
Cancer cell states and emergent properties of the dynamic tumor system
- 1Institute for Computational Medicine, NYU Langone Health, New York, New York 10016, USA;
- 2Department of Biochemistry and Molecular Pharmacology, NYU Langone Health, New York, New York 10016, USA
-
↵3 These authors contributed equally to this work.
Abstract
Phenotypic heterogeneity within malignant cells of a tumor is emerging as a key property of tumorigenesis. Recent work using single-cell transcriptomics has led to the identification of distinct cancer cell states across a range of cancer types, but their functional relevance and the advantage that they provide to the tumor as a system remain elusive. We present here a definition of cancer cell states in terms of coherently and differentially expressed gene modules and review the origins, dynamics, and impact of states on the tumor system as a whole. The spectrum of cell states taken on by a malignant population may depend on cellular lineage, epigenetic history, genetic mutations, or environmental cues, which has implications for the relative stability or plasticity of individual states. Finally, evidence has emerged that malignant cells in different states may cooperate or compete within a tumor niche, thereby providing an evolutionary advantage to the tumor through increased immune evasion, drug resistance, or invasiveness. Uncovering the mechanisms that govern the origin and dynamics of cancer cell states in tumorigenesis may shed light on how heterogeneity contributes to tumor fitness and highlight vulnerabilities that can be exploited for therapy.
Advances in single-cell technologies have revealed the extensive heterogeneity that exists within solid tumors (Suvà and Tirosh 2019). In addition to the range of nonmalignant cell types that make up the tumor microenvironment—most notably fibroblasts, macrophages, and lymphocytes—a variety of malignant cell subpopulations have been observed and characterized molecularly and phenotypically (Runa et al. 2017). Single-cell RNA sequencing (scRNA-seq) has enabled the unbiased profiling of tumors and the identification of sets of transcriptionally similar cells, leading to an inventory of cancer cell subpopulations (Table 1). The recurrence of these subpopulations across cancer types suggests that their emergence in tumors is a key component of tumor progression (Marusyk et al. 2012). Indeed, studies have shown that heterogeneity increases with tumor progression and predicts poor prognosis, supporting the hypothesis that cancer cell state diversity is advantageous to the tumor (Oh et al. 2019; Marjanovic et al. 2020; Ramón y Cajal et al. 2020). In particular, phenotypic heterogeneity caused by transcriptional variability may favor certain cellular states under specific environmental pressures, such as better survival during drug treatment (Frank and Rosner 2012; Lim and Ma 2019). Selective pressures may thus promote multifarious phenotypes within the tumor resulting in growth and therapeutic resistance.
Studies identifying cancer cell states in human solid tumors using single-cell transcriptomics
Although transcriptional heterogeneity appears to be recurrent in many cancers, its sources can be genetic or nongenetic (Marusyk et al. 2012). Genetic intra-tumoral heterogeneity has been extensively studied and is one of the prerequisites for tumor evolution resulting in therapeutic failure (Hu et al. 2017; McGranahan and Swanton 2017). However, recent work has revealed the prevalence of nongenetic heterogeneity as a vital driver of phenotypic variation during tumorigenesis as well as resistance to treatment (Sharma et al. 2010; Tirosh et al. 2016b; Neftel et al. 2019). This nongenetic heterogeneity can be caused by epigenetic differences, lineage determinants, or development hierarchies, or can arise through interactions with the tumor microenvironment.
Although the ability to characterize thousands of individual cells in a tumor has revolutionized the study of intra-tumoral heterogeneity, we still lack a comprehensive picture of how these cells function collectively to form the tumor system. The success of immunotherapy, for example, underscores the importance and therapeutic potential of understanding how cells within the tumor interact (Heinrich et al. 2021). To develop more effective therapies, we require a more complete understanding of how malignant cells in different states interact to cooperate or compete, resulting in improved tumor fitness. Here we review the emergence of cancer cell states and their functional properties and outline the possible sources of heterogeneity. Finally, we discuss the tumor as a system of heterogeneous cells and postulate the occurrence of adaptive interactions between diverse malignant cell states.
