
Transcript assembly improves expression quantification of transposable elements in single-cell RNA-seq data
- 1Department of Genetics, Washington University School of Medicine, St. Louis, Missouri 63110, USA;
- 2Edison Family Center for Genome Sciences and Systems Biology, Washington University School of Medicine, St. Louis, Missouri 63110, USA;
- 3McDonnell Genome Institute, Washington University School of Medicine, St. Louis, Missouri 63108, USA
Abstract
Transposable elements (TEs) are an integral part of the host transcriptome. TE-containing noncoding RNAs (ncRNAs) show considerable tissue specificity and play important roles during development, including stem cell maintenance and cell differentiation. Recent advances in single-cell RNA-seq (scRNA-seq) revolutionized cell type–specific gene expression analysis. However, effective scRNA-seq quantification tools tailored for TEs are lacking, limiting our ability to dissect TE expression dynamics at single-cell resolution. To address this issue, we established a TE expression quantification pipeline that is compatible with scRNA-seq data generated across multiple technology platforms. We constructed TE-containing ncRNA references using bulk RNA-seq data and showed that quantifying TE expression at the transcript level effectively reduces noise. As proof of principle, we applied this strategy to mouse embryonic stem cells and successfully captured the expression profile of endogenous retroviruses in single cells. We further expanded our analysis to scRNA-seq data from early stages of mouse embryogenesis. Our results illustrated the dynamic TE expression at preimplantation stages and revealed 146 TE-containing ncRNA transcripts with substantial tissue specificity during gastrulation and early organogenesis.
Transposable elements (TEs) occupy a large proportion of eukaryotic genomes, representing ∼50% of the human genome and 40% of the mouse genome (International Human Genome Sequencing Consortium 2001; Mouse Genome Sequencing Consortium 2002). Although once regarded as nonfunctional parasitic sequences, increasing evidence suggests that TE-derived sequences play pivotal roles in gene regulation. During evolution, TEs rewire host transcription networks through transposition and co-option, resulting in a wide variety of TE-derived regulatory elements, including promoters, enhancers, transcription terminators, and chromatin loop anchors (for reviews, see Feschotte and Gilbert 2012; Rebollo et al. 2012; Cowley and Oakey 2013; Garcia-Perez et al. 2016; Chuong et al. 2017; Sundaram and Wysocka 2020). In the present day, despite losing most of their transposition abilities, TE-derived sequences continue to impact host genomes through transcription, which generates protein-coding TE chimeric RNAs as well as noncoding RNAs (ncRNAs) that are involved in normal and cancer development (for reviews, see Gifford et al. 2013; Hadjiargyrou and Delihas 2013; Hutchins and Pei 2015; Anwar et al. 2017; Rodriguez-Terrones and Torres-Padilla 2018).
TEs are major contributors of ncRNAs in both human and mouse. More than two-thirds of mature long ncRNAs contain at least one TE and almost half of the total base pairs of long ncRNA are derived from TEs (Kelley and Rinn 2012; Kapusta et al. 2013). TE-containing ncRNAs show substantial developmental stage and tissue specificity and participate in embryonic stem cell (ESC) maintenance and early embryogenesis. For instance, endogenous retroviruses (ERVs) are highly expressed in ESCs and ERV-derived transcripts are involved in the maintenance of pluripotency (Macfarlan et al. 2012; Santoni et al. 2012; Fort et al. 2014; Lu et al. 2014; Ohnuki et al. 2014; Wang et al. 2014). During mouse and human embryogenesis, a large number of TEs, including ERVs, long interspersed nuclear element-1 (LINE-1), and short interspersed elements (SINEs) become active and contribute to a significant proportion of total RNAs before blastocyst stage (Kigami et al. 2003; Peaston et al. 2004; Svoboda et al. 2004; Maksakova and Mager 2005; Fadloun et al. 2013; Göke et al. 2015; Grow et al. 2015; De Iaco et al. 2017; Ge 2017; Hendrickson et al. 2017; Jachowicz et al. 2017; Whiddon et al. 2017; Percharde et al. 2018). Moreover, knocking down specific TE families, including LINE-1 and MuERV-L, results in clear developmental defects (Kigami et al. 2003; Huang et al. 2017; Jachowicz et al. 2017; Percharde et al. 2018).
Despite the importance of TEs, quantifying TE expression using high-throughput sequencing data has been challenging. Owing to the repetitive nature of TEs, sequencing reads that overlap with TEs are often discarded as a result of ambiguous mapping. Several software tools have been developed to address this issue, and they enabled TE expression quantification in bulk RNA-seq data (Criscione et al. 2014; Jin et al. 2015; Lerat et al. 2017; Jeong et al. 2018; Bendall et al. 2019; Kong et al. 2019; Yang et al. 2019). To quantify the expression of repetitive elements, these tools often aggregate multialigned reads at TE subfamilies/families or redistribute them to individual TEs based on heuristic or statistical rules. Although proven to be successful in a range of biological systems, applications of the current TE quantification strategies were mostly limited to bulk RNA-seq, which lacks the ability to distinguish cell type–specific TE expression.
