
Applications of single-cell genomics and computational strategies to study common disease and population-level variation
- 1Graduate Group in Genomics and Computational Biology, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania 19104, USA;
- 2Department of Biostatistics, Epidemiology and Informatics, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania 19104, USA;
- 3Division of Cardiology, Department of Medicine, Columbia University Irving Medical Center, New York, New York 10032, USA
Abstract
The advent and rapid development of single-cell technologies have made it possible to study cellular heterogeneity at an unprecedented resolution and scale. Cellular heterogeneity underlies phenotypic differences among individuals, and studying cellular heterogeneity is an important step toward our understanding of the disease molecular mechanism. Single-cell technologies offer opportunities to characterize cellular heterogeneity from different angles, but how to link cellular heterogeneity with disease phenotypes requires careful computational analysis. In this article, we will review the current applications of single-cell methods in human disease studies and describe what we have learned so far from existing studies about human genetic variation. As single-cell technologies are becoming widely applicable in human disease studies, population-level studies have become a reality. We will describe how we should go about pursuing and designing these studies, particularly how to select study subjects, how to determine the number of cells to sequence per subject, and the needed sequencing depth per cell. We also discuss computational strategies for the analysis of single-cell data and describe how single-cell data can be integrated with bulk tissue data and data generated from genome-wide association studies. Finally, we point out open problems and future research directions.
Human physiology is shaped by trillions of cells. Although all cells contain nearly identical genomes, cells are programmed via the complex rules of genomic regulation, which requires the consideration of many variables, such as chromatin conformation, DNA methylation, histone modifications, etc., to take on unique cell states. These cell states, such as those associated with our common notions of cell types, enable cells to perform specific functions. Through the interaction of cells within local structures defined by tissues and across different local structures in organ systems, cells generate higher level functions of human physiology, for example, serum glucose regulation via cells of the pancreas, liver, and skeletal muscle.
Human diseases are often marked by abnormalities in high-level functions of human physiology that are caused by abnormalities in subpopulations of cells. One fundamental goal of human disease research is to identify the appropriate perturbations, for example, taking a drug or eating a certain diet, that will produce molecular changes in the subpopulation of cells to fix aberrant behavior; in doing so, such perturbations should produce downstream changes in higher-level physiology that will achieve improvement in health status. Moreover, precision medicine aims to achieve this goal by considering the influence of genetics (Ashley 2016). Achieving the ability to predict the effect of perturbations in humans to improve health will require an unmasking of the complex regulation of the cell and improved understanding of how cell interactions shape human physiology.
The advent of high-throughput single-cell genomics technologies has brought the scientific community one step closer toward meeting this fundamental goal (Linnarsson and Teichmann 2016). As single-cell RNA-sequencing (scRNA-seq) has been adopted earliest by the scientific community, its use has now become widespread and the technology has improved rapidly. At present, it is now common for laboratories to assay genome-wide transcriptomes of thousands of cells in a single scRNA-seq experiment (Aldridge and Teichmann 2020). Recent years have brought on continued development of single-cell technologies. The cost of single-cell experiments continues to cheapen. Technologies that enable the measurement of new information about single cells—for example, chromatin accessibility (Cusanovich et al. 2015; Lake et al. 2018; Preissl et al. 2018), protein quantification (Oikonomou et al. 2020; Brunner et al. 2021; Specht et al. 2021), spatial location (Moffitt et al. 2018; Eng et al. 2019; Takei et al. 2021), and RNA velocity (Qiu et al. 2020)—have been developed. Further, it has now become possible to profile multiple molecular modalities simultaneously within the same cell (Macaulay et al. 2017; Stoeckius et al. 2017; Cao et al. 2018; Chen et al. 2019; Zhu et al. 2019; Fiskin et al. 2020; Ma et al. 2020; Swanson et al. 2021; Xiong et al. 2021).
In this article, we first review the current state of single-cell studies in common disease and then discuss factors that need to be considered when designing a large-scale population-based single-cell study. We then describe computational strategies for the analysis of population-scale single-cell data. We conclude by summarizing lessons learned so far from existing single-cell studies of human disease and point out open questions and new opportunities for future research.
Applications of single-cell genomics to characterize cell state abnormalities in human disease
Although recent years have seen the development of single-cell technologies to survey new molecular modalities such as proteins and chromatin accessibility, scRNA-seq has been mostly widely used to study human disease because of its maturity. Since the first transcriptome-wide profiling of mRNA by high-throughput sequencing in a single cell was reported in 2009 (Tang et al. 2009), scRNA-seq has increasingly gained popularity owing to its ability to survey cell state diversity in an unbiased fashion. In the past few years, we have witnessed rapid development of scRNA-seq technology both in throughput and in detection sensitivity (Svensson et al. 2018). In particular, sample multiplexing and droplet-based approaches allow several thousands of cells to be assayed simultaneously. These technological advances and the increased adoption of scRNA-seq approaches have begun to shift the application of this method from descriptive analyses of cell heterogeneity closer toward the understanding of disease mechanisms.
scRNA-seq has been used in several contexts to characterize cell state differences between diseased and nondiseased individuals in cross-sectional designs. Type 2 diabetes (T2D) is among the disease fields that has adopted scRNA-seq since the earliest stages of scRNA-seq technology. In 2016 alone, there were six published studies that used scRNA-seq to profile the transcriptomes of pancreatic islets in healthy and T2D donors. Although the initial study only had less than 100 cells (Li et al. 2016), later studies have increased the number of cells as well as the number of donors (Baron et al. 2016; Segerstolpe et al. 2016; Wang et al. 2016; Xin et al. 2016; Lawlor et al. 2017). Most notable among these studies, Segerstolpe et al. (2016) profiled more than 2200 cells in six healthy and four T2D donors, the largest single-cell study in T2D at that time. Using the Smart-seq2 protocol, they generated transcriptional profiles of individual pancreatic endocrine and exocrine cells of healthy and T2D donors and simultaneously defined the transcriptional signatures of both abundant and rare cell types in the pancreas, including delta, gamma, epsilon, stellate, immune, and endothelial cells. Further analyses revealed cell type–specific gene expression and novel subpopulations, as well as gene correlations to body mass index and gene expression alterations in diabetes. After assigning cells to cell types, they observed that cell types grouped according to donor, yet they were able to identify subpopulations and cellular states after correcting for donor differences. Their computational analyses showed the power of cell type–resolved analyses and revealed cell type–specific gene expression programs, subpopulations, and transcriptional alterations in T2D. scRNA-seq has shown broad use and impact in other disease areas as well, such as Alzheimer's disease (AD). By analyzing single-nucleus RNA-seq (snRNA-seq) data from the prefrontal cortex of 48 individuals with varying degrees of AD pathology, Mathys et al. (2019) identified transcriptionally distinct subpopulations, as well as cell type–specific disease-associated gene expression changes from 80,660 cells. Notably, they found that female cells were overrepresented in disease-associated subpopulations and that transcriptional responses were substantially different between sexes in several cell types. The relatively large number of subjects enabled the investigation of sex effects on AD for the first time.