Delineating cancer cell states
Technological advances, and particularly scRNA-seq, have enabled groups to systematically identify and characterize subpopulations of cells in diverse cancer types, including melanoma, head and neck cancer, glioblastoma, pancreatic adenocarcinoma, colon cancer, and others (Table 1). Although this review focuses on solid tumors, patterns of intra-tumoral transcriptional heterogeneity have also been observed in blood cancers (Granja et al. 2019; Jang et al. 2019; van Galen et al. 2019; Chen et al. 2020; Liu et al. 2021). The observation of transcriptional variation across malignant cells in many cancer types suggests that transcriptional heterogeneity is a consistent property of tumors. Moreover, this transcriptional heterogeneity is rooted in coherent patterns of expression that underpin phenotypic properties, constituting cell states (Table 1).
Cell states have been identified primarily by clustering cells with similar transcriptional profiles and performing differential gene expression analysis to annotate their distinctive features (Neftel et al. 2019; Ji et al. 2020). However, the separation between clusters of malignant cells is not generally as clear-cut as the differences between distinct cell types. Rather, transcriptional profiles appear to vary continuously along certain axes. This observation has prompted the characterization of cancer cells based on gene modules, where the genes of a particular module are coexpressed and differentially expressed between the cells of a tumor (Fig. 1A; Patel et al. 2014). Computational approaches can be used to identify those gene modules by searching for shared factors of variation across cells, including gene–gene correlation, principal component analysis (Patel et al. 2014; Tirosh et al. 2016b), and nonnegative matrix factorization (Puram et al. 2017). Despite being agnostic to known pathways, these methods have recovered many programs described in other contexts, including development (e.g., epithelial-to-mesenchymal transition [EMT]) and cell–cell interactions (e.g., antigen presentation). This suggests that cancer cell states arise from the co-option of existing gene regulatory modules rather than the de novo construction of a module. Recent methods also use the underlying gene regulatory framework, adding the constraint that gene modules share a transcription factor binding motif (Aibar et al. 2017; Rambow et al. 2018). We expect that computational methods aimed at delineating cell states—whether newly devised or adapted from existing tools—will advance alongside our understanding of the nature and organization of cell states (Tanay and Regev 2017; Lähnemann et al. 2020).
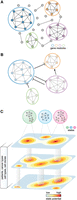
A gene module framework to characterize cancer cell states. (A) Gene modules emerge from the underlying gene regulatory network. Nodes and edges represent genes and coexpression, respectively. Colors indicate distinct gene modules. (B) Coherent gene modules interact with other modules to define a malignant cell's state. (C) The spectrum of states available to a cell can be described as a state potential map, where some states are shared across patients, cancer types, cell types, or clones for example, whereas others are accessible only in particular contexts.
Collectively, these approaches allow for the description of a cancer cell state according to the expression of one or more gene modules (Fig. 1B). Unlike the programs underlying cell types, which are more stably defined by mutually exclusive differentiation determinants, these modules can be expressed in combinations, potentially explaining the continuous range of possible states that can be adopted (Fig. 1C). This can be considered analogous to developmental states that exist along a trajectory, or physiological states in which cells of the same type can vary continuously in their level of activation. Thus, cancer cell states can be thought of as instantiations of the underlying gene regulatory network, depending on the interplay between intrinsic cell plasticity and environmental cues.
Phenotypic properties of cancer cell states
Before scRNA-seq, extensive studies of inter-tumor transcriptional heterogeneity using bulk RNA sequencing (RNA-seq) led to the identification of molecular and histological subtypes with distinct phenotypic properties and strong correlation with clinical prognosis (Koboldt et al. 2012; Kandoth et al. 2013; Hoadley et al. 2018). With the high-resolution view of scRNA-seq, it is now clear that cells within a tumor vary along several of these same dimensions (Patel et al. 2014; Tirosh et al. 2016a). For example, individual breast tumors harbor cells with transcriptional profiles corresponding to all four molecular subtypes: “basal,” “luminal A,” “luminal B,” and “HER2” (Chung et al. 2017; Gao et al. 2017), highlighting the importance of studying tumors at cellular resolution.