Recent developments in single-cell RNA-seq (scRNA-seq) provide unprecedented opportunities for examining cell type–specific TE expression. However, effective TE quantification tools optimized for scRNA-seq data are lacking. Although the assessment of genome-wide transcriptional activity of TEs in single cells has been attempted by counting signals at individual TE fragments or TE subfamilies/families (Göke et al. 2015; Ge 2017; Boroviak et al. 2018; Brocks et al. 2018; Yandım and Karakülah 2019; He et al. 2020; Jonsson et al. 2020), such approaches are not optimal. Compared with bulk RNA-seq, scRNA-seq signal is much noisier and often shows 5′ or 3′ end enrichment along the transcripts. Counting reads at individual TEs or subfamilies/families fails to take into account the structures of the full-length transcripts, which can consist of multiple TEs from different subfamilies/families. Consequently, different expression values will be assigned to individual TEs within the same transcript. This caveat is especially obvious when dealing with scRNA-seq data sets in which sequencing reads are enriched at either the 5′ or 3′ end of the RNA. Counting reads without the knowledge of the full-length transcripts will only capture TEs near the 5′ end or the poly(A) signal, resulting in an inaccurate picture of the genome-wide TE expression pattern (O'Neill et al. 2020).
In this work, we present an analytical framework tailored to TE expression quantification in scRNA-seq data sets. We systematically evaluated scRNA-seq reads that mapped to TEs and showed that quantifying TE expression in single cells using transcripts assembled from bulk RNA-seq effectively reduces noise. Applying our strategy to mouse early embryogenesis illustrated the dynamic TE expression during preimplantation stages and revealed TE-containing ncRNAs with substantial tissue enrichment during gastrulation and early organogenesis.
Results
A higher percentage of reads are mapped to TEs in scRNA-seq compared with bulk RNA-seq
Owing to the biological significance of TE-containing ncRNAs, we decided to focus our analysis on TEs that are not part of the exons of protein-coding genes. First, to determine the fraction of reads that can be mapped to these TEs in scRNA-seq data, we processed 36 publicly available single-cell data sets (Supplemental Table S1). These data sets contain both human and mouse samples and were generated using seven different scRNA-seq protocols. Bulk RNA-seq data sets from the same study or derived from the same cell line were included as controls. To preserve reads that originate from repetitive regions, multiple mapping was implemented during the alignment step. Reads that mapped to multiple locations or overlapped with more than one feature were distributed equally for signal quantification. Calculating the number of mappable reads based on genomic locations revealed that a large proportion of reads overlap with TEs in all tested scRNA-seq data sets, suggesting that TE expression can be captured by scRNA-seq (Fig. 1A). We also observed that a higher percentage of reads was mapped to TEs in scRNA-seq compared with bulk RNA-seq. This phenomenon was consistent across different scRNA-seq platforms even when only uniquely mapped reads were considered (Fig. 1A; Supplemental Fig. S1A), suggesting that the high TE mapping ratio is prevalent in scRNA-seq data sets and not introduced by nonunique mapping.

Counting scRNA-seq signal at individual TEs results in large numbers of false positive candidates. (A) Distribution of mappable reads in 16 bulk RNA-seq and 36 scRNA-seq data sets. Compared to bulk RNA-seq, scRNA-seq data have a higher percentage of reads mapped to TEs. Samples were arranged by studies. Data sets used in this figure are summarized in Supplemental Table S1. (PC) Protein-coding exons defined by RefSeq; (TE) transposable elements that do not overlap with protein-coding exons; (Other) other genomic locations; (mESC) mouse embryonic stem cell; (PBMC) human peripheral blood mononuclear cell; (GM12878 and GM12891) human lymphoblastoid cell lines. (B) Number of expressed (counts per million, CPM ≥ 1) protein-coding genes and TEs in mESC bulk RNA-seq and Smart-seq samples. On average, 12,000 protein-coding genes and 6000 TEs were detected in each bulk RNA-seq sample. In contrast, scRNA-seq captured 7000 protein-coding genes and 20,000 TEs per cell. (C) Number of candidates as a function of cell number cutoff. (Cell number cutoff) Minimum number of cells each candidate is expressed in; (expression cutoff) CPM ≥ 1. A cell number cutoff of 10 requires a candidate to have at least 1 CPM in at least 10 cells. Although the majority of protein-coding gene candidates were consistently detected in mESC Smart-seq data, a large number of TE candidates were detected in fewer than 10 cells. (D) Correlation between bulk RNA-seq and averaged scRNA-seq signal at protein-coding genes and TEs (Teichmann laboratory, mESC). Low correlation between bulk RNA-seq and averaged Smart-seq signal was observed at TEs regardless of expression cutoff. (Cell cutoff) Minimum number of cells each candidate is expressed in; (CPM cutoff) minimum CPM value for one candidate to be considered as expressed. Color scale represents the number of candidates. (E) TE-family enrichment analysis using TE candidates identified from mESC bulk RNA-seq and Smart-seq. Enrichment of ERV elements was observed with bulk RNA-seq data, but not in single cells. Smart-seq data of four single cells with different percentage of TE reads and merged Smart-seq data from 10 cells were included.
To evaluate whether the high TE mapping percentage in scRNA-seq was associated with data quality per cell, we further compared scRNA-seq data generated using Smart-seq, 10x Genomics Chromium, SCRB-seq, and Drop-seq and examined the relationships between the percentage of TE reads per cell and the two key parameters indicative of scRNA-seq quality: sequencing depth and the percentage of mitochondrial reads (Ilicic et al. 2016). Our analysis revealed that a high TE mapping percentage was observed across individual cells with limited correlation to sequencing depth (Supplemental Fig. S1B,C). Similarly, no strong correlation was detected between the percentage of TE reads and that of the mitochondrial reads (Supplemental Fig. S1C). These results suggest that the high TE mapping ratio in scRNA-seq is unlikely to be an artifact caused by variations in sequencing depth or cell death.
