data set. Reads were mapped to both the hg18 and mm9 reference genome using Bowtie 2. Cells were identified using the procedure described above, in which the top-ranked barcodes based on the number of reads corresponds to cells. We also used the same cutoff of 1000 reads per cell. We identified human and mouse collision as those that had less than a 15× enrichment over the minor genome. The collision rate is estimated as two times the number of human and mouse collisions over the total number of cells. Note that we have mapped the reads of some samples to both mm9 and mm10 and observed that their mappability and genome-wide profiles are highly similar. Thus, we believe that the use of mm9 in this study would not affect the conclusions.
Clustering analysis for the iscChIC-seq data
Expression matrix
Single cells with reads more than 3000 (4000) were first selected. This resulted in 7798 and 9207 single cells for H3K4me3 and H3K27me3, respectively. Second, it was required that the fraction of reads in peaks higher than 0.15 (0.15) were selected for clustering analysis for H3K4me3 (H3K27me3) single-cell data. This resulted in 6021 and 7038 single cells for H3K4me3 and H3K27me3, respectively.
For each cell in H3K4me3 (H3K27me3), reads located within the 52,978 (79,100) combined H3K4me3 (H3K227me3) were counted. We
applied a consensus clustering approach, similar to SC3 (Kiselev et al. 2017), to the iscChIC-seq data. First, we computed a read count matrix R, in which the columns correspond to cells and rows correspond to the peaks. Rij indicates the number of reads at the ith peak from the Jth cell. Each column in the read count matrix was divided by the library size and multiplied by a factor of 106. The resulting matrix is denoted as M. The log2 transformation was further applied, resulting in M′ where M′ = log2 (M + 1). For filtering the noninformative bins, a binary matrix Mb was obtained from M′ and defined as
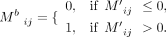 The ith row (peak) in the matrix M′ would be selected if
The ith row (peak) in the matrix M′ would be selected if 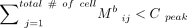 , where Cpeak is a cutoff value equal to 100 for both H3K4me3 and H3K27me3, respectively. The filtering of these bins is based on the assumption
that reads at a bin should be found in more single cells if the bin is more informative. We denoted the expression matrix
after the deletion of rows (peaks) as M′′.
, where Cpeak is a cutoff value equal to 100 for both H3K4me3 and H3K27me3, respectively. The filtering of these bins is based on the assumption
that reads at a bin should be found in more single cells if the bin is more informative. We denoted the expression matrix
after the deletion of rows (peaks) as M′′.
Calculation of the Laplacian matrix
Consider mj to be a vector equal to the jth column (cells) of M′′. First, we computed the similarity between cells using Pearson's correlation and resulting in a correlation matrix C. In particular, Cij is the Pearson's correlation value between the vectors mj and mi. Thus, the rows and columns of the matrix C correspond to single cells. The Laplacian matrix L is defined by L = I − D−1/2AD−1/2, where I is the identity matrix. A is a similarity matrix where A = e−(2−C)/max(2−C). Note that D is the degree matrix of A, a diagonal matrix that contains the row-sums of A on the diagonal 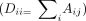 . We computed the eigenvectors of the Laplacian matrix and formed a matrix V where each column represents an eigenvector. The columns of V from left to right are sorted in ascending order based on their corresponding eigenvalues.
. We computed the eigenvectors of the Laplacian matrix and formed a matrix V where each column represents an eigenvector. The columns of V from left to right are sorted in ascending order based on their corresponding eigenvalues.