Although tumors are typically characterized by rapid growth and dedifferentiation, individual malignant cells differ in their degree of proliferative and stem-like behavior (Jögi et al. 2012). In many cancer types, including adenocarcinoma (Dalerba et al. 2011), melanoma (Rambow et al. 2018; Baron et al. 2020), and glioma (Tirosh et al. 2016b), studies have used scRNA-seq to show that fully differentiated cells coexist with more undifferentiated cells. This is consistent with the notion that tumor maintenance may follow principles of normal adult tissue homeostasis, in which slow-cycling stem cells give rise to rapidly-cycling progenitors and finally to nondividing differentiated cells (Pellettieri and Sánchez Alvarado 2007). In gliomas, this hierarchical model seems to hold true at least in part, with cancer cells in a proliferative state giving rise to two differentiated states, oligodendrocyte-like and astrocyte-like, supporting a complex landscape of differentiation within a single tumor (Tirosh et al. 2016b; Venteicher et al. 2017; Filbin et al. 2018). Furthermore, the existence of a quiescent, stem-like transcriptional program has been shown in several cancer types. In melanoma, for example, cells exist along a continuum between dormant AXL-high cells and proliferative MITF-high cells, with the former being less sensitive to MAPK inhibitors (Tirosh et al. 2016a). In a single-cell study of breast cancer, early metastases contained dormant low-proliferative cells, whereas later metastases were highly proliferative, suggesting a role for each of these properties at different stages of tumor progression (Lawson et al. 2015). Thus, characterizing how cells vary in their expression of quiescence, proliferation, and differentiation programs is necessary for a comprehensive understanding of tumorigenesis.
The EMT is a well-established process occurring in epithelial tumors that mimics normal development and wound healing (Hay 1995; Kalluri and Weinberg 2009; Haensel and Dai 2018; Ganesh et al. 2020; Laughney et al. 2020; Wouters et al. 2020). Within an epithelial tumor, cells can be identified at varying stages of this process, thus defining another axis of intra-tumoral heterogeneity (Pastushenko et al. 2018). Indeed, studies using scRNA-seq in patient tumors have identified gene modules indicative of partial (Puram et al. 2017) and complete (Aiello et al. 2018; Lin et al. 2020) mesenchymal phenotypes. Functional studies have implicated the mesenchymal state in invasion (Puram et al. 2017) and metastasis (Revenco et al. 2019) and shown that the reverse process, mesenchymal-to-epithelial transition (MET), enables metastatic cells to establish a secondary tumor with epithelial characteristics (Rothenpieler and Dressler 1993; Lawson et al. 2015; Shibue and Weinberg 2017).
Intra-tumoral heterogeneity is a critical barrier to treatment in oncology, as drug-naive tumors harbor a fraction of cells that are not eliminated by treatment, enabling them to seed tumor relapse. Increasing evidence has shown that drug tolerance is mediated by broad reversible transcriptional changes, suggesting that one or more states may underlie this phenotypic property (Sharma et al. 2010; Kim et al. 2016; Shaffer et al. 2017). Early studies in lung cancer showed that drug-tolerant cells express high levels of CD24 and PROM1 (also known as CD133), two markers associated with stemness and quiescence (Sharma et al. 2010). More recent work has also established higher drug tolerance in slow-cycling cells in many cancer types, including breast cancer (Kim et al. 2018) and melanoma (Roesch et al. 2013; Shaffer et al. 2017), supporting a link between quiescence and drug tolerance (Singh and Settleman 2010). Additional states have been associated with drug tolerance, including a stress-response state in melanoma (Baron et al. 2020) and mesenchymal state in carcinomas (Shibue and Weinberg 2017; Viswanathan et al. 2017).