Single-cell genomics has also impacted many aspects of cancer research. Cancer cell populations are subject to high mutation rates and show high epigenetic plasticity, making tumor cell populations heterogeneous and especially sensitive to selective pressures. Understanding the landscape of genetic and epigenetic heterogeneity, as well as characterizing downstream effects on expression and cell state, will be crucial to better understand tumor initiation and progression. Single-cell genomics has shown utility for understanding both aspects of cancer. To characterize genetic heterogeneity, Navin et al. (2011) conducted the first single-cell DNA-seq (scDNA-seq) study in cancer. With the analysis of hundreds of single cells collected from two breast cancer patients, they identified a genetically diverse subpopulation of cells that do not travel to the metastatic site and revealed a punctuated model of clonal expansion. Since then, many other studies have used scDNA-seq to investigate cancer clonal evolution and have consistently corroborated the genetic plasticity of cancer (Wang et al. 2014; Garvin et al. 2015; Bakker et al. 2016; Kim et al. 2018; Laks et al. 2019; Andor et al. 2020; Velazquez-Villarreal et al. 2020). More recently, novel computational methods have enabled the study of copy number alterations in an allele- and haplotype-specific manner. For example, Zaccaria and Raphael (2021) developed CHISEL, a method for allele-specific copy number analysis relying on external phasing, and applied it to a detailed lineage reconstruction of a breast cancer; Wu et al. (2021a) developed Alleloscope, and through the analysis of multiple types of cancer, they found pervasive haplotype-specific copy number changes seeding minor subclones throughout the course of cancer evolution. Furthermore, Alleloscope allows the detection of haplotype-differentiated subclones in single-cell ATAC-seq (scATAC-seq) data to examine the interplay of genetic and epigenetic evolution. It is the first time that scATAC-seq has been used to study cancer clonal evolution in an allele- and haplotype-specific manner, which enables the dissection of the contributions of chromosomal instability and chromatin remodeling to tumor evolution.
Single-cell approaches have also been applied to better understand tumor initiation and progression through the lens of transcription. Multiple studies have used scRNA-seq to identify tumor progenitor cells and to study their transition toward malignant cell states. For example, Kim et al. (2020) collected 208,506 cells of cancerous and noncancerous lung tissue and used these data to map the trajectory of normal epithelial cells toward malignant cell states in lung adenocarcinoma. Couturier et al. (2020) collected 53,586 adult glioblastoma and 22,637 normal fetal brain cells to map the developmental lineages of glioblastoma cells, which identified glial progenitor–like cells within the tumor that are highly proliferative. Crucially, such analyses can enable the identification of candidate molecular signaling pathways and regulators underlying the transition toward malignant cell states, which may form the basis for therapeutic development. For example, Couturier et al. (2020) identified E2F4 pathway activation in glial progenitor–like cells and showed that inhibition of this pathway more effectively targets these cells than does traditional temozolomide chemotherapy used in glioblastoma.
Beyond characterizing intrinsic cell state changes toward malignant phenotypes in cancer, single-cell approaches have also been used to help better understand nonintrinsic immune factors associated with the malignant tumor microenvironment. In lung adenocarcinoma, Kim et al. (2020) identified exhausted CD8+ T cells and identified macrophages and dendritic cells that express markers associated with immunosuppression, which may both play a crucial role in tumor progression. In kidney carcinoma, Zhang et al. (2021) used scRNA-seq data from normal and tumor tissue to identify tumor epithelial expression associated with aberrant myeloid recruitment, and they further used cell–cell communication analyses to characterize mediators of myeloid recruitment.
Single-cell genomics can also be deployed as a powerful diagnostic or prognostic tool in human disease. Particularly, in cancer, tumor heterogeneity may underlie differential survival and response to therapy. This suggests measurements of the tumor cell state distribution from single-cell genomics assays may offer novel insights needed to better diagnose, prognose, and treat cancer. Indeed, Zhang et al. (2021) identified associations between the presence of macrophage subtypes and patient survival in renal cell carcinoma, and they further suggested the fraction of endothelial cells has prognostic value in therapy response. Through integrative analysis of scRNA-seq data at transcriptomic, genotypic, molecular, and phenotypic levels, Wang et al. (2021) identified two subtypes of peritoneal carcinomatosis that were prognostically independent of clinical variables, and they further constructed a 12-gene prognostic signature that was predictive of cancer survival and validated the signature in large-scale gastric adenocarcinoma cohorts.
Existing efforts to study the population-level germline genetic determinants of cell state abnormalities in human disease
As scRNA-seq has become cheaper and more widespread, more groups have shown interest in understanding the role that germline genetic variation plays as a determinant of gene expression. The pioneering work by Wills et al. (2013) illustrated how single-cell analyses can provide mechanistic insights of genetic variants on gene expression variation. Through innovative analysis of 92 genes in the Wnt signaling pathway in 1440 cells from 15 individuals, the investigators showed, for the first time, that many parameters of gene expression, such as expression mean, burst size, burst frequency, and coexpression between cells, are genetically heritable and are masked when examining whole-tissue expression across cells. Later studies by Jiang et al. (2017) and Larsson et al. (2019) further provided evidence of genetically determined bursting kinetics. In particular, through genome-wide analysis of allele-specific bursting kinetics in mouse blastocyst cells and human fibroblast cells, Jiang et al. (2017) showed that a noticeable fraction of genes shows cis-dependent burst frequency. Larsson et al. (2019) further showed that burst frequency is primarily encoded in enhancers, whereas burst size is encoded in core promoters. These studies show the power of allelic scRNA-seq for investigating the genetic impact on transcriptional kinetics. One of the main approaches to identify causal factors in human disease is GWAS, and eQTL analysis has been pivotal for the functional interpretation of disease-associated loci. However, as shown by Jiang et al. (2017) , traditional eQTL analysis with bulk RNA-seq misses many associations that are bursting related. Thus, scRNA-seq can be used to identify a more complete set of genetic variants influencing expression and, specifically, can identify GWAS variants with functional effects on bursting parameters.