Optimal number of clusters
We applied the silhouette analysis to determine the optimal number of clusters. First, we created a matrix 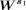 , which is a submatrix of V and
, which is a submatrix of V and 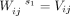 . Note that i is from one to the total number of bins and j = 1,…,s1. s1 is fixed to be 12 for both H3K4me3 and H3K27me3. Note that we have tried s1 to be larger than 12, up to 20. We observed that the results are the same. Here, we showed the results up to 12. We applied
the k-means method to the matrix
. Note that i is from one to the total number of bins and j = 1,…,s1. s1 is fixed to be 12 for both H3K4me3 and H3K27me3. Note that we have tried s1 to be larger than 12, up to 20. We observed that the results are the same. Here, we showed the results up to 12. We applied
the k-means method to the matrix 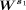 for clustering single cells into k clusters and computed the silhouette coefficient for the clusters. By varying the number of clusters k from four to 12, we determine the optimal k value by selecting the case of k having the largest silhouette coefficient value. The optimal k is equal to six for both H3K4me3 and H3K27me3.
for clustering single cells into k clusters and computed the silhouette coefficient for the clusters. By varying the number of clusters k from four to 12, we determine the optimal k value by selecting the case of k having the largest silhouette coefficient value. The optimal k is equal to six for both H3K4me3 and H3K27me3.
Clustering
A binary matrix E was considered in which its rows and columns correspond to single cells. The k-means method was applied to the matrix 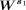 to cluster the single cells with k = 6. If cells i and j belong to the same cluster, Eij = Eji = 1; otherwise, 0. We consider s1 is between two and 15. For each s1, we repeated the clustering analysis 10 times, thus obtaining 10 different Es. A final matrix Ec is calculated by averaging all binary matrices from each individual clustering.
to cluster the single cells with k = 6. If cells i and j belong to the same cluster, Eij = Eji = 1; otherwise, 0. We consider s1 is between two and 15. For each s1, we repeated the clustering analysis 10 times, thus obtaining 10 different Es. A final matrix Ec is calculated by averaging all binary matrices from each individual clustering.
t-SNE visualization
We applied the dimension reduction method t-SNE to the matrix Ec. The position of single cells is visualized in the two-dimensional t-SNE representative space. Note that t-SNE was used for visualization and cells were clustered by applying the k-means method to Ec.
Hypergeometric test
A hypergeometric-based test was used to assess the significance of peaks that are both cluster-specific and cell type–specific. The rationale behind using the hypergeometric test was that, if a cluster-specific peak had any biological and/or functional association to a cell type, they would also be cell type–specific in a higher number of samples than expected by chance. We tested the null hypothesis that the properties for a peak that are cluster-specific and cell type–specific are independent.
Clusters annotation for both H3K4me3 and H3K27me3
Cluster annotations
After clustering single cells from the single-cell H3K4me3 or H3K27me3 data, we annotated the clusters to cell types using
the bulk-cell ENCODE data. First, we downloaded the H3K4me3 and H3K27me3 ENCODE data for B cells, monocytes, T cells, and
NK cells. There were at least two replicates for each histone mark and each cell type. For both H3K4me3 and H3K27me3, the
density matrices with log2 transformation (VB, Vmono, VT, VNK), which was similar to M′′, were computed for the four cell types, respectively. The number of rows was equal to the number of peaks, and the number
of columns was equal to the number of replicates. Note that the peaks in (VB, Vmono, VT, VNK) were the same as those in M′′. The two-sided Student's t-test was used to compute the cell type–specific peaks from the four density matrices (VB, Vmono, VT, VNK). The ith row vector of the matrix VZ (Z = B, mono, T, or NK) was denoted as 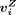 . The ith peak (row) was specific to a cell type Z if
. The ith peak (row) was specific to a cell type Z if 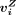 is significantly higher than all
is significantly higher than all 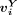 with a P-value of 0.05 and
with a P-value of 0.05 and 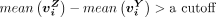 (0.4 for H3K27me3, and 0.2 for H3K4me3), where Y = B, mono, T, NK and Y ≠ Z. Different cutoff values were selected because we assumed that the bulk-cell and the pooled single cells should give a similar
number of peaks. For the purpose of cluster annotation, the sets of cell type–specific peaks (specific to cell type Z) were denoted as S4,an,Z and S27,an,Z for the H3K4me3 and H3K27me3 bulk-cell data, respectively. Note that the statistical significance of cell type–specific peaks
and cluster-specific peaks were determined by P-value instead of the FDR because very few cell type–specific peaks were discovered from ENCODE ChIP-seq data when using FDR.
