
Tagmentation-based single-cell genomics
Abstract
It has been just over 10 years since the initial description of transposase-based methods to prepare high-throughput sequencing libraries, or “tagmentation,” in which a hyperactive transposase is used to simultaneously fragment target DNA and append universal adapter sequences. Tagmentation effectively replaced a series of processing steps in traditional workflows with one single reaction. It is the simplicity, coupled with the high efficiency of tagmentation, that has made it a favored means of sequencing library construction and fueled a diverse range of adaptations to assay a variety of molecular properties. In recent years, this has been centered in the single-cell space with a catalog of tagmentation-based assays that have been developed, covering a substantial swath of the regulatory landscape. To date, there have been a number of excellent reviews on single-cell technologies structured around the molecular properties that can be profiled. This review is instead framed around the central components and properties of tagmentation and how they have enabled the development of innovative molecular tools to probe the regulatory landscape of single cells. Furthermore, the granular specifics on cell throughput or richness of data provided by the extensive list of individual technologies are not discussed. Such metrics are rapidly changing and highly sample specific and are better left to studies that directly compare technologies for assays against one another in a rigorously controlled framework. The hope for this review is that, in laying out the diversity of molecular techniques at each stage of these assay platforms, new ideas may arise for others to pursue that will further advance the field of single-cell genomics.
A rich history of scientific achievements precedes the use of Tn5 transposase for sequencing applications. The initial discovery of the bacterial Tn5 transposase came out of a study to investigate kanamycin resistance (Berg et al. 1975), which then led to years of efforts to characterize the molecular basis of transposition. This work was largely driven by William S. Reznikoff, who has dedicated his career to the study and characterization of Tn5 transposase, without which none of the technologies detailed in this review would have been possible. Seminal advancements included the detailed characterization of the “cut & paste” mechanism of the Tn5 transposase (Reznikoff 2003); identification of the Tn5 recognition sequence, referred to here as the “mosaic end” (ME) sequence (Johnson and Reznikoff 1983); development of a method to purify monomeric Tn5 (York and Reznikoff 1996); description of the crystal structure of the protein and synaptic complex, which forms when the transposome complex binds to target DNA (Davies et al. 2000); the identification and characterization of a variety of mutations to reduce target sequence preference (Zhou and Reznikoff 1997) and increase activity (Naumann and Reznikoff 2002), both of which were pivotal advancements for the use of Tn5 for sequencing applications; the ability to perform transposition in vitro (Goryshin and Reznikoff 1998); and the general establishment of Tn5 transposase as a model system for understanding DNA transposition (Reznikoff 2003). This is an abbreviated list of a staggering number of detailed and rigorous studies to characterize the Tn5 transposase that are best summarized in the 2008 review “Transposon Tn5” (Reznikoff 2008).
This wealth of biochemical and molecular research led to the establishment of Tn5 transposition as a primary system for performing transposon mutagenesis and transposon insertion sequencing methods to probe gene fitness contributions (Cain et al. 2020). Then, as high-throughput sequencing burst onto the genomics scene, it was inevitable that this efficient and powerful enzyme would play a major role, which came in the form of the tagmentation reaction, developed as an industry–academia partnership with Epicentre Biotechnologies (Adey et al. 2010).
The anatomy of the tagmentation reaction
The first step in tagmentation is the formation of the transposome complexes, composed of a hyperactive variant of the Tn5 transposase homodimer complexed with sequences that contain the 19-bp double-stranded ME sequence recognized by the enzyme. In a traditional transposition reaction, Tn5 would be loaded with a single, continuous stretch of double-stranded transposon DNA, often containing an antibiotic-resistance gene, and flanked by ME sequences; whereas in tagmentation, the transposon DNA is discontinuous, with two, unlinked adapter sequences. The adapter itself (Fig. 1A) is composed of the ME sequence with an additional 5′ overhang of single-stranded DNA on the transfer strand (i.e., the strand that becomes covalently bound to the target DNA) that is a mix of either forward or reverse adapter sequences to be used as PCR handles in subsequent processing steps. The single-stranded component is to prevent the action of the enzyme on the actual adapter complexes themselves. Tn5 has a high propensity to insert into free double-stranded DNA, and making the only double-stranded portion the ME, which is protected by the Tn5 enzyme, prevents this “self-tagmentation” from happening. On a related note, the in vitro assembly of transposome complexes should be performed in the absence of Mg2+, which is required for the tagmentation reaction to occur, in order to prevent tagmentation within the 19-bp double-stranded ME region of adapters that has not yet formed a complex. The other major aspects of adapter design include the use of a 5′ phosphorylated ME reverse complement. This bottom strand can also be reduced in length from the full 19-bp segment, with 16-bp versions (trimmed from the 3′ end) providing comparable efficiency (Adey and Shendure 2012). In standard tagmentation assays, transposome assembly is composed of mixing a 1:1:2 ratio of the forward and reverse adapters and purified Tn5 monomer (Fig. 1B). The Tn5 protein can be produced using published methods (Picelli et al. 2014a; Kia et al. 2017), although enzymes produced by individual laboratories may not have the same consistency or level of activity as commercially-available variants. One important note is that what may appear to be a poor quality Tn5 preparation, may in fact be driven by the use of poor-quality oligonucleotides. As such, it is critical to always use HPLC-purified oligonucleotides and perform activity-based quantification using standard adapters and benchmarking against commercially-available options. Other modes of failure include protein that has not properly folded or inaccurate quantification of active enzyme, the latter of which can be addressed by performing activity-based quantification by titrating across several possible concentrations and benchmarking against commercially-available options.