scRNA-seq has also been used in contexts to study single-cell expression effects of known genetic risk variants. For example, GWAS has identified more than 30 AD genetic risk loci, many of which appear to be related to innate immunity and microglial function, including APOE and TREM2 variants, which are associated with high genetic risks for sporadic AD (Guerreiro et al. 2013; Jonsson et al. 2013; Lambert et al. 2013; Efthymiou and Goate 2017; Neu et al. 2017; Kunkle et al. 2019; Bellenguez et al. 2021; Schwartzentruber et al. 2021). The TREM2 R47H variant is associated with an approximately threefold increased risk for AD, whereas the APOE E4 variant is associated with an approximately three- to fourfold increased risk with one copy and an approximately 10- to 12-fold increased risk with two copies. How genetic risk factors, like APOE and TREM2, intersect with cellular responses to AD pathology in human tissues is not understood. Using snRNA-seq of 131,239 nuclei obtained from 15 postmortem human brains with varied APOE and TREM2 genotypes and neuropathology, Nguyen et al. (2020a) identified distinct microglia subpopulations, including a subpopulation of CD163-positive amyloid-responsive microglia that are depleted in AD cases with APOE and TREM2 risk variants. These results were validated in an expanded cohort of AD cases, showing that APOE and TREM2 risk variants are associated with a significant reduction in CD163-postive amyloid-responsive microglia. This study showcased how genetic information, when integrated with single-cell transcriptomics, can advance our understanding of how genetic risk factors influence cellular responses to underlying pathologies.
Other studies have taken genome-wide approaches to identify the genetic determinants of disease-associated expression via single-cell eQTL studies. Sarkar et al. (2019) generated scRNA-seq data from induced pluripotent stem cells derived from 53 Yoruba individuals and investigated how genetic variants control gene expression variations both at the mean and the variance levels. Their analyses suggest that although the variance of gene expression is genetically controlled, the corresponding QTLs explain less phenotypic variance than eQTLs that control the mean expression. Although Wills et al. (2013) examined the relationship between coexpression and genetic variants, their study was limited by the small number of individuals and genes. Recently, van der Wijst et al. (2020) performed a similar study, but with 45 individuals and approximately 25,000 peripheral blood mononuclear cells. Through the construction of personalized coexpression networks, they identified genetic variants that significantly impact the coexpression of genes, implying that gene regulatory networks (GRNs) may vary across individuals. Because hundreds of genetic variants located in a few key regulatory pathways can contribute to complex diseases (Westra et al. 2013; Fagny et al. 2017), constructing personalized cell type–specific GRNs is a crucial step toward the understanding of genetic contributions to complex diseases. The recently formed Single-Cell eQTLGen Consortium will conduct GRN-based QTL analysis to examine genetic differences that change the architecture of the networks. Findings from such an analysis will enhance our basic understanding about the genetic contributions in gene expression and its regulation.
Efforts to detail the contribution of germline genetics to cell dynamics have also been made. Cuomo et al. (2020) studied the genetic determinants of iPS endoderm differentiation efficiency from 36,044 cells collected from 125 patient samples. More recently, in one of the largest scRNA-seq studies of humans to date, Jerber et al. (2021) studied the genetic determinants of iPS dopaminergic neuron differentiation from over 1 million cells collected from 215 human samples. Expanded efforts to study the genetic influence over other dynamic processes, such as differentiation, cell cycle, and circadian cycle, will greatly enhance our understanding of the context in which genetic variants exert their influence in disease.
Study design considerations for population-based single-cell studies
Although current studies have shown the power of single-cell technologies, these studies have been limited by the number of subjects. Nguyen et al. (2020a) were able to study the impact of APOE and TREM2 with a limited number of subjects owing to the use of a genetic risk variant–enriched study design. Their success in identifying risk variant–dependent microglia subpopulations underscored the importance of study design. As the field is now moving into large-scale population-based single-cell studies, it becomes even more important to consider study design–related issues. Given a fixed budget, a key question to ask is how to allocate the limited budget while maximizing the information gain. Parameters that need to be considered include the number of subjects, the number of cells per subject, and the sequencing depth per cell. Determination of these parameters will depend on the goals of the study and in the selection of study subjects.
Sample selection
When the goal is to investigate the interaction between known genetic risk factors and cellular responses to disease, an appealing design is the genetic risk factor–enriched design as was performed by Nguyen et al. (2020a). When genetic information is available, selecting genetic risk factor–enriched individuals can substantially reduce the number of needed subjects (Fig. 1A). Although DNA genotyping needs to be performed for a large number of individuals when studying rare variants, the cost of DNA genotyping is much lower than that of scRNA-seq. When genetic risk factors are unknown, an alternative design is the extreme phenotype sampling design, which selects individuals that cover both extreme ends of a disease spectrum (Fig. 1B). There is a well-established inverse relationship between the allelic frequency of a given variant and its effect size on the phenotype (Lander and Botstein 1989; Peloso et al. 2016), and many studies have shown that extreme phenotypes tend to occur in extreme cases with an excess of rare variants. The extreme phenotype sampling design offers a cost-effective strategy for studying the interaction between rare variants and cellular responses.
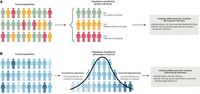
Sample selection strategy for population-based single-cell studies. (A) Genetic risk variant–enriched design in which individuals with the genetic risk variant are oversampled in order to achieve enough number of individuals that carry the genetic risk variant. (B) Extreme phenotype sampling design in which individuals with extremely low or extremely high phenotypes are selected. These extreme phenotype individuals are expected to carry more rare genetic risk variants than are individuals with intermediate phenotypes.
Number of cells and sequencing depth per cell
After study subjects are determined, the next consideration is how many cells to sequence and the sequencing depth per cell. Shall we sequence a large number of cells with shallow sequencing depth per cell or deeply sequence a few cells for each subject? Although common cell types can be detected and their gene expression levels reliably measured with a relatively small number of cells (Heimberg et al. 2016; Zhang et al. 2020), to reliably detect rare cell types, a larger number of cells is needed. Thus, the number of cells per subject is largely determined by the frequency of the rarest cell type of interest. A number of software packages have been developed to estimate the number of cells that must be sampled in a single-cell sequencing experiment. For example, based on the user-specified frequency of the rarest cell population and the number of populations with approximately this frequency, SCOPIT (Davis et al. 2019) can estimate the number of cells for planning single-cell sequencing experiments. Schmid et al. (2020) developed scPower, a more general framework for single-cell power calculation, in which they showed that, for a fixed budget, the number of cells per individual is the major determinant of power of detecting rare cell types and differentially expressed genes, followed by the number of subjects and read depth. In general, shallow sequencing of high numbers of cells per individual leads to a higher overall power than does deep sequencing of fewer cells.