(0.4 for H3K27me3, and 0.2 for H3K4me3), where Y = B, mono, T, NK and Y ≠ Z. Different cutoff values were selected because we assumed that the bulk-cell and the pooled single cells should give a similar
number of peaks. For the purpose of cluster annotation, the sets of cell type–specific peaks (specific to cell type Z) were denoted as S4,an,Z and S27,an,Z for the H3K4me3 and H3K27me3 bulk-cell data, respectively. Note that the statistical significance of cell type–specific peaks
and cluster-specific peaks were determined by P-value instead of the FDR because very few cell type–specific peaks were discovered from ENCODE ChIP-seq data when using FDR.
For each histone mark, pseudo-bulk log2 density matrices 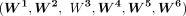 were computed for clusters 1, 2, 3, 4, 5, and 6, respectively. In each of these matrices, the number of columns was equal
to the number of peaks, and the number of rows was equal to the number of pseudo-bulk replicates. Note that the peaks in
were computed for clusters 1, 2, 3, 4, 5, and 6, respectively. In each of these matrices, the number of columns was equal
to the number of peaks, and the number of rows was equal to the number of pseudo-bulk replicates. Note that the peaks in 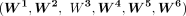 were the same as those in M′′. To generate
were the same as those in M′′. To generate 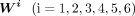 , six subsamples of cells were randomly selected from the cells belonging to cluster i, in which the size of each subsample was equal to one-third of the number of cells belonging to cluster i. By pooling the cells in each subsample, the log2 density for each peak was calculated for obtaining Wi. The jth row of Wi was denoted as
, six subsamples of cells were randomly selected from the cells belonging to cluster i, in which the size of each subsample was equal to one-third of the number of cells belonging to cluster i. By pooling the cells in each subsample, the log2 density for each peak was calculated for obtaining Wi. The jth row of Wi was denoted as 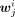 . The jth peak was specific to a cluster i if
. The jth peak was specific to a cluster i if 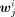 was significantly higher than all
was significantly higher than all 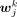 , where k = 1, 2, 3, 4, 5, 6 and k ≠ i. Note that the P-value computed by the two-sided Student's t-test was required to be smaller than 0.05 and
, where k = 1, 2, 3, 4, 5, 6 and k ≠ i. Note that the P-value computed by the two-sided Student's t-test was required to be smaller than 0.05 and 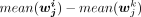 was higher than a cutoff (0.1 for both H3K4me3 and H3K27me3). The sets of cluster-specific peaks (specific to cluster i) for the use of cluster annotation were denoted as X4,an,i and X27,an,i for the H3K4me3 and H3K27me3 bulk-cell data, respectively.
was higher than a cutoff (0.1 for both H3K4me3 and H3K27me3). The sets of cluster-specific peaks (specific to cluster i) for the use of cluster annotation were denoted as X4,an,i and X27,an,i for the H3K4me3 and H3K27me3 bulk-cell data, respectively.
The set of cluster-specific peaks and cell type–specific peaks were compared. For H3K4me3 data, the P-value for the intersect between a cell type Z and a cluster i 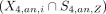 was computed by the hypergeometric test. A cluster i was considered to be annotated validly to a cell type Z if the P-value for
was computed by the hypergeometric test. A cluster i was considered to be annotated validly to a cell type Z if the P-value for 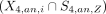 is smaller than 1 × 10−5 and the P-value for other comparisons
is smaller than 1 × 10−5 and the P-value for other comparisons 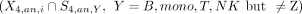 is greater than 1 × 10−5.
is greater than 1 × 10−5.