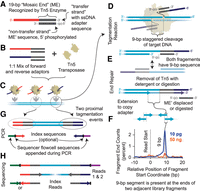
The anatomy of a tagmentation reaction. (A) The structure of the tagmentation adapter, which includes the double-stranded 19-bp mosaic end sequence recognized by Tn5 transposase, as well as a single-stranded overhang on the transfer strand that contains an adapter used for subsequent processing. This ssDNA overhang can be any length; however, shorter sequences improve efficiency of tagmentation. (B) The Tn5 enzyme is loaded with a mix of adapters with forward or reverse adapter overhangs. For standard tagmentation workflows, this includes a 1:1 molar ratio of the two adapter species and a 1:1 molar ratio of the total adapter content to Tn5 monomer. (C) The tagmentation reaction involves the binding of transposome complexes to the target DNA at high density, that is, one insertion every ∼500 bp. (D) Each tagmentation event results in the cleavage of the DNA backbone on both strands staggered by 9 bp. The 3′ end of the transfer strand is then covalently appended to the 5′ end of the nick in the target DNA backbone at each of the cut sites. (E) After the tagmentation, the Tn5 enzyme remains tightly bound to the target DNA and must be removed to enable end repair, where the bottom strand acts as a priming site to copy through the adapter on the transfer strand. (F) End repair results in the copying of the 9-bp region between the two cuts in the target DNA backbone, where the pair of adjacent library fragments produced by a single tagmentation event overlap by the 9-bp segment when aligned to a reference genome. In low input libraries, instances of the ends of two reads from separate, adjacent read pairs can be observed as the overlap between two separate, adjacent read pairs. (G) PCR amplification is then performed, using the adapter overhangs of the transfer strand as priming sites. The primers required for cluster generation or other means of sequencing along with optional sample indexes are appended here. (H) Sequencing is performed using primers that include the mosaic end and adapter sequence to obtain paired reads of target DNA as well as index sequences.
Purified DNA is then exposed to these transposome complexes within a buffer that contains Mg2+, which is required for the transposition reaction to occur (Fig. 1C). The complexes act on the target DNA by binding tightly and completing cleavage and strand transfer at two positions that are 9 bp apart. The result is a break in the target DNA at both strands with a 9-bp space in between (Fig. 1D). At each of these nicks, the transfer strand oligonucleotide containing the ME sequence and either a forward or reverse adapter is covalently attached. From a single tagmentation event, adapters are incorporated in an outward-facing manner; thus, in order to form a viable sequencing library molecule, a second tagmentation event must have been completed successfully nearby (i.e., within a length suitable for PCR and sequencing, typically <1000 bp). Additionally, forward and reverse adapters are incorporated randomly, resulting in only half of the produced molecules as having both a forward and reverse adapter, with the remaining having either two forward or two reverse adapters that cannot be carried through subsequent processing. During PCR, molecules that have two of the same adapter form a hairpin structure which has a favorable Tm over primer annealing owing to a longer region of homology, thus inhibiting amplification. Furthermore, molecules with the same adapter on both sides cannot form sequencing colonies. This 50% maximum efficiency is something that has been addressed by several strategies detailed below to improve yields of single-cell libraries, where every molecule counts. However, even with these limitations, the overall process was proven to be more efficient than traditional ligation-based library preparation methods at the time and even enabled the production of libraries from as little as 10 pg of starting material in its initial description, approaching the single-cell range of input (Adey et al. 2010).
After the transposition reaction itself, a process referred to here as end repair must be performed before denaturation of the template DNA for subsequent PCR amplification (Fig. 1E). This process first involves the removal of the Tn5 protein, which remains tightly bound to the target DNA (Goryshin and Reznikoff 1998; Amini et al. 2014) in order to free up the DNA present at the site of tagmentation. The specifics of the mechanism that release the Tn5 during in vivo transposition are not fully understood but likely involve cell machinery other than the Tn5 itself and do not occur when transposition is performed in vitro (Goryshin and Reznikoff 1998). For sequencing applications, Tn5 removal is facilitated by a cleanup procedure or treatment with a detergent (SDS). Skipping the Tn5 removal step is possible, although it results in a much lower efficiency of end repair, which may be acceptable for applications in which efficiency is of less value than a rapid workflow.
The removal of Tn5 effectively releases the two end fragments from one another that were generated during the reaction, each receiving one of the adapters from the transposome complex and retaining one strand of the 9-bp region in between the two cut sites. Extension using a DNA polymerase from the 3′ end of the strand that was not subjected to strand transfer then copies the 9-bp overlap region and the ME sequence, terminating at the end of the adapter. In standard tagmentation-based library preparation, this is the reason for the initial extension step in the PCR reaction before initial denaturation. The 9-bp region is effectively copied and is the sequence present at the outermost ends of sequencing library molecules, where two adjacent library molecules each overlap at the same 9-bp segment. This overlap between adjacent library fragments can be detected in low input libraries, where the probability of sequencing two reads that shared a tagmentation event to produce one of each of their respective ends is relatively high. This results in reads from separate read pairs that align in opposite directions and overlap by 9 bp (Fig. 1F; A Adey, HG Morrison, Asan, et al., unpubl.). The use of this overlap has been explored to link read pairs for haplotype resolution or genome assembly; however, the capture efficiency of fragments has to be extremely high to obtain any useful number of linkages, which is not possible with standard tagmentation workflows that are hampered by the 50% fragment dropout detailed above.
For applications that require a high efficiency, such as single-cell assays, the end repair step can be the key to improving yields and should not be overlooked. This single extension reaction is required to produce fragments that terminate in an adapter sequence at both ends. After end repair, templates are denatured and carried through PCR with primer sequences corresponding to the forward and reverse adapters that contain an overhang with an optional index sequence and terminate in the sequences used for cluster generation on a sequencer flowcell (Fig. 1G). Libraries are then sequenced using primers that correspond to the full forward or reverse adapters to provide reads of the intervening genomic DNA (i.e., Reads 1 and 2), as well as the reverse complement to sequence in the other direction to capture the library indexes, if they were included (Fig. 1H).
The versatility of tagmentation in genomic assays
Although the initial description of tagmentation-based library preparation focused on the production of whole-genome sequencing libraries; it also included exome capture, PCR-free library preparation, and direct library preparation (DLP) from bacterial colonies, showing the versatility of the approach from the start (Adey et al. 2010). Shortly after, the variety of tagmentation applications exploded, with strategies that leveraged the increased efficiency to reduce input requirements of existing assays including RNA-seq (Gertz et al. 2012; as well as a recent description of direct tagmentation of RNA/DNA hybrids [Di et al. 2020]), whole-genome bisulfite sequencing for DNA methylation analysis (Adey and Shendure 2012), ChIPmentation for ChIP-seq workflows (Schmidl et al. 2015), and HiChIP for protein-directed chromatin folding (Mumbach et al. 2016), just to name a few. Notably, the large number of assays that leverage tagmentation as a component in the workflow, whether minor or substantial, could constitute a review on its own. Of the variety of techniques, the one that has undoubtedly had the largest impact and is likely the most widely used is the assay for transposase accessible chromatin, or ATAC-seq (pronounced “attack-seq”) (Buenrostro et al. 2013).