Although scRNA-seq has contributed to the characterization of well-established states at the molecular level, it has also highlighted cell states and gene modules that were previously overlooked. Several studies have found a stress-response module that is differentially expressed within cells of the same tumors and is characterized by DNA-damage, unfolded protein, and TNF-signaling response genes (Tirosh et al. 2016a; Baron et al. 2020; Izar et al. 2020; Moncada et al. 2020). Gene modules associated with metabolism, including oxidative phosphorylation (Moncada et al. 2020) and hypoxia (Patel et al. 2014; Neftel et al. 2019; Baron et al. 2020), may be differentially expressed as a result of spatial variation in oxygen and metabolite availability within the tumor. An independent classification of cancer cells from glioblastoma based on pathway enrichment coincided with previously characterized lineage-specific cellular states, suggesting a link between developmental states and metabolic activity (Garofano et al. 2021). Cancer cells also appear to vary in their level of interferon response and antigen presentation (Patel et al. 2014; Izar et al. 2020). Understanding how these transcriptional changes affect interactions with immune cells is critical to the growing field of immunotherapy. Indeed, these changes appear to mediate increased immunogenicity as the result of antigen presentation but also, paradoxically, immune tolerance through expression of immune checkpoints (Thibaut et al. 2020; Williams et al. 2020). Thus, mapping transcriptionally defined cancer cell states to phenotypic properties remains an active area of investigation, and will help establish their in vivo relevance.
Origin and dynamics of cancer cell states
The consistent identification of distinct intra-tumoral cancer cell states raises the question of how they arise during tumorigenesis (Fig. 2). As tumors also harbor genetic heterogeneity, it is tempting to search for links between cancer cell states and genetic alterations. Methods to simultaneously measure genome and transcriptome in single cells make it possible to directly link genetic alterations and transcriptional states (Macaulay et al. 2015). Several studies have identified specific mutations leading to widespread transcriptional and phenotypic differences, including drug resistance (Lim and Ma 2019; Sachs et al. 2019), growth factor independence (Rubinfeld et al. 1996; Dempke and Heinemann 2010; Kim et al. 2019), and high proliferative rates (Marusyk et al. 2014). In a model of small cell lung carcinoma, a mesenchymal state was shown to arise as a result of an oncogenic HRAS mutation (Calbo et al. 2011). Similarly, Wnt1-driven mammary gland tumors can evolve two clones, with the Hras-mutated clone acquiring a basal-like proliferative phenotype (Cleary et al. 2014).
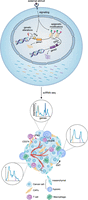
Cell- and system-level view of intratumor heterogeneity. (Top) Intrinsic factors (genetic alterations or epigenetic changes) and extrinsic factors (stimuli from the environment) lead to transcriptional changes, represented here with the expression of two genes (orange and purple). (Bottom) Transcriptional heterogeneity is revealed by scRNA-seq, which gives the gene expression profile of each cell. Histograms depict expression profiles corresponding to different cancer cell states in which two genes (orange and purple lines) are expressed at different levels. Within the tumor, cancer cells may also compete or cooperate with each other and interact with other cells of the tumor microenvironment. Intrinsic epigenetic factors may induce the EMT. Low vascularization and oxygen levels (O2) may induce the hypoxic state, which in turn promotes angiogenesis through VEGFA secretion. Interferon gamma (IFNG) secretion by T cells may lead to an interferon response state with high CD274 (also known as PDL1) expression.
Increasingly, however, there is evidence that genetically encoded states are the exception rather than the rule. In brain tumors, each clone contributes to all identified states in patient tumors (Tirosh et al. 2016b). Furthermore, in vivo studies show that sorted cells of a single state—or indeed single cells—are able to reconstitute the full range of states observed in the original tumor (Dirkse et al. 2019; Neftel et al. 2019). In other cancer types as well, a high degree of plasticity between cancer cell states within the tumor has been identified (Gupta et al. 2011; Kreso et al. 2013; Cleary et al. 2014; Seino et al. 2018; Kinker et al. 2020; Marjanovic et al. 2020). This plasticity appears to play a particular role during metastasis: EMT first enables dissemination and seeding, and MET leads the metastasis to regain the original epithelial states (Lawson et al. 2015). In a study of triple-negative breast cancer combining single-cell DNA and RNA sequencing, resistance to chemotherapy was associated with pre-existing genetic clones present before therapy (Kim et al. 2018). However, transcriptional signatures typically involved in drug tolerance were detected only in posttreatment patients, suggesting that induced transcriptional changes play a role in the resistance phenotype even in the presence of genetic clonal selection. Thus, similar to how different cell types emerge from a genetically identical population during development, it appears that cancer cells in different states can arise without genetic differences. The sources of nongenetic heterogeneity may then be intrinsic to the cell or caused by external signals from the microenvironment.