Reproducibility of cluster annotations
To check how reproducible the cluster annotations are, we repeated the computations 100 times and the cluster density matrices were regenerated each time via the same subsampling procedures. The mean and the standard deviation of the P-value in the comparisons were computed and are shown in Figs. 3B and 4E. Also, the frequency for a cluster to obtain a valid annotation was recorded and shown in Supplemental Figure S5, B and D. To consider that a cluster annotation is valid finally, we required that the frequency is greater than 0.9.
Matching the clusters between H3K4me3 and H3K27me3 marks
For either single-cell H3K4me3 or H3K27me3 data, six clusters were found where four of them were annotated as monocytes, T cells, B cells, and NK cells, respectively. If a cluster obtained from single-cell H3K4me3 data annotated with a cell type, this cluster was expected to correlate with the cluster obtained from single-cell H3K27me3 data annotated with the same cell type.
Bivalent domains were defined as regions where both H3K4me3 and H3K27me3 peaks obtained from ENCODE data were overlapped (command:
BEDTools intersect -a ‘H3K27me3 peak file’ -b ‘H3K4me3 peak file’) (Supplemental Fig. S12), and at least a 100-bp overlap was required for the estimation of one nucleosome overlap. In this manner, 25,951 bivalent
domains were obtained, in which 7989 bivalent domains were overlapped with the TSS regions. For both single-cell H3K4me3 and
H3K27me3 data, we computed the pseudo-bulk log2 density matrices (WB,4, Wmono,4, WT,4, WNK,4 and WB,27, Wmono,27, WT,27, WNK,27) for clusters annotated to B cells, monocytes, T cells, and NK cells, respectively. To generate WZ,4 or WZ,27, six subsamples of cells were randomly selected from the cells belonging to a cluster annotated to cell type Z, in which the size of each subsample was equal to two-thirds of the number of cells belonging to that cluster. By pooling
the cells in each subsample, the log2 density for each peak was calculated for obtaining WZ,4 or WZ,27. The jth row of WZ,4 was denoted as 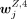 , and the jth row of WZ,27 was denoted as
, and the jth row of WZ,27 was denoted as 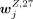 . A peak was specific to an H3K4me3 cluster annotated to cell type Z if
. A peak was specific to an H3K4me3 cluster annotated to cell type Z if 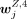 was significantly higher than all
was significantly higher than all 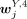 , where Y = B, mono, T, NK but Y ≠ Z. Note that the FDR of the P-value (computed by the two-sided Student's t-test) was required to be smaller than 0.05 and
, where Y = B, mono, T, NK but Y ≠ Z. Note that the FDR of the P-value (computed by the two-sided Student's t-test) was required to be smaller than 0.05 and 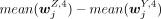 larger than 0.3. A peak was specific to an H3K27me3 cluster annotated to cell type Z if
larger than 0.3. A peak was specific to an H3K27me3 cluster annotated to cell type Z if 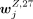 was significantly lower than all
was significantly lower than all 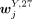 , where Y = B, mono, T, NK but Y ≠ Z. Note that the FDR for the P-value was required to be smaller than 0.05 and
, where Y = B, mono, T, NK but Y ≠ Z. Note that the FDR for the P-value was required to be smaller than 0.05 and 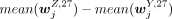 smaller than 0.3. The sets of cluster-specific peaks (specific to cluster annotated to cell type Z) for the use of matching H3K4me3 and H3K27me3 clusters were denoted as X4,mat,Z and X27,mat,Z for the H3K4me3 and H3K27me3 clusters, respectively. The P-value for the intersection
smaller than 0.3. The sets of cluster-specific peaks (specific to cluster annotated to cell type Z) for the use of matching H3K4me3 and H3K27me3 clusters were denoted as X4,mat,Z and X27,mat,Z for the H3K4me3 and H3K27me3 clusters, respectively. The P-value for the intersection 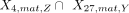 was computed by a hypergeometric test, where Z, Y = B, mono, T, NK.
was computed by a hypergeometric test, where Z, Y = B, mono, T, NK.