ATAC-seq is the latest version in a rich history of assays that leverage the steric hindrance of chromatin to prevent enzymatic action in regions of DNA wrapped around histones or bound by other nuclear proteins. Originally based on the use of micrococcal nuclease to digest DNA at the linker region between nucleosomes, leading to the early formulation of the principles of chromatin structure (Noll 1974), and then to map nucleosome-free regions by leveraging DNase I (Weintraub and Groudine 1976), these methods shifted to high-throughput sequencing with the advent of DNase-seq for genome-wide chromatin accessibility mapping (Boyle et al. 2008). In spite of the high sensitivity afforded by DNase-seq and the ability to accurately map the footprints of transcription factors within these regions of open chromatin (Vierstra et al. 2020), the method is generally regarded as difficult and requires a large amount of starting material, although single-cell variants do exist (Jin et al. 2015). ATAC-seq largely solved these challenges by the direct and efficient tagmentation of native chromatin. Much like DNase I, Tn5 transposase has extremely low efficiency on DNA wrapped around nucleosomes and limited efficiency at the linker regions in-between them, making open chromatin loci the only DNA readily available for tagmentation. Although early implementations suffered from a high proportion of mitochondrial DNA reads (later developed into a feature for single-cell ATAC assays) (Fiskin et al. 2020; Lareau et al. 2020), the original description of the assay was able to produce useful chromatin accessibility profiles from as few as 500 cells (Buenrostro et al. 2013), and the proportion of mitochondrial DNA was reduced by extensive protocol and buffer optimization in a protocol referred to as Omni-ATAC (Corces et al. 2017). In further developments, fluorescently labeled transposomes were used to visualize the global accessibility of cells by tagmenting permeabilized nuclei within intact tissue followed by recovery of the fragments and sequencing (Chen et al. 2016). Taken further, the direct tagmentation of intact cells and use of imaging techniques were used to perform direct in situ sequencing of genomic DNA off of the tagmented adapter, enabling the mapping of sequence reads in 3D space within the nucleus (Payne et al. 2020).
The next major advancement in the tagmentation space has been the use of strategies that direct tagmentation to occur at regions targeted by an antibody to enable ChIP-seq like assays (Ai et al. 2019) without the need to perform immunoprecipitation. Although several strategies have been developed, including an approach referred to as chromatin integration labeling, or ChIL-seq (Harada et al. 2019), the method that has seen wide adoption using a fusion protein between the Tn5 transposase and Protein A (Tn5-pA), a surface protein with a high binding affinity to immunoglobulins. These strategies, CUT&Tag and ACT-seq (Carter et al. 2019; Kaya-Okur et al. 2019; Bartosovic et al. 2020), involve the binding of antibodies to their target histone modifications with or without secondary antibody staining, followed by the binding of the Tn5-pA loaded with sequencing adapters to the antibody in the absence of Mg2+, thus preventing the tagmentation activity. After washing away unbound Tn5-pA transposomes, Mg2+ is then added to activate the transposase, leading to the tagmentation and production of sequencing library fragments only at regions that were originally bound by the targeting antibody. Much like other tagmentation-based assays, the simplicity and efficiency of the workflow is the most appealing factor, including workflows for CUT&Tag that can be performed on a workbench at home (Henikoff et al. 2020) or on single cells (Carter et al. 2019; Wang et al. 2019; Bartlett et al. 2021), in what is likely the early stage of a wave of technology advancements on this front, including strategies that capture both RNA and histone modifications in the same cells for which two methods were recently described (Xiong et al. 2020; Zhu et al. 2021).
Tagmentation of individual cells
The initial use of tagmentation for single-cell applications took advantage of the workflow simplicity as opposed to the high efficiency afforded by tagmentation. This simplicity enabled the high-throughput multiplexed production of sequencing libraries of cDNA constructed from single cells as the final stage of the original Smart-seq technology (Ramsköld et al. 2012; Picelli et al. 2014b), and was subsequently accompanied by a detailed workflow for the production of Tn5 enzyme and preparation for its use in sequencing assays (Picelli et al. 2014a). However, the efficiency of conversion of genomic DNA into sequencing library fragments ultimately became the driving factor for the use of tagmentation for single-cell workflows, largely based on the initial descriptions of single-cell ATAC-seq and the wealth of technologies that have followed (Buenrostro et al. 2015; Cusanovich et al. 2015). This included a number of assays that leveraged a variety of strategies to isolate individual cells within their own reaction compartments for subsequent processing (Fig. 2A).
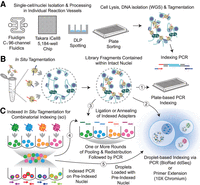
Indexing strategies for tagmentation-based single-cell assays. (A) Physical isolation of single cells into individual rection vessels that are then lysed and processed individually using the standard tagmentation workflows. Indexed PCR on each reaction compartment enables single-cell discrimination. (B) In situ tagmentation is performed by performing the tagmentation reaction in bulk to produce a single cell's library contained within the nucleus of that cell. These preprocessed nuclei can then be subjected to indexing via several techniques, including sorting and indexing PCR in plates (arrow 1), droplet encapsulation with indexing PCR or primer extension (arrow 2), or combinatorial indexing strategies using ligation (arrow 3). (C) Tagmentation with a large set of indexed adapters corresponding to individual reaction wells enables the IST nuclei to then be pooled and redistributed for a second round of indexing, enabling single-cell discrimination using the combination of indexes. The preindexed nuclei can also be carried through additional rounds of indexing using ligation-based methods (arrow 4) or droplet-based methods to enable increased throughput (arrow 5).
For single-cell ATAC-seq, this involved individual cell isolation using a microfluidics chip (Fluidigm C1) to produce up to 96 single-cell libraries per experiment, which for some time boasted the highest coverage that could be obtained per cell, although hampered by relatively low throughput (Buenrostro et al. 2015). This was subsequently addressed by the use of a nanoliter chip instrument (Takara iCell8) that enabled scaling to over 1000 cells in a single preparation (Mezger et al. 2018), although still provides lower throughput than most in situ tagmentation (IST) approaches, which have largely supplanted methods that isolate individual cells. Other strategies have also been developed that take advantage of the flexibility of a single reaction per cell system to capture additional properties and are detailed later in this review.