The coexistence in a tumor of mature and immature cells of the same lineage suggests that differentiation and dedifferentiation processes play a role in cell state diversification (Dalerba et al. 2011). Epigenetic encoding of cancer cell state identity appears to be less hardwired than in normal cells, although lineage identity is retained to some extent (Flavahan et al. 2017). In melanoma, for example, the neural crest lineage identity is not lost, as cells vary only between neural crest and differentiated melanocyte-like states (Rambow et al. 2018; Baron et al. 2020). Similarly, cell states in glioblastoma include neural progenitor-like, oligodendrocyte progenitor-like, and astrocyte-like, all of which are derived from the neural lineage (Neftel et al. 2019). Throughout tumor progression, however, cells take on states of increasing regulatory distance from the original lineage of the cell type of origin. In a lung cancer model, for example, a state resembling lung progenitors appears first, followed by a primordial gut-like state (Marjanovic et al. 2020). Thus, lineage-related cancer cell states may follow from partial loss of epigenetic stability.
Beyond differentiation and dedifferentiation along lineages, other epigenetic mechanisms may explain the existence of distinct transcriptional states within the tumor (Kundaje et al. 2015). Intra-tumoral heterogeneity in the chromatin state, along the restricted-permissive axis, may dictate cell state not only by affecting gene module expression but also by silencing tumor-suppressor programs or allowing stochastic oncogene activation (Flavahan et al. 2017). In colon cancer, Meir et al. (2020) used a “Luria– Delbruck”-like experiment to show that the epithelial and mesenchymal states are inherited across several generations through epigenetic memory, specifically through DNA methylation. This is in line with the in vivo finding that the epigenetic state of the cell of origin determines the propensity of tumor cells to undergo EMT in squamous cell carcinoma (Latil et al. 2017). Drug tolerance in persister cells also appears to be based in a specific reversible chromatin state (Sharma et al. 2010). In glioblastoma, for example, pre-existing epigenetic composition dictated by histone demethylases KDM6A/B leads to a reversibly slow-cycling persister state that survives treatment (Liau et al. 2017). Furthermore, epigenetic changes occurring throughout tumorigenesis appear to mediate cell state diversification. In lung cancer, single-cell ATAC-seq (scATAC-seq) revealed distinct chromatin signatures underlying metastatic and highly-plastic cell states (LaFave et al. 2020; Marjanovic et al. 2020). In glioblastoma, epigenetic profiling through scATAC-seq uncovered distinct states within the self-renewing stem cell population, with an invasive state correlating with poor prognosis (Guilhamon et al. 2021). In addition, these epigenetic states were not associated with somatic copy number alterations. Taken together, these studies highlight the role of epigenetic mechanisms in the generation of cancer cell states, independent of clonal structure.