To address the concern that genomic DNA contamination may contribute to the majority of TE signal in scRNA-seq, we next quantified the number of total reads mapped to five nonoverlapping genomic regions: protein-coding exons, TEs within the introns of protein-coding genes, other intronic regions of protein-coding genes, intergenic TEs, and other intergenic regions. A higher percentage of total reads was mapped to the intronic regions of protein-coding genes in scRNA-seq, and the majority of TE overlapping reads were located within introns (Supplemental Fig. S1A), arguing against severe genomic DNA contamination.
Although additional experiments and analyses will be needed to pinpoint the origin of these intronic reads in scRNA-seq data, considering their presence across diverse scRNA-seq platforms, the limited correlation between the amount of intronic reads and sequencing quality, as well as previous successes in using intronic reads to infer transcription dynamics and cell states (Gaidatzis et al. 2015; La Manno et al. 2018), we hypothesize that these intronic reads originate from unprocessed RNA. Indeed, consistent with previous reports (La Manno et al. 2018; Selewa et al. 2020), we observed regions that are enriched for unspliced scRNA-seq reads and located within introns that tend to be flanked by AT-rich sequences, which could be involved in the poly(A) priming step during cDNA synthesis (Supplemental Fig. S1D).
Counting scRNA-seq reads at individual TEs leads to large numbers of false positive candidates
Current TE expression analyses often quantify RNA-seq signal at individual TE fragments or TE subfamilies/families (Criscione et al. 2014; Jin et al. 2015; Lerat et al. 2017; Jeong et al. 2018; Yang et al. 2019; He et al. 2020; Jonsson et al. 2020). Our observation that a large proportion of scRNA-seq reads map to TEs, especially intronic TEs, raises the concern that counting reads at single TEs or TE subfamilies/families will aggregate noise and fail to exclude TEs that are part of protein-coding genes, resulting in high numbers of false positive candidates. To test this, we applied a similar strategy and analyzed bulk and Smart-seq data sets generated using mouse embryonic stem cells (mESCs) cultured in 2i medium (Buettner et al. 2015; Kolodziejczyk et al. 2015). Because mESCs represent a population with limited heterogeneity and reads generated by Smart-seq and bulk RNA-seq share a similar distribution along the gene body (Ramsköld et al. 2012), we expected that the expression profiles obtained with scRNA-seq to be largely similar to those generated with bulk RNA-seq.
We first calculated the numbers of expressed protein-coding genes and expressed TEs (CPM ≥ 1) in these data sets. On average, 12,000 protein-coding genes and 6000 TEs were detected in bulk RNA-seq samples. In contrast, scRNA-seq captured an average of 7000 protein-coding genes and 20,000 TEs per cell (Fig. 1B). To evaluate the quality of these expressed candidates, we examined the following three parameters: (1) the number of cells each candidate is expressed in, (2) the correlation between the signal in bulk RNA-seq and the average signal across single cells, and (3) for TE candidates, the overrepresented TE families among all candidates. We reasoned that a candidate representative to the population should be expressed in a relatively large number of mESCs and show a strong correlation between its bulk RNA-seq and averaged scRNA-seq signal. However, only protein-coding genes matched this expectation (Fig. 1C,D; Supplemental Fig. S2A). A large proportion of TE candidates were only detected in a small number of cells and showed weak correlations between scRNA-seq and bulk RNA-seq signal regardless of the expression cutoff. This observation remained valid after we performed the same analysis by counting signals from individual exons. The exon length distributions were comparable to those of TEs, ruling out the possibility that length discrepancy between TEs and protein-coding genes contributes to false positive TE candidates (Supplemental Fig. S2B–D). We further compared overrepresented TEs within candidates identified from bulk RNA-seq and scRNA-seq by performing a TE-family enrichment analysis (Supplemental Fig. S3A). Although ERV1 and ERVK elements have been shown to be expressed in stem cells (Santoni et al. 2012; Fort et al. 2014; Lu et al. 2014; Ohnuki et al. 2014; Wang et al. 2014), they were only enriched in bulk RNA-seq in this analysis (Fig. 1E). scRNA-seq candidates obtained from this analysis were depleted of ERV1 and ERVK and instead enriched for SINEs (Fig. 1E; Supplemental Fig. S3B), which are often found near protein-coding genes and provide sequences that could act as reverse transcription priming sites (Medstrand et al. 2002).
In summary, the high number of TE candidates obtained from scRNA-seq, the weak signal correlation between individual cells, as well as the discordance between bulk and scRNA-seq strongly suggest that counting scRNA-seq reads at individual TEs will result in large numbers of false positive candidates.
Transcript assembly improves TE expression analysis
Transcript annotation serves as the cornerstone for expression quantification. Our ability to accurately assess the expression of protein-coding genes relies on well-annotated gene structures, which help to focus analysis on genomics regions with true signal. Although individual TEs are well annotated, it is usually unclear which TEs are expressed in a biological system and what the underlying transcript structures are. We reason that the large number of false positive candidates in scRNA-seq analysis is caused by counting sparse and noisy signal at millions of TE copies, of which only a small proportion are truly expressed (Supplemental Fig. S3C). Indeed, the signal correlation between averaged scRNA-seq and bulk RNA-seq is much stronger at TEs that overlap with the exons of RefSeq annotated ncRNA (Supplemental Fig. S4A). Therefore, we hypothesize that incorporating transcript structures into the analysis should help to reduce noise.