Beyond ATAC implementations, cell isolation and processing have also been applied to other genomic properties. Direct tagmentation of genomic DNA from single cells has emerged as an alternative to performing whole-genome amplification techniques before library construction. First described as “direct library preparation” (DLP), this approach leverages a nanoliter microfluidics or spotting system to isolate DNA from single cells and perform tagmentation and PCR-based indexing (Zahn et al. 2017; Laks et al. 2019). Other variants that use direct tagmentation of isolated genomic DNA from single cells include LIANTI, which uses a T7 promoter and in vitro transcription (IVT) (Chen et al. 2017), and META, also applied to produce 3D chromatin profiles (Dip-C) (Tan et al. 2018); both of which are described in more detail later owing to their innovative strategies for tackling the 50% efficiency cap of standard two-adapter tagmentation.
In situ tagmentation
The use of the nuclear scaffold itself has emerged as a primary way of achieving single-cell compartmentalization, whereby cells or nuclei can be prepared in bulk with tagmentation performed within intact nuclei (Fig. 2B). These preprocessed nuclei can then be carried through strategies that enable the unique indexing of library fragments from each individual nucleus. The core component that makes this technique viable is that the Tn5 enzyme remains tightly bound to target DNA after completing cleavage and strand transfer. This effectively “glues” the library fragments in place within the intact nucleus. This property was described some time ago (Goryshin and Reznikoff 1998) but was not used as an advantage until it was leveraged to produce long-range sequence information in the form of linked sequence reads: contiguity-preserving transposition and sequencing, or CPT-seq. In this workflow, Tn5 binding effectively stitches together long chains of library molecules for subsequent indexing. The resulting sets of reads can be used in haplotype resolution (Amini et al. 2014) or genome assembly (Adey et al. 2014). CPT-seq was also the first to include indexed transposase adapter sequences in lieu of the standard sequencing primers, along with pooling and redistribution for a combinatorial index space, laying the early groundwork for subsequent single-cell combinatorial indexing (sci) methods.
The adaptation of the tight Tn5 binding to IST for single-cell assays was borne out of an attempt to produce haplotype-resolved ATAC-seq by using the linked read CPT-seq workflow. In these experiments, the nuclear scaffold proved resilient to the gentle lysis workflows that were used and resulted in the containment of library fragments within the nucleus, inadvertently producing single-cell profiles. After substantial refinement and optimization, this resulted in the first description of IST, as well as single-cell combinatorial indexing, to produce single-cell ATAC-seq profiles in a technology now referred to as sci-ATAC-seq (Cusanovich et al. 2015).
IST has since become the most popular strategy for single-cell technologies that leverage tagmentation (Table 1). A typical IST workflow for ATAC-seq involves the isolation and permeabilization of nuclei, which are then carried through the tagmentation reaction. This process is typically performed at either 37°C, as in standard bulk ATAC-seq workflows, or 55°C, which is typical for other tagmentation workflows. The higher tagmentation temperature can increase transposition efficiency and produce greater reads per cell, although often at the expense of decreased specificity for open chromatin and reduced nuclear integrity. However, the magnitude of these differences is highly sample-type dependent and warrants initial testing at the initiation of any study that is applying IST on tissues or cell types that have not yet been characterized for these effects. At either temperature, a large proportion of nuclei inevitably rupture as a result of the tagmentation process, with yields typically falling in the 50% range; however, recovery varies substantially and is influenced primarily by cell or tissue isolation, preservation, or other processing. The IST nuclei then serve as the input for downstream processing steps to achieve single-cell indexing.
Tagmentation-based single-cell technologies
The dominant use of IST is to obtain single-cell ATAC-seq profiles; however, strategies have been developed to enable the acquisition of other molecular properties. These approaches use a preprocessing step in which nuclei or cells are first cross-linked with formaldehyde followed by exposure to SDS to disrupt or deplete nucleosomes. This effectively makes the entire genome accessible to the tagmentation reaction as opposed to being restricted to regions of natively accessible chromatin. Nucleosome disruption strategies have been applied to assess genome sequence (Vitak et al. 2017; Yin et al. 2019; Mulqueen et al. 2021), DNA methylation (Mulqueen et al. 2018), and chromatin folding (Mulqueen et al. 2021).
Single-cell indexing of IST nuclei
The most direct strategy to index individual preprocessed nuclei is to stain them with DAPI and perform fluorescence-assisted nuclei sorting (FANS) of individual nuclei into wells of a plate, where each well can then be carried through PCR with well-specific indexed primers (Chen et al. 2018). One advantage of this approach is that it does not require any specialized equipment and can use off-the-shelf tagmentation reagents; however, throughput is limited owing to each cell requiring its own PCR. In theory, this approach could also be applied in a chip-based platform to increase throughput, similar to what has been previously described (Mezger et al. 2018). However, instead of both tagmentation and PCR within the wells of the chip; tagmentation would be performed in situ before distribution of the nuclei into the chip for indexed PCR.
To further increase throughput, droplet-based workflows that were originally developed for single-cell RNA-seq applications have been leveraged for single-cell ATAC-seq. In these technologies, pretagmented nuclei are loaded into individual droplets along with a bead that contains indexed primers, similar to transcriptome methods, except instead of reverse transcription to extend the indexed primer, either PCR (Lareau et al. 2019) or rounds of linear extension (Satpathy et al. 2019) are performed. This includes two commercially-available options, the Bio-Rad ddSeq and 10x Genomics (2021) chromium instruments; however, custom instrumentation built in the laboratory could also be used, such as the Drop-seq or inDrop platforms (Klein et al. 2015; Macosko et al. 2015), or use of other commercially available single-cell platforms that do not currently include tagmentation assays, for example, the Mission Bio Tapestri or 1CellBio inDrop systems. The major challenges for the implementation of droplet-based compartmentalization for tagmentation-based assays are that the PCR or linear extension reactions require higher temperatures than reverse transcription or primer annealing (∼95°C vs. ≤42°C; which can be solved with optimized oil and surfactant chemistry), and the removal of bound Tn5 to enable the end repair to copy the transfer strand adapter (Fig. 1E). For plate or chip-based workflows, tagmented nuclei are deposited into a buffer that facilitates Tn5 removal; these buffers are then diluted or inactivated before the addition of PCR reagents, which is a trivial process. In droplet-based workflows, the addition of reagents to already-formed droplets is either extremely difficult or not possible. This necessitates that Tn5 removal, end repair, and amplification must all be performed in the same buffer, potentially limiting efficiency of either component; however, the published workflows on these platforms have achieved a balance that enables the production of quality data sets (Satpathy et al. 2019; Lareau et al. 2020), producing some of the highest coverage single-cell ATAC-seq profiles to date.