Heterogeneity in the malignant compartment may stem from different interactions with other factors of the tumor microenvironment, in the form of cell–cell interactions (Bagley 2010), metabolite availability (Le 2018), or drug concentrations (Marusyk et al. 2020). Cancer-associated fibroblasts (CAFs) are known to play a crucial role in pancreatic cancer (von Ahrens et al. 2017), and recent work has shown that CAFs induce proliferative and EMT states (Ligorio et al. 2019), as well as a Wnt-independent state (Seino et al. 2018). Spatial transcriptomics, which comprehensively map cell types and states within the tumor, can yield insight into these interactions (Rao et al. 2021). Using this technology, CAFs were shown to colocalize with a partial EMT state in squamous cell carcinoma (Ji et al. 2020) and inflammatory fibroblasts, with a stress-response state in pancreatic cancer (Moncada et al. 2020). However, other methods are required to capture interactions mediated by secreted factors rather than contact-dependent signaling. For example, although the interferon-response state appears to be elicited by CD8+ T cells, there is no significant colocalization between the two cell populations (Thibaut et al. 2020). Additionally, varying concentrations of drugs and metabolites within the tumor may also play a role in heterogeneity (Wu and Dai 2017). In glioblastoma, treatment with receptor tyrosine kinase inhibitors induces genetic and epigenetic changes, leading to the emergence of a drug-tolerant persister state (Eyler et al. 2020). By altering gene expression or epigenetic state of a malignant cell, drug treatment can also lead to cell state transitions of pre-existing primed subpopulations within the tumor (Shaffer et al. 2017; Kim et al. 2018). These studies suggest that tolerance is likely facilitated by intrinsic transcriptional variability that primes cells for further induced adaptations during therapy.
A system-level view of cancer cell states
The diversity of states within a single tumor raises the question of how they interact within the tumor system (Barkley and Yanai 2019). In a context of limited resources, competition between states would be expected to result in decreased heterogeneity, as the most-fit states overtake the tumor population (Parker et al. 2020). In contrast, studies have found an increase in cell state diversity throughout tumorigenesis. In a model of esophageal tumorigenesis, new states were found to appear at each stage of progression without loss of the earlier states (Yao et al. 2020). Similarly, an increase in the diversity of cell states was observed during lung adenocarcinoma progression, with some cells retaining the original alveolar identity concurrently with the appearance of new states reminiscent of earlier developmental stages (Marjanovic et al. 2020). These observations suggest that state diversity is a consistent property of advanced tumors.
Tumors are reminiscent of developmental systems in their capacity to recapitulate normal developmental and differentiation hierarchies, analogous to progenitor cells maintaining tissue structure in normal organs (Yan and Owens 2008; Biteau et al. 2011; Gehart and Clevers 2019). For example, mammary gland tumors can be maintained by a bipotent progenitor, giving rise to basal and luminal cells (Cleary et al. 2014; Tammela et al. 2017), and oligodendrogliomas by a pool of stem-like cells that differentiate into oligodendrocyte-like and astrocyte-like cells (Tirosh et al. 2016b). Furthermore, signaling niches also play an important role in tumorigenesis similar to development. It has been shown that a Wnt-secreting state maintains the stem cell niche of Hras-mutated proliferative cancer cell state in breast cancer (Cleary et al. 2014; Tammela et al. 2017). This relationship was not transient, supporting the view of the tumor as a system rather than a set of independent or competing cells (Cleary et al. 2014). Furthermore, this two-state system was shown to emerge either clonally, with the basal-like cells acquiring a Hras mutation, or hierarchically, with both cell states sharing a common progenitor and genetic background (Cleary et al. 2014). Although genetic differences do not necessarily delineate cell states, studies have found that certain genetic mutations, such as amplification of EGFR, PDGFR, or CDK4, are associated with different frequencies of malignant cell states (Neftel et al. 2019). This convergence of genetic and nongenetic mechanisms of state segregation strongly supports the functional importance of cell state heterogeneity in tumor progression.