Several recent studies took advantage of well-studied TE transcription units such as full-length ERVs or LINEs for TE expression quantification, but did not consider transcripts that are composed of TEs from different families or classes (Tokuyama et al. 2018; Bendall et al. 2019; McKerrow and Fenyö 2020). To obtain a more comprehensive catalog of ncRNAs with exonized TEs, we performed transcript assembly using mESC bulk RNA-seq data. We selected transcripts with lengths exceeding 200 nt and identified 692 transcripts whose exons overlap with TEs but not the exons of RefSeq annotated protein-coding genes (Fig. 2A,B; Supplemental Fig. S5). These include transcripts that are entirely derived from TEs (e.g., some ERV transcripts and LINE transcripts) as well as transcripts derived from multiple fragmented TEs and TE–non-TE hybrid units. These transcripts were termed TE transcripts. To test the accuracy of our assembly, we focused on the promoters of assembled TE transcripts and examined several genomic signatures that are indicative of active transcription. Indeed, the majority of our TE transcript promoters overlap with FANTOM5 CAGE peaks (The FANTOM Consortium and the RIKEN PMI and CLST (DGT) 2014) and are enriched for ATAC-seq signal while depleted of CpG methylation (Fig. 2C).
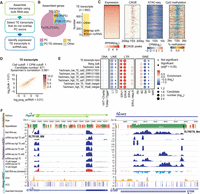
Transcript assembly improves scRNA-seq TE expression analysis. Data sets used in this figure are summarized in Supplemental Table S1. (A) Flowchart of scRNA-seq TE quantification pipeline. In short, transcript assembly was performed with bulk RNA-seq data, and transcripts that overlap with TEs but not protein-coding exons were used for expression quantification in scRNA-seq data. (B) Transcript assembly using three mESC bulk RNA-seq data (Wang laboratory) yielded 692 TE transcripts. Among these TE transcripts, 179 overlap with ncRNAs annotated by RefSeq. (C) FANTOM5 CAGE peaks, ATAC-seq signals, and CpG methylation signals at the promoter region of TE transcripts with RPKM ≥ 1 (reads per kilobase million). (D) Correlation between mESC bulk RNA-seq and averaged Smart-seq (Teichmann laboratory) signals at TE transcripts. Color scale represents the number of candidates. (E) TE-family enrichment analysis using expressed TE transcripts. Enrichment of ERV elements was observed with both bulk RNA-seq and Smart-seq samples. (F) Examples of TE transcript. Assembled TE transcripts, uniquely mapped reads of mESC bulk RNA-seq, Smart-seq, merged Smart-seq, ATAC-seq, and CpG methylation were included. (Left) A TE transcript that initiates from RLTR16b_MM. This TE transcript overlaps Platr14, a long ncRNA known to impact the mESC differentiation-associated genes. (Right) A TE transcript that initiates from RLTRETN_Mm. This transcript is largely composed of TEs and reflect the transcription unit of ERV.
Using these newly generated transcript models, we recalculated the expression of the TE transcripts. Because a major source of noise in measuring TE expression comes from intronic reads (Supplemental Fig. S4A) and intronic signals are products of passive cotranscription of mature RNAs (Lanciano and Cristofari 2020), we only quantified signals within the exonic regions of these TE transcripts. Quantifying TE expression at the TE transcripts led to a much stronger correlation between mESC bulk and Smart-seq data (Fig. 2D; Supplemental Fig. S4B). Furthermore, we obtained much more consistent TE-family enrichment results between bulk and scRNA-seq and were able to identify the expression of ERV elements at single cells (Fig. 2E,F).
Although Smart-seq-based protocols generate deeper sequencing depth with reads covering the entire transcript, other popular scRNA-seq strategies such as 10x Genomics Chromium, Drop-seq, and SCRB-seq produce shallow sequencing with reads biased toward the 5′ or 3′ end of the RNA (Ramsköld et al. 2012; Soumillon et al. 2014; Macosko et al. 2015; Zheng et al. 2017). Counting reads at individual TEs using data with 5′ or 3′ signal enrichment will only capture TEs that are located at either end of the transcripts, thus biasing our understanding about TE expression. We reason that our approach can help to overcome this limitation by using the annotation of full-length TE transcripts.
To support our reasoning, we analyzed the data set from a previously published mESC differentiation study, in which single-cell Smart-seq2, single-cell SCRB-seq, and bulk RNA processed with SCRB-seq protocol were performed (Semrau et al. 2017). Using individual TEs as reference, we observed a severe discordance of TE expression between SCRB-seq and Smart-seq2, likely resulting from differences in signal distribution along the transcripts (Supplemental Fig. S6A). Conversely, using full-length TE transcripts as reference led to significantly improved signal correlations (Supplemental Fig. S6B). Moreover, quantifying TE expression at transcript level allowed us to recover the enrichment of ERVs from all data sets, whereas only Smart-seq2 showed ERV enrichments when counting at individual TEs (Supplemental Fig. S6C).
Taken together, these results suggest that our analysis approach is applicable not only to scRNA-seq data with a high number of reads covering the entire transcript body, but also to other popular scRNA-seq strategies that feature shallow sequencing depth at the 3′ end of the transcripts.