Single-cell combinatorial indexing
As an alternative to indexing cells by isolating each individual nucleus within its own reaction compartment, technologies have been developed that leverage one or more rounds of indexing, pooling, and redistribution to achieve a unique set of indexes that are used to deconvolve single-cell profiles (Fig. 2C). This approach, single-cell combinatorial indexing, or sci (pronounced “sky”) was first developed for single-cell ATAC-seq, sci-ATAC-seq (Cusanovich et al. 2015), where a set of 96 transposome complexes, each with a unique indexed adapter, are used for IST to produce several thousand preindexed nuclei in each reaction. The nuclei are then pooled together across all indexed reactions such that if any random nucleus were assessed, it would be labeled with a random one of the 96 indexes. If a second nucleus were assessed, there is effectively a one in 96 chance that it is labeled with the same index as the first, and so on. A limited set of preindexed nuclei is then deposited into the wells of one or more new plates, such that the probability of having any two nuclei with the same tagmentation index within the same deposition well is low. End repair and PCR amplification with primers containing indexes specific to each individual deposition well are then performed. This produces libraries with two distinct indexes: one from the tagmentation reaction and one from the PCR reaction, which can be used for single-cell discrimination. The primary advantages of sci methods are that they can scale to large numbers of cells by expanding the tagmentation and/or PCR index set and do not require specialized equipment (Cao et al. 2017). Another major benefit of sci is that the initial tier of cell indexing can be used to encode sample ID. This enables large numbers of separate input samples to be multiplexed within a single experiment, minimizing batch effects of library preparation and enabling targeted proportions of cells to be profiled for each individual sample. The use of indexed IST and combinatorial indexing has since been expanded to a number of other assays beyond single-cell ATAC-seq (Table 1).
For typical sci workflows using indexed IST and indexed PCR, the throughput is in the tens of thousands of cells, similar to the throughput provided by droplet-based systems (Preissl et al. 2018). Recently, the high index space afforded by the Bio-Rad ddSeq droplet-based single-cell ATAC-seq platform was melded with upfront indexed IST, enabling the superloading of droplets and cell throughput an order of magnitude greater than the droplet platform on its own (Lareau et al. 2019). This technology, dsci-ATAC-seq, not only offers high throughput but also affords the sample multiplexing capabilities of traditional sci methods.
Although the use of two-stage combinatorial indexing (tagmentation and PCR) can achieve relatively high cell throughput, other strategies have been developed that incorporate additional tiers of indexing to increase throughput to even greater levels using ligation-based index addition, similar to what was developed previously for single-cell RNA-seq (Rosenberg et al. 2018). However, with each additional round of pooling, redistribution, and indexing, there is a reduction in overall efficiency because each processing step does not have perfect yield, which may be an important consideration depending on the goals of the experiment. Although one of these multitier assays includes indexed IST with a single tier of ligation-based indexing (Yin et al. 2019), the rest use off-the-shelf transposome reagents with two or more ligation rounds (or annealing), making their implementation much more accessible to the broader community. This includes one assay that profiles ATAC, sci-ATAC-seq3 (Domcke et al. 2020), as well as two similar assays to profile both chromatin accessibility and transcription in the same cells: Paired-seq (Zhu et al. 2019) and SHARE-seq (Ma et al. 2020), or histone modifications plus transcription, Paired-Tag (Zhu et al. 2021). These advancements enable the possibility to scale to very large numbers of cell profiles, with the capability to go far beyond what could reasonably be carried through sequencing.
It is important to note that although sci technologies have shown great promise, they have not been widely adopted. This is largely because of the challenge of procuring Tn5 transposase that can be loaded with specific adapters required for these assays, detailed later in this review. With the development of assays that use off-the-shelf tagmentation reagents, wider adoption may follow; however, currently no sci technologies are available in kit form, which is preferred for many groups that want to avoid extensive work to get the assays up and running. Furthermore, sci approaches have generally provided lower coverage than methods that isolate individual cells, although recent developments in transposome design may overcome this challenge (Mulqueen et al. 2021).
Capturing additional properties alongside tagmentation-based assays
One of the most promising areas of tagmentation-based assays has been the development of technologies to capture multiple properties from the same cell, with one or more of those properties encoded by the tagmentation reaction. These “multimodal” or “multiomic” assays hold immense potential for revealing the interactions between layers of genomic properties, particularly those that capture both RNA and ATAC, enabling the association of regulatory element activity with transcriptional state. Versions of these assays have leveraged IST along with in situ reverse transcription and combinatorial barcoding (sci-CAR [Cao et al. 2018], Paired-seq [Zhu et al. 2019], and SHARE-seq [Ma et al. 2020]); IST followed by droplet encapsulation for simultaneous indexed reverse transcription and indexing of ATAC fragments (SNARE-seq [Chen et al. 2019] and 10x multiome [10x Genomics 2021]), as well as with the inclusion of antibody-linked oligonucleotides to measure cell surface protein abundance (TEA-seq [Swanson et al. 2021]); or methods that do not include IST and rely instead on cell capture and processing in individual reaction vessels (scCAT-seq [Liu et al. 2019] and ASTAR-seq [Xing et al. 2020]). With the exception of the latter methods, which are hampered by low throughput owing to single-cell isolation (at least in their current form), all of these techniques face the challenge of performing IST on permeabilized cells or nuclei without loss of RNA, or cross-contamination of RNA from other cells (i.e., ambient RNA).
Another advancement in the multiomic space is the capture mitochondrial DNA sequence in single cells along with the ATAC profile (Lareau et al. 2020). In the initial description of ATAC-seq, the high proportion of mitochondrial reads was considered a problem, but their inclusion in single-cell ATAC-seq data presented the opportunity to track clonal lineages based on mitochondrial DNA variants, as well as heteroplasmy within individual cells. This strategy to capture mtDNA in parallel was also leveraged in a novel strategy to detect cell surface proteins using a phage-nanobody-display approach that introduces nanobody-target indexes that are processed along with ATAC fragments (Fiskin et al. 2020).