Sample collection design to mitigate batch effects
Like many high-throughput technologies (Leek et al. 2010), single-cell methods are susceptible to batch effects, which refer to systematic differences among samples processed in different batches (Hicks et al. 2018). Although batch effects can be minimized by a completely randomized experimental design (Bacher and Kendziorski 2016), such designs are often infeasible for studies that involve human tissues because practical considerations require tissue samples to be processed immediately to avoid tissue degradation. Furthermore, for studies that involve a large number of subjects, patients are recruited sequentially, and single-cell experiments may span several days, months, or years apart, introducing systematic nonbiological differences that can confound biological variations. Recently, Song et al. (2020) proposed two experimental designs, the reference panel and the chain-type designs, that can reduce the impact of batch effects from the study design stage. Under the reference panel design, one batch is required to include cells from all cell types to serve as the reference panel, whereas the other batches need to have at least two cell types. The requirement of a reference batch that includes all cell types makes it difficult to achieve in practice. An alternative and more practical design is the chain-type design, which requires two cell types to be shared between every two consecutive batches. A special form of this design is when two cell types are shared among all batches, a situation that is easy to meet in real studies. Song et al. (2020) mathematically proved that under these two experimental designs, true biological variability can be separated from batch effects.
Cost reduction by cell type deconvolution analysis in bulk RNA-seq
Although the cost of scRNA-seq has reduced in recent years, using scRNA-seq for all study subjects in a large-scale population-based study might still be cost prohibitive. Integrative analysis of scRNA-seq and bulk RNA-seq data offers an alternative approach that can substantially reduce the cost while returning cell type–specific gene expression information. Such integrative analysis relies on cell type deconvolution, which aims to infer cell type proportions from bulk transcriptomics data. Many methods have been developed that use scRNA-seq data to infer cell type proportions in bulk RNA-seq samples in the last few years (Newman et al. 2015, 2019; Du et al. 2019; Wang et al. 2019; Jew et al. 2020; Dong et al. 2021). The estimated cell type proportions can be treated as known, and further analyses that incorporate these proportions as covariates can infer cell type–specific gene expression in each subject, as is performed in CIBERSORTx (Newman et al. 2019); detect allelic expression imbalance, as is performed in BSCET (Fan et al. 2021); or detect cell type–interacting QTLs (Donovan et al. 2020; Kim-Hellmuth et al. 2020) or cell composition QTLs (Park et al. 2021). The estimated cell type proportions can also be used to compare cell type compositions between diseased cases and controls. Determining whether certain cell types are increased or decreased in proportion in a disease state is informative for understanding disease pathophysiology. For example, such analyses have detected the loss of beta cells in T2D (Wang et al. 2019; Dong et al. 2021), an increase of disease-associated microglia in AD (Buttner et al. 2020), and an increase of microglia in advanced age-related macular degeneration (Lyu et al. 2021).
Computational analysis and considerations for population-based single-cell studies
In this section, we describe analytical strategies of population-based single-cell studies. An overview of single-cell analysis workflow is shown in Figure 2. Analysis of single-cell data starts from data preprocessing and normalization. Imputation may also be performed when needed. As other papers have thoroughly reviewed these aspects (Bacher and Kendziorski 2016; Hie et al. 2020; Hou et al. 2020; Lytal et al. 2020; Wu and Zhang 2020; Zhang and Zhang 2020; Ahlmann-Eltze and Huber 2021; Melsted et al. 2021; Slovin et al. 2021), we will focus our discussion on the downstream statistical analyses.
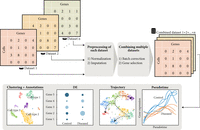
Overview of single-cell data analysis workflow. The typical workflow involves data preprocessing, combination of multiple single-cell data sets into a combined data set, clustering and cell type annotation, differential expression analysis, trajectory inference, and pseudotime analysis.
Correction of batch effects
Large-scale single-cell data sets with many subjects contain batch-specific systematic variations that present a challenge to data analysis. Batch effects are inevitable in analyses of human tissue and are prevalent in many single-cell studies (Hicks et al. 2018; Lähnemann et al. 2020). Failure to remove batch effects can not only generate false-positive signals but also obscure true biological variations. As such, many methods have been developed to remove batch effects in single-cell data analysis. Batch effect correction can be performed either in the original high-dimensional gene expression space or the low-dimensional embedding space, for example, gene expression data projected down onto principal components from principal component analysis. Batch effect correction methods such as LIGER (Welch et al. 2019), Conos (Barkas et al. 2019), Harmony (Korsunsky et al. 2019), BBKNN (Polanski et al. 2020), and DESC (Li et al. 2020) remove batch effects only for the embedding space. Although useful for profiling the overall characteristics of cells such as clustering and trajectory reconstruction, these methods cannot be used for downstream gene-level analysis like differential expression (DE) and coexpression.
To be useful for gene-level analysis, batch effects need to be removed in the original high-dimensional gene expression space. However, this task is much more challenging than batch effect correction in the embedding space (Lucken et al. 2020). Popular methods such as Seurat 3.0 (Stuart et al. 2019) rely on the mutual nearest-neighbor (MNN) approach (Haghverdi et al. 2018) to remove batch effects for each gene, but MNN can only analyze two batches at a time. Its performance is affected by the order in which batches are corrected, and it quickly becomes computationally infeasible when the number of batches gets large. Scanorama (Hie et al. 2019) overcomes the computational issue of MNN by finding matching elements among all batches at once, which also makes it invariant to batch order. A more desirable approach, however, should remove batch effects in gene expression for all batches simultaneously. A few neural network–based methods have been developed for this purpose. For example, scVI (Lopez et al. 2018) removes batch effects by conditioning on batch information in a variational autoencoder, which learns a nonlinear embedding of cells; SAVERCAT (Huang et al. 2020) uses a conditional variational autoencoder to remove batch effects through explicit modeling of batch information as covariates; and CarDEC (Lakkis et al. 2021) uses a joint autoencoder together with iterative clustering to remove batch effects. Beyond the ability to model all batches simultaneously, an additional advantage of these neural network–based methods is their flexibility in achieving multiple tasks within the same framework. These approaches can not only remove batch effects in the original high-dimensional gene expression space but also remove batch effects in the low-dimensional embedding space to facilitate cell clustering. Moreover, these methods can impute gene expression, which may be desirable for downstream gene-level analyses.