Finally, we examined whether the incorporation of an expectation–maximization (EM) algorithm will improve the accuracy of expression quantification at repetitive regions. We simulated RNA-seq reads at TE transcripts with a wide range of coverages and quantified the observed signal by redistributing multiple mapped reads with an EM algorithm using the amount of uniquely mapped reads as priors. In line with previous reports (Jin et al. 2015; Yang et al. 2019), we found that redistributing multiple mapped reads using the EM algorithm outperforms even-distribution, resulting in a higher percentage of observed reads matching the ground truth (Supplemental Fig. S7). Based on these observations, we implemented this EM-based algorithm in the expression quantification step.
Dynamic TE expression in preimplantation embryos
Encouraged by the results from the mESC data, we decided to apply our strategy to a more complex biological system: mouse embryogenesis. The dynamic regulation of the epigenome during development not only fine-tunes protein-coding genes, but also allows specific TE expression at different developmental stages (Rowe and Trono 2011; Gifford et al. 2013; Gerdes et al. 2016; Rodriguez-Terrones and Torres-Padilla 2018; Deniz et al. 2019). Several recent studies used scRNA-seq to profile the transcription landscape of mouse embryos from zygote to early organogenesis, providing valuable resources for dissecting the dynamic expression of TEs.
To facilitate TE expression quantification, we performed transcript assembly using 37 bulk RNA-seq samples (Supplemental Table S1) that cover a range of tissues and developmental stages and obtained 5299 TE transcripts (Fig. 3A,B; Supplemental Fig. S8A; Supplemental Table S2). Seven hundred seventy of these transcripts overlap with known ncRNAs annotated by RefSeq (Fig. 3A). Compared with assembled protein-coding transcripts, which show similar length and exon number as RefSeq protein-coding gene annotations, assembled TE transcripts are shorter in length and possess fewer exons, a pattern consistent with annotated ncRNAs (Supplemental Fig. S8B,C).
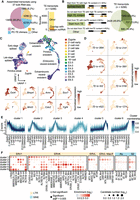
Dynamic TE expression in mouse preimplantation embryos. (A) Using 37 bulk RNA-seq samples, 5299 TE transcripts were constructed. Of these, 770 TE transcripts overlap with ncRNAs annotated by RefSeq. (B) More than half of all the assembled TE transcripts either initiate from TEs or have >50% of their exons composed of TEs. (C, upper) UMAP of scRNA-seq data from mouse zygote to E6.5 embryos. Cells were colored based on developmental stages. (Lower) Expression of cell type–specific markers. (D) Examples of developmental stage– and tissue-specific TE transcripts. (E) TE transcripts were grouped into six clusters based on their expression pattern across preimplantation stages. (F) TE subfamily enrichment analysis using TE transcripts within each of the six clusters.
Next, we analyzed three publicly available data sets in which the transcription landscape of mouse embryos from zygote to gastrulation was profiled using Smart-seq derived protocols (Deng et al. 2014; Mohammed et al. 2017; Cheng et al. 2019). In these data sets, a significant number of reads overlap with TEs, and ∼3% of the total reads are mapped to TE transcripts (Supplemental Fig. S9A,B). After data integration and dimension reduction using the top 4000 variable features, we observed clear clustering patterns that were driven by cell type and developmental stage (Fig. 3C). Among the top 4000 variable features, 410 are TE transcripts, suggesting that the expression of TE transcripts could be cell type– or developmental stage–specific (Supplemental Fig. S9C–E). Indeed, we were able to observe TE transcript expression with strong tissue or stage specificity (Fig. 3D).
To further investigate the dynamics of TE transcription, we focused on preimplantation stages, in which high TE expression was documented. Because of the limited number of cells, scRNA-seq signals of each TE transcript across all the cells with the same developmental stage were averaged to reduce noise. Grouping TE transcripts based on their expression patterns across preimplantation stages resulted in the following six clusters (Fig. 3E): TE transcripts that are maternally deposited (cluster 1), TE transcripts that are up-regulated during minor and major waves of zygotic genome activation (clusters 2 and 3), TE transcripts that are up-regulated during zygotic genome activation and keep accumulating until the blastocyst stage (cluster 4), and TE transcripts that are up-regulated in the early- and mid-blastocyst stage (clusters 5 and 6).
We next performed TE enrichment analysis and observed that TE transcripts with distinct expression profiles tend to be enriched for different TE subfamilies (Fig. 3F). For instance, IAP elements are highly enriched in cluster 4, consistent with a previous report that IAP expression initiates from the two-cell stage, accumulates, and then disappears at the blastocyst stage (Pikó et al. 1984; Poznanski and Calarco 1991; Svoboda et al. 2004). We also observed the enrichment of ERVL and ERVL-MaLR members in cluster 2, in line with previous studies suggesting that ERVL and ERVL-MaLR members are highly expressed during the two-cell stage and constitute ∼5% of the total transcripts (Kigami et al. 2003; Peaston et al. 2004; Svoboda et al. 2004). MTA_Mm-int and ORR1B1-int from the ERVL-MaLR family were also enriched in cluster 6, showing high expression during E4 blastocyst stage, an intriguing observation that is yet to be validated. Moreover, transcription factor binding site analysis using a 500-bp window upstream of TE transcripts identified footprints of transcription factors that were shown to be involved in mouse early embryogenesis such as Krupple-like factors, GABPA, and ELF5 (Ristevski et al. 2004; Donnison et al. 2005; Zhou et al. 2005; Bialkowska et al. 2017), suggesting shared regulatory networks between TE transcripts and protein-coding genes (Supplemental Fig. S10).