In addition to assays that capture multiple genome-scale molecular properties, tagmentation workflows have also been adapted to capture other forms of information pertinent to each cell. This has included the targeted capture of specific sequences alongside single-cell ATAC-seq profiles, including the TCR sequence of immune cells (T-ATAC-seq [Satpathy et al. 2018]), as well as the capture of guide RNA identity in CRISPR-Cas9 pooled perturbation experiments: Perturb-ATAC, Spear-ATAC, and CRISPR-sciATAC (Rubin et al. 2019; Liscovitch-Brauer et al. 2020; Pierce et al. 2021), as the ATAC equivalent to the transcriptional-based Perturb-seq (Dixit et al. 2016) or CROP-seq (Datlinger et al. 2017). Although these CRISPR perturbation approaches that profile ATAC or other DNA-based properties are still in their early stages, they hold incredible potential to enable the systematic dissection of regulatory circuits within a complex biological system, particularly if coupled to assays that capture both chromatin accessibility and transcription, which in theory should be possible with existing techniques that produce both ATAC and RNA molecular profiles detailed above. Furthermore, the use of a platform that scales to large cell number will be of particular value for any application in which a large perturbation space is probed in order to provide sufficient sampling of any individual condition.
Another data channel that has been developed in the transcriptional space is the capture of spatial information along with the genomic property of interest. In the recently described sciMAP-ATAC (Thornton et al. 2021), hundreds of 250-micron-diameter microbiopsy punches from tissue sections are processed using the tagmentation stage of sci-ATAC-seq indexing to record the punch from which the cell profiles were derived. Although not as high resolution as some of the methods to profile transcription with spatial resolution, it does provide multiple single-cell profiles for each position and is also theoretically applicable to any combinatorial indexing workflow. However, the future of capturing spatial information may lie in the use of the direct tagmentation of intact tissues, which was recently described to pinpoint the 3D position of sequences within the genome by performing in situ sequencing off of the adapter (Payne et al. 2020); by taking in situ tagmented tissue sections and capturing the fragments with spatially encoded barcode sequences, similar to spatial transcription methods (Ståhl et al. 2016; Rodriques et al. 2019); or by leveraging direct spatial barcoding of tissue sections and capturing those barcodes alongside the tagmentation-encoded property, similar to DBiT-seq (Liu et al. 2020). The latter of these may hold the greatest near-term promise owing to the ability to resolve true single-cell profiles, as opposed to purely spatial profiles, in which an index feature may overlap the boundary of two cells; however, the challenge of achieving high cell and spatial-barcode capture efficiency is nontrivial.
Improving efficiency for high-coverage single-cell assays
The primary advantage of tagmentation in single-cell assays is its efficiency. However, even with perfect yield in all subsequent steps, the tagmentation reaction itself is limited to a maximum of 50% recovery using standard workflows. This is because of the random incorporation of forward and reverse adapters: where 50% of molecules contain one of each and are viable and the remaining 50% is split between molecules that contain two forward or two reverse adapters and are not viable for subsequent processing (Fig. 3A). The second efficiency limitation is that two independent tagmentation events must occur within ∼1000 bp of one another in order to produce fragments that can be PCR amplified and sequenced. Addressing these challenges is key to advancing the utility of tagmentation-based single-cell assays by enabling the production of more-complete profiles for each cell.
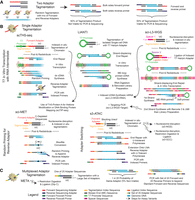
Transposome strategies to improve efficiency. (A) The standard tagmentation reaction randomly incorporates a mix of forward and reverse adapters. This results in 50% of the resulting molecules with both a forward and reverse adapter that can be carried through subsequent processing steps, with such fragments preferentially forming hairpin complexes rather than primer annealing during PCR and also being unable to form sequencing clusters. The remaining 50% are flanked by two forward or by two reverse adapter sequences and are not viable. This effectively caps the maximum efficiency of two-adapter tagmentation at 50%. (B) Several strategies have been developed that use single-adapter tagmentation with an alternative means of appending a reverse adapter. Three of these use tagmentation with a T7 promoter to enable linear amplification using in vitro transcription. The other two use either random priming or adapter switching strategies. Arrows indicate alternative processing workflows: (1) sciTIP-seq to obtain histone modification profiles, (2) sci-L3-WGS + RNA to capture RNA alongside DNA, (3) capture of targeted regions of the genome within the sci-L3-WGS workflow, (4) s3-WGS to capture whole-genome sequence with the s3 workflow, and (5) s3-GCC to capture both WGS and chromatin folding with the s3 workflow. (C) Tagmentation with an expanded set of adapters reduces the probability of producing fragments that terminate in the same adapter species from 50% to 1/n, where n is the number of adapter species present.
Over the years, several strategies have been leveraged to combat one or both of these inefficiencies for bulk and/or single-cell assays. Most of these approaches deploy a single adapter tagmentation strategy and use other means to incorporate the reverse adapter (Fig. 3B). The first of these was developed for low-input whole-genome bisulfite sequencing, Tn5mC-seq (Adey and Shendure 2012), where tagmentation was performed using only a forward adapter followed by the denaturation of a truncated ME bottom strand and replacement with the corresponding reverse adapter. Also in the DNA methylation space, sci-MET (Mulqueen et al. 2018) leveraged indexed IST with a single adapter followed by the incorporation of a reverse adapter using random priming to produce single-cell methylomes. This line of development led to the recently described “s3” technique that uses tagmentation for the forward adapter and incorporates the reverse adapter using a strand-switching technique (Mulqueen et al. 2021), which resulted in substantial improvements in the coverage obtained per cell, as high as 10-fold over predecessor sci-ATAC-seq technologies and 100-fold over sci-DNA-seq. The improvement beyond the expected twofold is owing to the increased efficiency of adapter switching using multiple rounds of extension over the single round of end repair that is used in standard tagmentation workflows.