Annotation of cell identities
Consistent annotation of cell identities is also a critical step in population-based single-cell studies. As such studies involve a large number of individuals, the data generation may span multiple years and across multiple laboratories. For such studies, it becomes infeasible to use unsupervised clustering algorithms as these algorithms require the reanalysis of all cells whenever new data become available. Moreover, unsupervised clustering algorithms may have difficulty resolving cell subtypes whose differences are biologically meaningful (Kiselev et al. 2019). One attractive approach to circumvent these issues is to rely on available, well-annotated single-cell data sets, such as those contained in Azimuth (Hao et al. 2021). Using these high-quality reference data, methods have been developed to identify and annotate cell types in new data. For example, scmap (Kiselev et al. 2018) projects cells in a query data set to a space determined by highly informative genes selected from a well-labeled labeled data set and then assigns cell identities for cells in the query data based on their correlation with average cell type–specific gene expression in the reference data. scANVI (Xu et al. 2021), a semisupervised variant of scVI (Lopez et al. 2018), annotates cell types in a query data set by leveraging any available cell annotations in a reference. Seurat 3.0 classifies cells in the query data by finding anchor cell pairs between a well-labeled reference and the unlabeled query data sets. Scmap learns cell type–specific gene expression information only in the reference but ignores useful information in the query data; thus, it is vulnerable to batch effects and platform differences between the reference and query data. Although Seurat 3.0 uses information both in the reference and the query data in the identification of anchor pairs, it does not specifically use cell type label information in the reference.
An ideal approach for cell identity annotation should be able to use cell type–specific gene expression information both in the reference and the unlabeled query data. Although reference data sets have become increasingly comprehensive, cell types/subtypes may exist in the unlabeled query data that were not previously detected in the reference, for example, owing to differences between the query and reference data in cell sample size or to differences in subject-specific covariates, etc. As such, approaches should carefully balance the contribution of each data type in cell type assignment annotation. As large single-cell references are continuously generating well-annotated reference data across various tissues, an ideal approach should also be able to combine multiple references together so that the users can learn from these comprehensive maps when annotating their own data. To address these issues, transfer learning–based approaches have been developed. For example, ItClust (Hu et al. 2020a) borrows ideas from supervised cell type classification algorithms but also leverages information in target data to ensure sensitivity in classifying cells that are only present in the target data through the use of an iterative transfer learning approach with neural networks. scArches (Lotfollahi et al. 2021) relaxed the requirement of having raw data from the reference. Through reusing neural network models by adding input nodes and weights and then fine-tuning those, it learns the joint latent representations of the reference and the query data, which allows the identification of rare cell states in the query data that is otherwise difficult to detect. As the scale of single-cell studies continues to grow, we anticipate these transfer learning–based approaches will automate the labor-intensive clustering and annotation tasks and facilitate comparative analyses across tissues and disease conditions.
DE analysis
After cell identities are annotated, an important next step is to identify genes that are differentially expressed between conditions, for example, healthy versus diseased, within the same cell subpopulation. Although methods have been developed for DE analysis in scRNA-seq (Kharchenko et al. 2014; Finak et al. 2015; Korthauer et al. 2016; Jia et al. 2017), these methods ignore the effect of subject-specific covariates. Although subject-to-subject variation may have little impact on the identification of subpopulation-specific marker genes, their impact on DE analysis between different conditions within the same cell subpopulation is unknown. Through simulations, Crowell et al. (2020) investigated the performance of various methods in detecting DE genes in this situation. Interestingly, they found that the simple “pseudobulk” approach outperforms methods that are specifically designed for scRNA-seq. In such “pseudobulk” analysis, cell-level counts from a subpopulation are aggregated into a single observation per subject, which are then used as input for DE analysis using traditional bulk RNA-seq methods such as edgeR (Robinson et al. 2010), DESeq2 (Love et al. 2014), or limma-voom (Law et al. 2014). These aggregation-based DE methods not only are fast but also show a stable high performance across various scenarios, making them an appealing choice for large-scale scRNA-seq studies that involve many subjects. Notably, cell-level mixed models performed comparably to pseudobulk approaches in DE analysis, as the DE gene sets identified were similar. However, cell-level mixed models severely underestimated the expression differences of certain genes between different cell populations. For these genes, this is likely owing to the abundance of cells with zero counts, for which the gene's maximum likelihood estimate of the mean will be equal to zero for that cell. This becomes more likely for lowly expressed genes under sparse data settings, underscoring the need to model expression uncertainty. Perhaps future cell-level approaches can improve upon this issue by modeling gene expression uncertainty directly.
Differential splicing analysis
Previous studies have shown that genes showing changes in alternative splicing may reflect different biological processes from those with DE. For example, a recent scRNA-seq study in the adult mouse cortex found differences in splicing dynamics across cells were not explained by neuronal cell type definitions based on differences in isoform-agnostic transcript expression levels, suggesting that alternative splicing regulation might be orthogonal to transcriptional regulation in specifying neuronal identity and function (Feng et al. 2021). Therefore, differential alternative splicing may complement DE analysis in characterizing gene regulation. However, low sequencing depth, technical noise, and the lack of appropriate computational methods have precluded the investigation of splicing heterogeneity in most scRNA-seq studies. To date, only a few methods have been designed specifically for splicing analysis in scRNA-seq. Huang and Sanguinetti (2017) detected differential exon-usage by performing a pairwise comparison between every two cells. Song et al. (2017) quantified exon-inclusion levels based on junction-spanning reads. Qiu et al. (2017a) and Ntranos et al. (2019) detected differential transcript usage based on pre-estimated cell-specific isoform expressions or transcript compatibility counts. Hu et al. (2020b) detected differential alternative splicing by accounting for technical noise and low sequencing depth through grouping exons that originate from the same isoform(s). Although these methods have shown promising performance, they still have limited power for data without full-length transcript sequencing. Most single-cell studies use droplet-based technologies, for example, 10x Genomics (https://www.10xgenomics.com) or Drop-seq (Macosko et al. 2015), which have inherent limitations for splicing analysis owing to their sequencing of only the 3′ or 5′ end of the gene following fragmentation. Although Smart-seq2 (Picelli et al. 2014) can generate full-length transcripts, the lack of unique molecular identifiers (UMIs) makes it difficult to remove amplification bias. To fully characterize the splicing complexity of single cells, technologies with full-length transcriptome coverage and UMIs, such as ScISOr-Seq (Gupta et al. 2018; Joglekar et al. 2021) and SMART-seq3 (Hagemann-Jensen et al. 2020), are needed.