Tissue-specific TE expression during mouse gastrulation and early organogenesis
Comparing to preimplantation stages, TE expression during gastrulation and organogenesis is much less well studied, and a comprehensive catalog of tissue-specific TE transcripts is lacking. To address this, we analyzed a 10x scRNA-seq data set in which more than 100,000 cells were assayed using mouse E6.5 to E8.5 embryos (Pijuan-Sala et al. 2019). Comparing with mESC or mouse preimplantation data analyzed in previous sections, this E6.5 to E8.5 10x data contains considerably fewer TE overlapping reads, with ∼1% of the UMI mapping to TE transcripts (Supplemental Fig. S11A,B). Although TE transcripts are in general lowly expressed and lack the high standardized variance observed at some protein-coding genes, they still constitute a small proportion of the top 1000 variable features that can be used to recapitulate the clustering pattern in the original study (Fig. 4A,B; Supplemental Fig. S11C). Furthermore, we were able to observe TE expression patterns that are enriched in small clusters of cells, suggesting that TE transcripts display considerable tissue specificity during these stages (Fig. 4C).
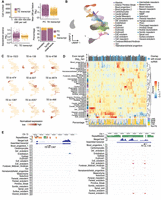
Tissue-specific TE expression during mouse gastrulation and early organogenesis. (A, upper left) Fewer unique molecular identifiers (UMIs) were mapped to TE transcripts than to protein-coding genes. (Upper right) The averaged expression level of TE transcripts across all the cells was lower compared to protein-coding genes. (Lower left) TE transcripts lack the extreme standardized variance observed at protein-coding genes. (Lower right) TE transcripts account for 73 of the top 1000 variable features. (B) UMAP of scRNA-seq data. Cells were colored based on tissue information provided by the original study. (C) Examples of tissue-specific TE transcripts. (D) Normalized expression pattern (center, heatmap) of 146 TE transcripts (columns) across 37 tissues (rows). Transcript length, annotation status (top, bar plot), and TE composition (bottom, bar plot) were shown for each TE transcript. (E) Genome browser view of two TE transcripts with strong tissue enrichment. Assembled TE transcripts, uniquely mapped reads of merged bulk RNA-seq (from 37 samples that were used for transcript assembly), and scRNA-seq signal for selected tissues were shown. (Left) A TE transcript that is initiated from an L2a element, the second exon of this transcript is composed of non-TE sequences. (Right) A TE transcript that is almost exclusively composed of ERV sequences.
Next, we systematically examined the dynamic TE expression and obtained 146 TE transcripts that show substantial tissue enrichment (Supplemental Table S3). Hierarchical clustering analysis using the expression of these TE transcripts showed that tissues with similar origins are grouped together (Fig. 4D). For instance, tissues within the hematoendothelial lineage including hematoendothelial progenitors, endothelium, blood progenitors, and erythroids are adjacent to each other, and tissues linked to the neuronal lineage including neuromesodermal progenitor, spinal cord, forebrain/midbrain/hindbrain, and neural crest are clustered together.
Although 10x reads were enriched at the 3′ end of the transcript, all TEs located along the transcripts were captured using our assembled full-length TE transcripts (Fig. 4E). Among the 146 TE transcripts, 84 initiated from TE or have >50% of their exonic sequences contributed by TEs (Fig. 4D; Supplemental Fig. S11D). Overlapping these 146 TE transcripts with annotated ncRNAs revealed that 51 have been annotated by RefSeq. Although these known ncRNAs tend to contain a lower percentage of TEs, transcripts that are almost exclusively composed of TEs are largely unannotated, demonstrating the value of our approach in capturing transcripts that originated from highly repetitive regions. Moreover, we observed that TE transcripts enriched in different tissues display distinct sequence composition. For instance, TE transcripts enriched in extraembryonic ectoderm, extraembryonic endoderm, parietal endoderm, and visceral endoderm are almost exclusively composed of LTRs, although we did find that several highly expressed transcripts in extraembryonic ectoderm are derived from LINEs (Supplemental Fig. S12).
Discussion
Current genome-wide TE expression quantification tools often count signal at individual TEs or TE subfamilies/families. This strategy has been widely adopted in bulk RNA-seq and inspired similar analyses with scRNA-seq data. However, we caution that compared with bulk RNA-seq, a higher percentage of scRNA-seq reads are mapped to TEs, making it challenging to identify bona fide TE expression. Moreover, quantifying signal at individual TEs or TE subfamilies/families leads to the false impression that transcripts originated from repetitive regions are mostly composed of a single TE or TEs from the same subfamilies/families. Although this is true for some well-studied examples such as full-length ERVs, in most other cases, TEs only contribute to fragments of the full-length transcript, and TEs from different families can be incorporated into the same transcript.