One of the most compelling advances to overcome both the 50% bottleneck and the requirement of two proximal tagmentation events is in the use of a single Tn5 adapter that contains the T7 promoter sequence. First deployed as a means of obtaining chromatin accessibility information via transposome hypersensitive site sequencing (THS-seq) (Sos et al. 2016), the incorporation of the T7 promoter sequence enables subsequent amplification via IVT extending out from the site of tagmentation. The RNA can then be converted into cDNA and sequenced to identify all sites of Tn5 insertion, which indicates open chromatin in a way similar to that of ATAC-seq. THS-seq has since evolved to include IST and the use of indexed adapters, such that the Tn5 adapter index is included in the IVT, enabling combinatorial indexing (Lake et al. 2017). A similar transposome structure was also used with Tn5 fused with Protein A to map histone modifications and CTCF binding in recent work: sciTIP-seq (Bartlett et al. 2021). Similarly, the use of T7 promoter incorporation was developed for single-cell genome sequencing, with a similar workflow performed on DNA isolated from single cells in an approach called linear amplification via transposon insertion (LIANTI), which also leveraged hairpin transposase adapters that enabled self-priming for an efficient conversion of the RNA intermediate back into cDNA (Chen et al. 2017). Taken further, the nucleosome disruption and IST strategies of sci-DNA-seq coupled to the linear amplification provided by LIANTI and the use of an additional layer of indexing using barcoded adapter ligation resulted in sci-L3-WGS (Yin et al. 2019). This technique provided increased throughput potential with the third layer of indexing, as well as a substantial increase in reads obtained per cell compared with sci-DNA-seq, along with variants to obtain WGS plus targeted sequencing, and a coassay capable of obtaining both WGS and RNA from the same cells.
In a starkly different direction, the use of an expanded set of adapter sequences has also been leveraged to address the same problem (Fig. 3C). This technique, multiplexed end-tagging amplification (META) was first described to achieve high read counts for the assessment of the 3D chromatin structure in single cells (Dip-C) (Tan et al. 2018) and then later for single-cell whole-genome sequencing (Xing et al. 2021). The chromatin conformation component is performed much like Hi-C techniques, with restriction digestion and then ligation to produce a proportion of chimeric ligation junctions that can be used to infer chromatin contacts in three dimensions; however, in Dip-C, there is no enrichment for these ligation junctions, which would be the case for typical Hi-C technologies. Once the processed DNA for a single cell has been produced, tagmentation is performed with Tn5 that has been loaded with a set of 20 different adapter sequences, thus making the probability of producing a fragment terminating in the same sequence only one in 20, as opposed to one in two. Two PCR reactions are then performed to append the required pair of sequencing primers. This technology was able to produce very high coverage maps of chromatin folding, with contacts per cell exceeding 2 million, well beyond other platforms. This enabled the contacts to be split on the basis of haplotype, using heterozygous variants to produce haplotype-resolved single-cell maps of chromatin folding. However, the throughput of META and Dip-C is relatively low, with tens of cells produced in any given experiment, although this tradeoff may be worthwhile for certain applications when considering the high coverage that can be obtained.
The future of tagmentation-based single-cell assays
Tagmentation has already become the core component for a number of single-cell technologies. In line with all single-cell efforts, the continued push to improve cell throughput, cell coverage, and expansion of the catalog of properties that can be assayed using tagmentation-based single-cell technologies will remain relevant; however, a number of these challenges have largely been achieved. This includes the ability to profile very large numbers of cells using inexpensive assays, driven primarily by the use of combinatorial indexing. Similarly, several of the tagmentation techniques that improve efficiency have achieved high cell coverage for certain properties, which will inevitably be applied to other tagmentation-based single-cell technologies. Finally, the span of molecular properties that can be profiled using tagmentation methods is extensive, with the capability to assay a substantial portion of the regulatory machinery that drives cell state. Some of the more recent variants, for example, the profiling of histone modifications using antibody-targeted tagmentation, are still in their early stages, although are sure to be further optimized and likely extended to enable the profiling of transcription factor binding. However, a number of properties that can be profiled in bulk cell populations have not fully made their way to the single-cell space, such as the assessment of lncRNA–DNA interactions (Chu et al. 2011; Simon et al. 2011); of protein–protein interactions along with DNA targets (Mohammed et al. 2013), which may reveal dynamic changes in regulatory complex formation; or of other bulk assays that already use tagmentation, such as for assessing protein-mediated chromatin folding (Mumbach et al. 2016). Although it is likely that assays to target these and other individual properties will eventually be developed, the next frontier of single-cell assays is in technologies that profile two or more properties simultaneously, including cell metadata such as spatial position or perturbation.
The advancements so far have focused on the inclusion of transcription profiling along with chromatin accessibility, which makes sense given the value of information that can be obtained from that combination. Many of these workflows can, in theory, be readily adapted to capture transcription along with other tagmentation-based assays. One of the most direct adaptations is the use of antibody-targeted tagmentation to profile histone modifications alongside transcription; however, the challenge of retaining mRNA within permeabilized cells or nuclei during the antibody washing steps may prove challenging, although it was recently accomplished with the Paired-Tag and CoTECH technologies (Xiong et al. 2020; Zhu et al. 2021). Similarly, chromatin conformation assays that leverage tagmentation could be adapted to these multiomic workflows, although again facing the challenge of mRNA retainment during pretagmentation processing steps, in this case restriction digestion and ligation of proximal genomic fragments. As the field continues to develop, it can be expected that these assays, as well as those that capture more than two properties, will become the primary focus of development in the single-cell space as a whole, with tagmentation playing an important role.
Although challenges remain on the molecular biology and biochemistry front for assay development, realistically there is a major barrier that does not pertain to the assays themselves, sequencing. Many of the assays detailed in this review can produce profiles for very large numbers of cells or provide very high unique read counts per cell (or both), necessitating a massive amount of sequencing. Furthermore, a number of these assays are quite inexpensive to perform, further shifting the cost burden of any experiment to the sequencing side of the equation, where sequencing can account for >90% of the total costs of an experiment (Mulqueen et al. 2018, 2021; Sinnamon et al. 2019; Thornton et al. 2021). As such, any advancements that reduce the cost per read will have an immediate impact on the design of single-cell studies.