Relationship between cell-to-cell variation in H3K4me3 and H3K27me3
Different from the procedures of matching the H3K4me3 and H3K27me3 clusters, all bivalent domains were considered. Also, instead
of calculating the pseudo-bulk log2 density matrices, the vectors of coefficients of variation (cvB,4, cvmono,4, cvT,4, cvNK,4 and cvB,27, cvmono,27, cvT,27, cvNK,27) were calculated for the H3K4me3 and H3K27me3 clusters annotated to B cells, monocytes, T cells, and NK cells, respectively.
Similar to the single-cell log2 density matrices M′′, the log2 density matrices for single cells in H3K4me3 and H3K27me3 clusters were denoted as (MB,4, Mmono,4, MT,4, MNK,4, MB,27, Mmono,27, MT,27 and MNK,27), referring to H3K4me3 and H3K27me3 clusters annotated to B cells, monocytes, T cells and NK cells, respectively. Each of
these density matrices has the dimensions of the number of bivalent domains multiplied by the number of single cells in the
clusters. The vectors of coefficients of variation were computed using these density matrices over the single cells. For the
purpose of finding the relationship between cell-to-cell variation in H3K4me3 and H3K27me3, the jth bivalent domain was specific to an H3K4me3 cluster annotated to cell type Z if the value of 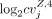 −
− 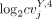 is larger than a cutoff (0.2) for any Y, where Y = B, mono, T, NK and Y ≠ Z, and the number of nonzero elements in the jth row of MZ,4 MB,4 is larger than 5% of the mean of the number of nonzero elements over all rows in MZ,4. The second requirement is to only include those relatively more confident CV values for each cluster. The same calculation
was applied to obtain the bivalent domains that were specific to an H3K27me3 cluster annotated to cell type Z. The sets of cluster-specific peaks (specific to a cluster annotated to cell type Z) for the use of finding the relationship between cell-to-cell variation in H3K4me3 and H3k27me3 were denoted as X4,CV,Z and X27,CV,Z for the H3K4me3 and H3K27me3 clusters, respectively. By considering the bivalent domains in the set of
is larger than a cutoff (0.2) for any Y, where Y = B, mono, T, NK and Y ≠ Z, and the number of nonzero elements in the jth row of MZ,4 MB,4 is larger than 5% of the mean of the number of nonzero elements over all rows in MZ,4. The second requirement is to only include those relatively more confident CV values for each cluster. The same calculation
was applied to obtain the bivalent domains that were specific to an H3K27me3 cluster annotated to cell type Z. The sets of cluster-specific peaks (specific to a cluster annotated to cell type Z) for the use of finding the relationship between cell-to-cell variation in H3K4me3 and H3k27me3 were denoted as X4,CV,Z and X27,CV,Z for the H3K4me3 and H3K27me3 clusters, respectively. By considering the bivalent domains in the set of 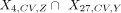 , the Spearman's correlation between cvZ,4 and cvY,27 was calculated and Y, Z = B, mono, T, and NK.
, the Spearman's correlation between cvZ,4 and cvY,27 was calculated and Y, Z = B, mono, T, and NK.
Software availability
The source code and scripts are available as Supplemental Code and at GitHub (https://github.com/wailimku/iscChIC-seq).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE139857.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank the National Heart, Lung, and Blood Institute DNA Sequencing Core Facility for sequencing the libraries and the National Heart, Lung, and Blood Institute Flow Cytometry Core facility for sorting the cells. The work was supported by the Division of Intramural Research, National Heart, Lung and Blood Institute.
Author contributions: K.Z. conceived the project. L.P. performed the experiments. W.L.K. analyzed the data. Y.C. contributed to data analysis. W.G. contributed to the design of the experiments. W.L.K., L.P., and K.Z. wrote the paper.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.260893.120.
-
Freely available online through the Genome Research Open Access option.
- Received January 8, 2020.
- Accepted March 1, 2021.
This is a work of the US Government.