Trajectory analysis
A substantial portion of cell state variation can be explained by treating states as discrete; differences in our notion of “cell type” underlie large differences in cell morphology, function, and molecular composition across cells. Although treating cell states as discrete may be appropriate in many settings, cell state variation is best described as a continuum. Cells undergo gradual changes during cellular differentiation, as they transition from one cell type to another. Further, cell states can follow a continuum within a given cell type: Cell states are perturbed by both constant factors, such as the circadian clock, as well as asynchronous factors, such as the cell cycle. Characterizing continuous aspects of cell state and understanding the dynamics that give rise to them will be crucial to understand how cells function and how these functions can go awry in human disease.
Single-cell technologies provide a powerful tool to study continuous cell state variation. In particular, scRNA-seq has seen widespread use to characterize state differences owing to cell differentiation in both human developmental and adult-life contexts, for example, smooth muscle cell phenotypic switching during atherosclerosis (Wirka et al. 2019; Pan et al. 2020), subtype switching of macrophages during pathological cardiac hypertrophy (Ren et al. 2020), the transition of myeloid cells during the progression and regression of kidney disease (Conway et al. 2020), the transition from homeostatic microglia to amyloid-responsive microglia or motile microglia during AD progression (Nguyen et al. 2020a), and iPS-based models of cell type maturation (Cuomo et al. 2020; Jerber et al. 2021).
A key step in such analyses is the computational assignment of cells to continuous states, often referred to as trajectory or pseudotime assignment. For example, in the simple case of cells differentiating from one cell type into another, cells could be assigned a continuous value from zero to one, where zero indicates the starting cell state, one indicates the final cell state, and intermediate values indicate intermediate states. After the assignment of cells to continuous states, researchers can characterize molecular changes, such as mRNA expression associated with changes in cell state, and generate candidate mechanisms underlying these changes, for example, changes in transcription factor (TF) activity.
The growth of single-cell technologies has been accompanied by the development of several computational tools for trajectory inference (Saelens et al. 2019). The choice and use of such tools designed for scRNA-seq require careful consideration. Before trajectory inference, the high-dimensional gene expression data may be transformed into a dimensionality-reduced representation. In the context of large-scale human disease studies, compressed representations can confer benefits as a tool for noise reduction when cells are shallowly sequenced and for improvement of the computational efficiency of trajectory assignment methods. Using this input, trajectory inference can be performed. A crucial consideration in selecting a trajectory method is the user's expectation of the underlying trajectory topology. For settings in which the user has no expectation of the trajectory topology, flexible tools have been developed that can detect a wide range of topologies, such as linear, circular, trees, and disconnected components (Ji and Ji 2016; Qiu et al. 2017b; Street et al. 2018; Wolf et al. 2019).
One consideration in the use of trajectory reconstruction methods is the relatively high degree of uncertainty of the trajectory shape and cell ordering (Saelens et al. 2019). This consideration becomes more crucial when the underlying trajectory has not been adequately sampled, that is, too few cells, which may produce unstable results owing to the similar likelihood of multiple topological hypotheses. Continued progress in trajectory inference methods to incorporate RNA velocity information (Lange et al. 2020) and the quantification of trajectory uncertainty (Lin et al. 2021) may aid in resolving such ambiguities and in interpreting results, respectively. Given the challenges associated with flexible trajectory models, for cases in which users have expectations of the trajectory topology, it is recommended they use methods with inductive biases that reflect this expectation. For example, variation across cells owing to cell cycle variation should be modeled by methods designed to detect circular topologies, such as reCAT (Liu et al. 2017).
Outlook and future research
Thus far, single-cell technologies have seen use in characterizing cell state differences among diseased and healthy individuals. Incorporating genetic information, groups have now begun to identify variants influencing cell states. Nonetheless, although single-cell technologies have rapidly advanced our ability to survey multiple molecular modalities describing cellular behavior, we remain far from the ability to predict how molecular and/or behavioral perturbations will influence high-level physiological features to improve human health. We believe the following four areas will see great strides toward this goal in the near future.
Modeling the effect of genotypic variation on transcriptional regulation
The development of precision medicine therapies will benefit from predictive models to interpret how genetic variants influence gene expression. At present, eQTL studies have largely modeled variants as having additive, linear effects on the expression of individual genes. In the presence of small sample sizes, this is a reasonable approach. However, as regulatory element interactions influence transcription, for example, enhancer–promoter interactions (Schoenfelder and Fraser 2019; Fitz et al. 2020) and enhancer cooperativity (Huang et al. 2018), models that consider regulatory element variants to contribute independently to changes in transcription kinetics are likely misspecified. Moreover, the lack of variance explained by eQTL models assuming additive linear effects (Price et al. 2011; Lloyd-Jones et al. 2017) suggests substantial model improvements are required not only to identify variants with effects on gene expression but also to faithfully capture how they affect gene expression.
Moving beyond purely additive linear models, convolutional neural networks (CNNs) appear to be a promising approach toward modeling the role of genomic variants in cis regulatory logic. In particular, CNNs have already shown great promise in modeling the contribution of promoter genetic variation on mean gene expression levels. Agarwal and Shendure (2020) first introduced their algorithm, Xpresso, a CNN designed to predict steady-state mean expression levels using sequence features of gene promoters and gene bodies. Motivated by the high correlation of gene expression across cell types, Xpresso first demonstrated an ability to detect sequence features describing expression variation in a cell type–agnostic fashion. This suggests that rules exist that generalize across cell type–specific contexts, and indeed, inspection of the model identified a number of genomic features associated with steady state expression including ORF exon density, 5′ UTR GC content, and promoter CpG content. Nonetheless, promoter-based models cannot explain all genetically determined expression variation. Notably when Xpresso was trained on cell type–specific expression with accompanying chromatin accessibility data, genes with the largest prediction residuals were those adjacent to stretch enhancers. This suggests that an ideal model of cis regulatory expression likely will require the consideration of multiple layers of regulatory control, such as the role of enhancer sequences, 3D genome configuration, and chromatin accessibility. In a recent preprint, Avsec et al. (2021) introduced a novel model architecture, dubbed Enformer, which jointly considers distal and proximal regulatory sequences in gene expression prediction. When applied to bulk human expression data from GTEx, Enformer shows a substantial improvement in our ability to predict expression from sequences. Moreover, the investigators point toward the use of in silico perturbations of the model to yield candidate trans regulators of distal regulatory activity. Although promising, future work remains to incorporate other layers of transcriptional control into genetically determined models of gene expression. Innovation in computational method development will be essential for the advancement in our understanding of the transcriptional regulation effects of variants in the context of human disease.