A major difference between the expression quantification of protein-coding genes and TEs is that the transcript structures of protein-coding genes are usually well annotated and readily available. Gene annotation guides expression analysis toward genomic regions with true signal and facilitates accurate expression quantification with scRNA-seq data. In this study, we showed that transcripts constructed from bulk RNA-seq can serve as references for TE-containing ncRNAs and improve the accuracy of TE expression analysis in scRNA-seq data generated across multiple sequencing platforms. In comparison to individual TEs or TE subfamilies/families, TE transcripts more accurately reflect the natural transcription units. These transcripts contain not only previously annotated TE transcription units, but also novel ncRNAs that are partially composed of TEs. Of the 5299 TE transcripts that we assembled, 98 closely resemble the well-studied transcription units of ERVs. These transcripts have >80% of their exonic sequences contributed by TEs. They start from 5′ LTR, transcribe through internal sequences, and end at 3′ LTR. In addition, we also obtained 104 TE transcripts that initiate from TEs and are within 100 bp away from FANTOM5 CAGE peaks (The FANTOM Consortium and the RIKEN PMI and CLST (DGT) 2014). Of these, 84 transcripts were not previously annotated, highlighting the value of our approach in identifying TEs that potentially function as promoters.
Using our analytical pipeline, we dissected the expression dynamics of TE transcripts during mouse early embryogenesis and identified 146 TE-containing ncRNAs with strong tissue specificity. A close examination of these candidates revealed intricate interaction between TE transcripts and protein-coding genes. For instance, we were able to identify a ChIP-seq peak of regulatory factor X, 3 (RFX3) at the promoter region of the TE transcript TE-tx-3856 (Supplemental Fig. S13A). RFX3 is a transcription factor essential for brain development (Baas et al. 2006; Benadiba et al. 2012; Magnani et al. 2015). Our observation that TE-tx-3856 is highly expressed in mouse neuronal tissues suggests that this TE-containing ncRNA is a potential downstream target of RFX3. In another example, the TE transcript TE-tx-3715 overlaps with sonic hedgehog (Shh), a secreted signaling molecule produced by the notochord (Placzek 1995; McMahon et al. 2003). The expression pattern of TE-tx-3715 strongly resembles that of Shh, indicating that they are under the control of a common regulatory circuit (Supplemental Fig. S13B). In addition, we also captured TE transcripts TE-tx-3178 and TE-tx-2841, both of which are strongly expressed in the epiblast (Supplemental Fig. S13C). Both candidates initiate from TEs and were previously annotated as pluripotent associated transcripts (Platr10 and Platr14) (Bergmann et al. 2015). Earlier reports suggested that Platr10 transcript physically interacts with the promoter of pluripotent transcription factor Sox2, whereas the depletion of Platr14 alters the expression of differentiation- and development-associated genes in stem cells (Bergmann et al. 2015; Zhang et al. 2019). Our observation that Platr10 and Platr14 are expressed in the epiblast suggests that they may play similar roles during mouse early embryogenesis. Taken together, we dissected the dynamic TE expression during mouse early development and provided a curated list of promising TE candidates for future functional studies.
In summary, we established an effective TE quantification pipeline for scRNA-seq data and illustrated the dynamic TE expression during mouse early embryogenesis. In contrast to commonly used bulk RNA-seq tools that evaluate reads at single TEs or TE subfamilies/families, our pipeline emphasizes the importance of full-length TE transcript structures in scRNA-seq TE quantification. Furthermore, our work provides an initial set of TE-containing long ncRNAs during mouse early development, laying the foundation for future work on constructing a more comprehensive TE transcript database across distinct tissue types and developmental stages and encompasses different types of TE transcripts, such as small RNAs and nonpolyadenylated long ncRNAs (McCue and Slotkin 2012; Dumesic and Madhani 2014; Lanciano and Cristofari 2020). Additionally, exploring effective techniques for quantifying TE-derived intronic reads (Chung et al. 2019; Kong et al. 2019; Navarro et al. 2019), as well as developing isoform-specific quantification tools for TE protein-coding gene chimeras (Wang et al. 2016; Pinson et al. 2018; Attig et al. 2019; Jang et al. 2019) will further expand the TE analysis toolkit for scRNA-seq and greatly advance our knowledge on the expression and the function of TE transcripts.
Methods
Data processing of bulk RNA-seq data sets
Raw sequencing files were downloaded from NCBI Sequence Read Archive and EMBL-EBI ArrayExpress (Supplemental Table S1) and aligned to the mouse (mm10) or human (hg38) genomes using STAR (Dobin et al. 2013). To retain reads derived from repetitive regions, “‐‐outFilterMultimapNmax” was set to 500. To facilitate downstream transcript assembly “‐‐outSAMattributes” was set to “NH HI NM MD XS AS.” After alignment, signal quantification at regions of interests was performed using featureCounts. See “Read assignments” for details.
Data processing of scRNA-seq data sets
scRNA-seq data generated with Smart-seq-derived protocols were processed and quantified using the same procedures as bulk RNA-seq data. scRNA-seq data generated with the other protocols were processed using zUMIs (Parekh et al. 2018) with the following modifications: (1) To retain reads derived from repetitive regions, “‐‐outFilterMultimapNmax” was set to 500 during STAR alignment. (2) To quantify reads that were mapped to multiple locations or features, “allowMultiOverlap” and “countMultiMappingReads” were set to TRUE for function “.runFeatureCount.” BAM files with cell barcode, UMI, and the name of overlapping features were reported. (3) A customized R script (R Core Team 2017) was used to process the BAM file generated in step 2. See “Read assignments” for details.
Constructing TE transcripts
Transcript assembly of each RNA-seq sample was performed using StringTie2 (Kovaka et al. 2019). “-j 2 -s 5 -f 0.05 -c 2” was used to improve the specificity of the assembly results. To generate the master reference file, assembled transcripts from multiple RNA-seq samples were merged using TACO with default parameters (Niknafs et al. 2017). Transcripts shorter than 200 nt were excluded. Transcripts whose exons that overlapped with TEs but not the exons of RefSeq protein-coding genes were named as TE transcripts and used for TE expression quantification.
Read assignments
FeatureCounts (Liao et al. 2014) was used to obtain the features to which each of the reads was mapped. BAM files with read information (read name, UMI, cell barcode) and overlapping features were reported. An EM algorithm was implemented to redistribute reads that mapped to multiple features. During the initial round of assignment, the n numbers of features without uniquely mapped reads first receive fractions of reads inversely proportional to the total number of features (N) to which each read is mapped. The remainder (1−n/N) read is then assigned to the remaining features proportionally to the amount of uniquely mapped reads after normalizing for feature length. During the subsequent rounds of assignment, reads mapped to multiple locations are reassigned to all the features proportionally to the amount of reads each feature received in the previous iteration after normalizing for feature length. The algorithm stops when the maximum read change per feature is smaller than 1 or the number of iterations reaches 50, whichever comes first.
RNA-seq simulation
To evaluate the performance of the EM algorithm, we simulated RNA-seq reads using Polyester (Frazee et al. 2015). One hundred base pair paired-end stranded RNA-seq reads were simulated using the 692 TE transcripts assembled from mESC data sets as templates. These simulated reads have a mean fragment length of 250 bp and sequencing error rate of 0.5%. TE transcripts were simulated to have a mean read coverage of 5× with expression deviation from the mean between twofold and 100-fold. We performed eight rounds of simulation with four independent expression designs (two replicates for each design). To evaluate the performance of the EM algorithm, we aligned the simulated reads to the mm10 genome and calculated the amount of observed reads at each TE transcript.
Mouse early embryogenesis scRNA-seq data set analysis
Reads of the scRNA-seq data sets from mouse zygote to gastrulation (Smart-seq-derived protocols) were quantified at protein-coding genes (RefSeq annotation, n = 20,779) and TE transcripts (assembled from bulk RNA-seq, n = 5299). Only cells that had 200–18,000 features and <10% mitochondria reads were kept. To remove batch effect and visualize all three data sets in the same UMAP, data integration was performed using Seurat with the top 4000 variable features (Stuart et al. 2019). Cell type was determined using the stage information provided by the original studies, the expression patterns of cell type–specific markers, and Seurat clustering results.
UMIs of the 10x scRNA-seq data set from mouse gastrulation to early organogenesis were quantified at protein-coding genes (RefSeq annotation, n = 20,779) and TE transcripts (assembled from bulk RNA-seq, n = 5299). Sample_25 was removed owing to higher batch effect. Only cells that have more than 200 features and were annotated by the original study were kept. Cell type information provided by the original study was used for identifying tissue-specific markers. The 146 TE transcripts with strong tissue enrichment were obtained by combining and filtering Seurat-defined markers and customized markers. Seurat-defined markers were obtained by running “FindAllMarkers” with “only.pos = T, min.pct = 0.10” and selecting for TE transcripts with adjusted P-value < 0.05. Customized markers were obtained by identifying TE transcripts with at least 1 UMI in at least 10% of the cells in any tissue and selecting candidates that were expressed in, at most, three tissues. After combining Seurat-defined markers and customized markers, manual curation was performed to remove candidates that were highly expressed in a large number of tissues or with suboptimal transcript structures.
TE transcript clustering in mouse preimplantation stages
Because of the limited number of cells, scRNA-seq signals of each TE transcript across all the cells with the same developmental stage were averaged to reduce noise. TE transcripts were then grouped into six clusters using soft clustering (R package TCseq) based on their expression patterns across preimplantation stages.
TE subfamily/family enrichment analysis
For each TE-family, its enrichment was calculated using the following equation: The observed frequency of TEs belonging to this family in all candidates divided by the expected frequency of TEs belonging to this family in genomic regions that do not overlap with protein-coding genes. The significance for the observed frequency was calculated with Fisher's exact test and corrected for multiple testing with the Benjamini–Hochberg method. Only TE families with more than 20 members in the candidates and more than 100 members in the background were included in the figures. TE subfamily enrichment analysis was performed similarly. Only TE subfamilies that were significantly enriched in the candidates had more than 10 members in the candidates and more than 100 members in the background and were plotted.
Other statistical analysis and figure generation
All the statistical analyses and associated figures were done using R (R Core Team 2017). Genome browser view was generated with WashU epigenome browser (https://epigenomegateway.wustl.edu/). A browser session showing the expression patterns of TE transcripts during mouse gastrulation and early organogenesis is available (https://epigenomegateway.wustl.edu/browser/?sessionFile=https://raw.githubusercontent.com/wanqingshao/TE_expression_in_scRNAseq/master/datahub/Gottgen_eg-session‐‐da9eced0-e71d-11ea-be04-31bc80338b33.json).
Publicly available data sets used in this study
Descriptions and accession IDs of all the data sets used in this manuscript are provided in Supplemental Table S1.
Software availability
A detailed description of our analysis framework and customized scripts used for this work are publicly available at GitHub (https://github.com/wanqingshao/TE_expression_in_scRNAseq) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Kara Quaid and Shuonan He for comments on the manuscript; we thank all the Ting Wang laboratory members for discussions and critical input. This work is supported by National Institutes of Health grants R01HG007175, U24ES026699, U01CA200060, U01HG009391, and U41HG010972.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.265173.120.
- Received April 27, 2020.
- Accepted November 24, 2020.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