A final barrier to the further development and refinement of tagmentation-based technologies has been the availability of the Tn5 enzyme in sufficient quantities. The Tn5 enzyme can be purchased preloaded with the standard forward and reverse adapter sequences compatible with standard Illumina sequencing workflows. Tn5 is also commercially available in limited quantities as unbound enzyme, although often at a high price point. This is particularly challenging for assays that use indexed tagmentation, where often 96 or more individually indexed transposome complexes are assembled, thus requiring a substantial volume of enzyme. As a result, many have turned to producing the enzyme themselves using various published workflows (Picelli et al. 2014a; Kia et al. 2017). However, Tn5 production often is challenging, time-consuming, and not the desired route because the quality of the Tn5 enzyme and assembled transposome complexes is pivotal to the success of tagmentation assays.
Concluding remarks
With over 10 years of tagmentation behind us, the use of Tn5 transposase in genomic assays appears to be here to stay, at least in one form or another. Tagmentation is a fixture of a wide range of single-cell technologies, motivated in large part by ATAC-seq methods to assay chromatin accessibility, although the catalog of assayed properties extends well beyond. This is driven by the same facets of Tn5 that made it a useful tool before single-cell assays and even preceding its use in high-throughput sequencing: efficiency and versatility. These assets are exemplified by the plethora of technological innovations that have leveraged tagmentation, many of which are detailed in this review and many others that are either not relevant or have not yet been applied at the single-cell level. Beyond the technology component, these methods have enabled profound biological insight. This has included atlases of chromatin accessibility across a range of tissues and species, including the developing fly (Cusanovich et al. 2018b), adult mouse (Cusanovich et al. 2018a), mouse cerebrum (Li et al. 2020), human immune cell development (Satpathy et al. 2019), and the developing human fetus (Domcke et al. 2020) to name a few. These methods have also proven incredibly valuable for the dissection of gene regulatory circuitry and impact of cis-regulatory elements on transcription during dynamic state transitions. Such strategies have been applied to understand state changes in myogenesis (Pliner et al. 2018), the stimulation of bone-marrow-derived cells (Lareau et al. 2019), or drug response (Torkenczy et al. 2020), often using computational strategies to coembed chromatin accessibility data with associated single-cell RNA-seq data sets (e.g., Stuart et al. 2019; Welch et al. 2019). However, the greatest promise for biological discovery is in the deployment of assays that capture multiple properties in the same cell. The catalog of technologies that capture a tagmentation-based property alongside transcription continues to expand (Table 1), and the current literature has only scratched the surface of what will be possible with these technologies, enabling detailed breakdowns of chromatin state within individual cell populations in heterogeneous tissues (Zhu et al. 2021) or during cell state changes (Ma et al. 2020). Ultimately, these platforms have largely been made possible owing to the efficiency and versatility of tagmentation-based workflows. Looking to the next 10 years, it is hard to imagine the field of single-cell genomics without a substantial tagmentation component.⇓
Glossary of key terms
Adapter—In this review, adapter refers to the oligonucleotides that are loaded onto the Tn5 transposase to form transposome complexes, specifically the sequence portion other than the ME region that is used as a priming or ligation site for subsequent processing.
End repair—The term “end repair” covers a broad range of ways that the ends of double-stranded DNA are extended or digested to produce blunt ends. In this review, the term is used to specifically refer to the use of polymerase to extend the 3′ end of tagmented DNA to displace or digest the nontransferred ME sequence through the end of the adapter that was present on the transfer strand. This process is critical before denaturation during PCR amplification for the majority of tagmentation-based assays.
IST—The process of performing the tagmentation reaction on intact cells or nuclei, whereby the resulting cells or nuclei contain precursor sequencing library molecules contained within them owing to the tight binding of Tn5 transposase to target DNA.
Mosaic end (ME)—The 19-bp sequence that is recognized by the Tn5 transposase. It must be double-stranded for the majority of the sequence; however, a reduction to 16 bp on the nontransfer strand from the 3′ end does not show evidence of any major efficiency reduction, as long as the transfer strand remains the full 19 bp.
sci—Abbreviation for single-cell combinatorial indexing, which is one of the mechanisms to index cells through one or more rounds of pooling and redistribution in which in each round indexes are appended to the library molecules contained within the cell or nucleus.
Strand transfer—The process of covalently linking the 3′ end of transfer strand ME sequence with the 5′ end of the target DNA.
Synaptic complex—The term for the transposome bound to the target DNA before strand transfer.
Tagmentation—The process of using a transposase to both fragment DNA and tag it with adapter that were loaded onto the transposase enzyme.
Tn5—The Tn5 cut-and paste transposase enzyme. It is a 53.3-kDa enzyme that forms a transposome complex as a homodimer when it binds ME DNA sequences.
Tn5-pA—The Tn5 transposase enzyme fused to Protein A for use in antibody-targeted tagmentation assays. Other variants that include other antibody-binding proteins also exist, such as Protein AG to enable a broader range of antibody species targets.
Transfer strand—The DNA strand that is a part of the transposome complex that becomes covalently attached at its 3′ end of the ME sequence to the 5′ end of the nick that is introduced to the target DNA backbone. In tagmentation workflows, this strand contains a 5′ overhang of single-strand DNA beyond the ME sequence that is the forward or reverse adapter sequence that serves as a subsequent priming site for PCR. The nontransfer strand is typically the reverse complement of the 19-bp ME sequence.
Transposome—The transposase complexed with the DNA oligonucleotides. For the Tn5 transposase, this is composed of the Tn5 homodimer and two oligo sequences that contain the ME sequence at a minimum.
Competing interest statement
A.C.A. is an author on patents that pertain to several technologies described in this review—“Nucleosome depletion of intact nuclei for compartmentalized single-cell (epi)genomic profiling” (US20180355348A1), “Single-cell whole-genome libraries and combinatorial indexing methods of making thereof” (US20180023119A1)—and pending patents that pertain to improving sequencing library yields.
Acknowledgments
I thank Ryan Mulqueen for providing several illustrations that were modified for some of the figures in this review. I also thank the many groups and researchers around the world that have come up with innovative uses for tagmentation; it is a continued joy to see new and exciting takes on the technology, and I hope that this review may inspire additional new ideas. Finally, I thank William S. Reznikoff, who pioneered the characterization and use of Tn5 transposase. I am supported by the National Institutes of Health (NIH)/National Institute of General Medical Sciences R35GM124704 and the NIH/National Institute on Drug Abuse R01DA047237.
Footnotes
-
Article and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275223.121.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