The development of models to interpret the transcriptional regulation effects of human variants will also benefit from continued development in experimental assays. The largest existing single-cell eQTL studies have assayed hundreds of individuals (Cuomo et al. 2020; Jerber et al. 2021). Although an achievement, this is a limited sample size relative to the space of regulatory variation observed in humans. The development of high-throughput base editor mutagenesis technologies holds great promise to probe the role of genetic variation. Hanna et al. (2021) recently introduced a cytosine base editor to study the effect of 52,034 ClinVar variants in 3584 genes. Future efforts to pair base editor mutagenesis with scRNA-seq will greatly advance our ability to explore the space of regulatory variation from human cells at scale.
Construction of GRNs
Evidence suggests cis regulatory variation only modestly explains gene expression variation (Liu et al. 2019). Although this may partially reflect misspecified models of how cis regulatory variants affect expression, it also points to the need to model the role of trans effects. Gene expression is regulated by the interaction of cis regulatory elements with TFs. The activity of TFs, themselves, depends on their expression, which has its own regulatory logic. As such, faithfully modeling the role of trans effects on gene expression will require mapping cell type–specific GRNs that detail the gene targets of TFs. Mapping GRNs will enable researchers to better understand the underlying drivers of expression differences between cell states, such as differences in underlying TF levels. Further, they can inform predictions of how gene expression will change upon perturbations of TFs or upstream signaling pathways.
It has become increasingly common to estimate GRNs from steady-state scRNA-seq data, and several computational tools have been developed for this task. Although there are key nuances that distinguish each method, these tools generally construct GRNs by identifying gene pairs showing coexpression patterns within a given data set. GRNs are then represented as an undirected graph in which nodes represent genes and binary edges represent the presence or absence of relationships. To date, the application of GRN detection methods to scRNA-seq data has yielded results of mixed success. Using simulated scRNA-seq generated from ground truth GRNs, Nguyen et al. (2020b) recently showed that existing tools detect GRNs with success slightly better than random. This may reflect, in part, the inherent limitations of using scRNA-seq to detect GRNs. However, the general principles used by the best-performing GRN tools should form the basis for future computational developments. Notably, one of the earliest and most popular tools, SCENIC (Aibar et al. 2017), constructed networks most accurately across a variety of benchmarks. This is likely owing to SCENIC's inductive bias that predicted coregulated genes share motifs for an underlying TF, suggesting that methods incorporating domain knowledge may be better suited to construct GRNs using scRNA-seq. Nguyen et al. (2020b) also point out that existing tools assume GRN relationships are linear and that there are no interactions. This assumption may limit the power of GRN detection tools, as TF–TF interactions are known to significantly shape gene expression (Zeitlinger 2020). The interpretation of GRNs detected by existing tools is also challenging, as edges in GRN graphs are often undirected and may not represent functional relationships but, instead, correlations. Future approaches incorporating RNA velocity may help resolve the direction of GRN relationships from scRNA-seq.
Although GRN detection from steady-state scRNA-seq data has proved challenging, a promising alternative is the use of perturbation approaches paired with single-cell omics to map GRNs. Crucially, these approaches are high throughput in nature, allowing researchers to identify the regulatory targets of hundreds of TFs from a single tissue sample. Perturb-Seq (Dixit et al. 2016) first introduced the ability to generate a loss-of-function library of CRISPR guide RNAs to transfect a cell population and whose effects could be read out using single-cell transcriptomics. Using this technique, Dixit et al. (2016) were able to identify TF–gene regulatory relationships that were recapitulated using ChIP-seq. Depending on the loading concentration of guide RNAs, Perturb-Seq is also amendable to probing the transcriptional effects of higher order combinations of TF knockouts. A complementary approach to mapping GRNs is detailing how chromatin accessibility is perturbed by TF knockouts. Rubin et al. (2019) introduced Perturb-ATAC, which uses a loss-of-function library of CRISPR guide RNAs to assay their effects on single-cell chromatin accessibility. Using this approach, researchers may be able to preferentially identify TFs responsible for binding heterochromatin and promoting chromatin accessibility in particular cellular contexts. As such, Perturb-ATAC may be of particular relevance to help researchers identify pioneer factors that act as hubs in GRNs.
Integrative analysis of multiple molecular modalities and their correspondence with cell state
Although scRNA-seq has been predominantly used to characterize cell state differences between diseased and nondiseased individuals thus far, the emergence of single-cell multiomic technologies, wherein multiple molecular modalities are simultaneously profiled within the same cell, signifies an important next step in the study of human disease using single-cell approaches. Stoeckius et al. (2017) first introduced CITE-seq, an approach to jointly profile proteins and RNA in single cells. Since then, technological developments have made it possible to jointly profile the transcriptome in single cells with chromatin accessibility (Cao et al. 2018; Chen et al. 2019, Ma et al. 2020), DNA methylation (Gaiti et al. 2019; Luo et al. 2019), nucleosome occupancy (Pott 2017; Clark et al. 2018), chromatin occupancy (Xiong et al. 2021), or spatial location (Rodriques et al. 2019; Vickovic et al. 2019). Encouragingly, recent efforts show a trend toward increased detection sensitivity and cost reduction.
Single-cell multiomics will enable researchers to measure cell state on a more granular level, as different modalities may contain independent cell state information. Indeed, Hao et al. (2021) found protein information could segregate known T cell subtypes where mRNA could not, suggesting not only that multiomics can measure more granular aspects of cell state but also that these differences coincide with known aspects of biology that distinguish cell subtypes. Beyond independent information captured by individual modalities, multiomic data will also enable more meaningful measures of cell state via modeling of interactions between modalities that are known to modulate cell state, such as TF abundance and chromatin accessibility. Ultimately, the more granular cell state information provided by multiomic data will help researchers better distinguish between diseased and healthy cell states.