Coexistence of various cancer subpopulations within a tumor has led to the hypothesis that cancer cell states have distinct functions that together promote overall success of the tumor. This hypothesis has been extensively explored theoretically, borrowing from the field of game theory (Gatenby and Vincent 2003; Aktipis and Nesse 2013; Archetti and Pienta 2019). Jouanneau et al. (1994) showed that the presence of an FGF1-producing population within the tumor increases tumorigenic and metastatic potential. The resulting primary and metastatic tumors remained mixed, suggesting that the increase in fitness was owing to a community effect rather than a single population outcompeting the other. In small cell lung carcinoma, mixed tumors of neuroendocrine and nonneuroendocrine cells displayed increased proliferation in vitro and metastasis in vivo (Calbo et al. 2011). This effect was shown to be local rather than systemic, as it was not observed when the neuroendocrine and nonneuroendocrine cells were separate in contralateral flanks. In contrast, Polyak and colleagues showed a systemic effect in breast cancer, where subpopulations of IL18 or VEGFA-producing cells induce tolerance in neutrophils, leading to increased growth and metastasis of the tumor as a whole (Marusyk et al. 2014; Janiszewska et al. 2019). In melanoma, Campbell et al. (2020) found that subpopulations of tumor cells with a proliferative or invasive cell signature cooperate to seed metastases (Campbell et al. 2020). In the context of immunotherapy, it was shown that interferon-insensitive cells have a selective advantage over interferon-sensitive cells in the same tumor but that their survival depends on the interferon-sensitive cells (Williams et al. 2020). Taken together, these results suggest that indirect parasitism or cooperation between cancer cell states promotes tumor growth and metastasis.
Finally, heterogeneity within the tumor may be key to robustness in the face of environmental fluctuations, including drug treatment. The presence of proliferative and quiescent cells in the same tumor is reminiscent of survival strategies observed in unicellular species, including yeast and bacteria (Lewis 2007). A similar bet-hedging strategy may evolve in tumors, wherein the coexistence of these two states enables both growth and robustness to environmental changes, as the quiescent cells are less sensitive (Chen et al. 2016; Brown and Schober 2018). Such a strategy is further exemplified in the context of drug treatment, in which pre-existing transcriptional heterogeneity enables a fraction of cells—drug-tolerant persisters—to survive drug insult, thus providing a substrate for natural selection and, finally, emergence of genetically drug-resistant cells (Sharma et al. 2010; Emert et al. 2021). Increasing transcriptional variability may be one of the ways in which histone demethylases, which increase transcriptional heterogeneity, function as oncogenes (Roesch et al. 2013; Hinohara et al. 2019). Thus, heterogeneity may provide a framework for tumor cells to explore novel states that are advantageous to the cell itself but also to the tumor system as a whole.
Outlook
As the importance of intra-tumoral heterogeneity for tumor progression has become abundantly clear, the efforts to examine the tumor as a complex system have also come into focus. Although the existence of diverse cancer cell states has been known for many years, several challenges have thwarted efforts to understand their role and functional relevance on a deeper level. In particular, many molecular approaches were not available to dissect the relationships among the cell states as a part of the collective tumor system.
Recent advances in molecular biology, single-cell technologies, and computational methods promise to provide an integrative understanding of the tumor system. First, we can now study tumors at an unprecedented scale and resolution, with a high-dimensional, multiomic view at the level of individual cells. Integrating multiple modalities, including proteomic, transcriptomic, epigenomic, and genomic, will help to understand the origins of cancer cell states. Although each sample under study with these technologies is a snapshot, computational methods to infer dynamics make use of naturally occurring lineage tracing using copy-number alterations and mitochondrial mutations. Furthermore, leveraging spatial transcriptomics to understand how states mix or segregate within the tumor, as well as their colocalization with elements of the tumor microenvironment, may hint at how they arise and at their functional consequences.
Perturbation experiments in model systems will be required to rigorously establish the causal aspects of cancer cell states and move beyond correlative observations. With increased practicality and tractability, model systems like patient-derived organoids and genetic mouse models can be used to recapitulate the tumor and its microenvironment and mimic human disease. Single-cell CRISPR screens to perturb cell states may also lead to novel insights into the plasticity and dynamics of cell states as well shed light on the drivers of heterogeneous populations within the tumor. Computational strategies such as agent-based modeling can be incorporated to understand how tumor-level properties emerge from these components. Together, these two complementary approaches—holistic but correlative, and causal but reductionist—will enable us to understand how the tumor system emerges from its individual components and may highlight the system's vulnerabilities for treatment.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Deborah Liberman for a critical reading of the manuscript. This work was supported by a grant from the National Institutes of Health (R01 LM013522).
Footnotes
-
Article and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275308.121.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