The development of tools to estimate cell state from single-cell multiomic data will be essential to maximize its utility. In brief, most tools estimate latent factors that maximize the joint probability of the observed data. Using these tools, researchers can study differences in cell states associated with disease and health. One of the earliest tools, LIGER (Welch et al. 2019), deploys an integrative nonnegative matrix factorization approach to estimate latent factors describing cell state. More recently, Argelaguet et al. (2020) introduced MOFA+, a Bayesian matrix factorization approach that uses priors to encourage the learning of sparse latent factors and loading matrices to improve their interpretability. Moving beyond linear approaches, Wu et al. (2021b) introduced BABEL, a nonlinear joint autoencoder approach. Although existing approaches to estimating cell state have made meaningful contributions to the analysis of multiomic data, they suffer from two main issues. First, models that aim to purely maximize the probability of the data are more likely to learn spurious statistical associations under sparse data settings and are less equipped to generalize to unseen data from new cell states. Indeed, Wu et al. (2021b) highlight their method's difficulty in generalizing to unseen cell states. Second, latent factors may be uninterpretable, making the identification of testable hypotheses for experimental follow-up challenging. In both respects, we believe future cell state estimation tools would benefit from using latent variable models based on underlying explanatory factors that reflect known biology. For example, the protein abundance of TFs is known to partially determine both a cell's chromatin accessibility and transcriptomic states; as such, multiomic chromatin accessibility and transcriptomic data could be meaningfully described by a latent variable model wherein latent factors encode TF abundance. Evidence suggests such models are better equipped to deal with sparse data and generalize to unseen data and are more robust to learning spurious statistical associations (Bengio et al. 2013). Moreover, these more interpretable approaches can help identify testable hypotheses, such as the knockdown of a TF to perturb cells from a diseased to a healthy state.
Understanding how cells cooperate to give rise to tissue-level phenotypes
As we move closer toward understanding how processes are regulated within the cell and predict how molecular perturbations can direct changes in individual cell states, it is equally important to understand how changes in individual cells will contribute to changes in tissue-level and organismal-level physiology. For example, one goal in the treatment of atherosclerosis is the development of therapies to promote plaque stability. Atherosclerotic plaques are composed of a multitude of cell types, such as fibrochondrocytes, macrophages, smooth muscle cells, and lymphocytes (Wirka et al. 2019; Alencar et al. 2020; Pan et al. 2020). Although a great deal of work has detailed factors associated with plaque stability such as the role of inflammation, no working model exists of how cell types and their interactions relate to plaque stability. Such a working model would be valuable for identifying candidate molecular perturbations in specific cells to promote plaque stability. More broadly, in the future it may be fruitful to understand not just how individual cell states are perturbed in disease but also how these dysregulated cells jointly contribute to disrupted tissue-level physiology.
The advent of single-cell spatial transcriptomics (and other spatial omics methods) appears to be a promising experimental assay that will help researchers approach this task. In brief, sequencing-based spatial omic technologies deploy surfaces that are arrayed with barcodes corresponding to cellular position. After tissue permeabilization and sequencing, individual cellular locations can be ascertained based on the identity of the cell barcode. Using these data, researchers can build models predicting how cells interact to produce tissue-level physiological features. An ideal model for this task should take into account the spatial location of the cells, and interactions among cells should be a function of cell–cell proximity; namely, adjacent pairs of cells should be more likely to interact than distant pairs of cells (Hu et al. 2021). A natural choice to consider is the use of graph convolutional neural networks (GCNNs) in either regression-based or classification-based settings (Hu et al. 2020c). Using existing approaches to map cells’ gene expression to cell states, individual cell states can be represented as nodes on a graph, where edges between nodes indicate that two cells are physically adjacent to one another. GCNNs can then use this graph as an input to predict either continuous or discrete aspects of the tissue of interest. At present, single-cell spatial transcriptomics may not yet be practical to do in large-scale human studies. Nonetheless, single-cell spatial transcriptomics combined with cost-effective histology may be a practical alternative to generate hypotheses of molecular perturbations to improve tissue-level measures. Using patient samples with matching spatial transcriptomics and histology, generative models such as those using graph convolutions can be trained to learn the joint relationship between the histology, spatial transcriptomics, and tissue-level information such as the stability of an atherosclerotic plaque. Given larger data of subjects with only collected histology and tissue-level measures, the generative model can first be used to predict the expression of individual cells for each sample. Using this predicted expression and the generative models, users can perform in silico perturbations of gene expression that produce improvements in tissue-level measures for each subject. Perturbations predicted to improve tissue-level measures that are shared across subjects, or subgroups of subjects, should then be prioritized for experimental follow-up.
Conclusion
Single-cell technologies have proven to be a valuable tool to understand human disease. Single-cell resolution enables researchers to characterize differences in cell states associated with disease status. This can be a powerful approach toward building an understanding of disease pathogenesis and its effects. At present, scRNA-seq has accounted for a substantial body of single-cell data collected. Using these data and the substantial body of supporting computational tools for their analysis, many groups have effectively detailed cell state differences underlying differences in human disease status. As this technology has grown more widespread, efforts to understand the genetic underpinnings of cell state differences have begun and continue to grow. The future of single-cell technologies in studying human disease appears promising, as new single-cell technologies to capture additional modalities such as chromatin accessibility, proteins, and spatial location have matured (Moffitt et al. 2018; Chen et al. 2019; Eng et al. 2019; Zhu et al. 2019; Ma et al. 2020; Specht et al. 2021; Takei et al. 2021; Thornton et al. 2021) and will enable researchers to detail factors underlying cell state differences not described by mRNA alone. Moreover, these technologies may help researchers further our understanding of the interaction between these factors in cell regulation. Maximizing the impact of single-cell technologies will require continued development in both experimental approaches to perturb cell states and in computational approaches to better understand their effects. Doing so will hopefully bring us closer to a better understanding of disease and how to treat it.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Dr. Nancy Zhang for helpful discussions and comments on the manuscript. This work was supported by the following grants: National Human Genome Research Institute grant T32HG000046-21 (to B.J.A.); National Institute of General Medical Sciences grant R01GM125301 (to M.L.); National Eye Institute grants R01EY030192 (to M.L.), R01EY031209 (to M.L.), and R21EY031877 (to M.L.); and National Heart, Lung, and Blood Institute grants R21HL156234 (to M.L.), R01HL113147 (to M.L. and M.P.R.), and R01HL150359 (to M.L. and M.P.R.).
Author contributions: B.J.A. and M.L. wrote the manuscript with input from J.H. and M.P.R.
Footnotes
-
Article and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275430.121.
-
Freely available online through the Genome Research Open Access option.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵








