
Decrypting noncoding RNA interactions, structures, and functional networks
- 1University of Hawaii Cancer Center, Cancer Biology Program, Honolulu, Hawaii 96813, USA;
- 2Department of Oncology-Pathology, Cellular and Molecular Tumor Pathology, Karolinska Institute, and Karolinska University Hospital, Stockholm, 17164 Sweden;
- 3Department of Chemistry, University of Washington, Seattle, Washington 98195-1700, USA;
- 4Department of Experimental Therapeutics, Division of Cancer Medicine, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA;
- 5Center for RNA Interference and Non-Coding RNAs, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA
-
↵6 These authors contributed equally to this work.
Abstract
The world of noncoding RNAs (ncRNAs) is composed of an enormous and growing number of transcripts, ranging in length from tens of bases to tens of kilobases, involved in all biological processes and altered in expression and/or function in many types of human disorders. The premise of this review is the concept that ncRNAs, like many large proteins, have a multidomain architecture that organizes them spatially and functionally. As ncRNAs are beginning to be imprecisely classified into functional families, we review here how their structural properties might inform their functions with focus on structural architecture–function relationships. We will describe the properties of “interactor elements” (IEs) involved in direct physical interaction with nucleic acids, proteins, or lipids and of “structural elements” (SEs) directing their wiring within the “ncRNA interactor networks” through the emergence of secondary and/or tertiary structures. We suggest that spectrums of “letters” (ncRNA elements) are assembled into “words” (ncRNA domains) that are further organized into “phrases” (complete ncRNA structures) with functional meaning (signaling output) through complex “sentences” (the ncRNA interactor networks). This semiotic analogy can guide the exploitation of ncRNAs as new therapeutic targets through the development of IE-blockers and/or SE-lockers that will change the interactor partners’ spectrum of proteins, RNAs, DNAs, or lipids and consequently influence disease phenotypes.
A quarter century after the cloning of the first human noncoding RNA (ncRNA), H19 (Zemel et al. 1992), the number of annotated ncRNAs is continuously increasing and greatly exceeds that of protein-coding genes (Iyer et al. 2015; Hon et al. 2017). An even larger set of noncoding transcripts, many of which are primate-specific, still awaits annotation (Necsulea et al. 2014; Washietl et al. 2014; Rigoutsos et al. 2017). Over the last decade, advances in bioinformatics and deep sequencing technology have allowed the identification and annotation of tens of thousands of short and long ncRNAs (lncRNAs). These include endogenous microRNAs (miRNAs), small interfering RNAs (endo-siRNAs), PIWI-interacting RNAs (piRNAs), small nucleolar RNAs (snoRNAs), tRNA-derived small RNAs (tsRNAs), natural antisense transcripts (NATs), circular RNAs (circRNAs), long intergenic noncoding RNAs (lincRNAs), enhancer noncoding RNAs (eRNAs), transcribed ultraconserved regions (T-UCRs), or primate-specific pyknon transcripts (Lee et al. 2009; Haussecker et al. 2010; Esteller 2011; Rigoutsos et al. 2017; Smith and Mattick 2017), and more. These discoveries have created a compelling need to understand the structure–function relationships that underlie the biological roles of ncRNAs.
A very well studied class of ncRNAs is the family of small (19- to 24-nucleotide [nt]) miRNAs (Ambros 2003). Mature miRNAs are generated by two sequential enzymatic cleavage reactions from pri-miRNAs, primary transcripts ranging from hundreds to thousands of nucleotides in length through precursor miRNAs (pre-miRNAs), stem-loop structures of 60–110 nt. Functionally, a miRNA can regulate the expression of protein-coding or noncoding transcripts in a sequence-specific fashion mostly through the complementarity with the miRNA's specific “seed” sequence (the first 2–8 nt at the 5′ end) (Bartel 2018). As a result of these interactions, mRNA's stability and/or translation can be impaired, leading to a reduction in RNA or protein expression levels (Filipowicz et al. 2008). Yet, it is now apparent that the effects of miRNAs on gene expression are more varied than initially proposed (Dragomir et al. 2018). For instance, nuclear miRNAs can regulate transcription by acting at promoters (Hwang et al. 2007). Pri-miRNA processing to miRNA can be controlled by interactions with lncRNAs (Liz et al. 2014) that can also act as miRNA decoys, sequestering miRNAs or reducing their expression levels (Davis et al. 2017; Kleaveland et al. 2018) and thus increasing the expression of genes that would otherwise be specifically repressed (Poliseno et al. 2010).
LncRNAs (>200 nt in length) have cell-specific expression patterns and are mechanistically involved in many biological processes (Long et al. 2017). The length of lncRNAs, sometimes in the range of tens of kilobases, allows them to fold into potentially complex but poorly understood secondary and three-dimensional (3D) structures. It is generally believed that these structures affect the interaction of lncRNAs with regulatory DNA sequences; other lncRNAs, miRNAs, and messenger RNAs (mRNAs); various types of nuclear proteins, such as transcription factors, histones, or other chromatin-modifying enzymes; and perhaps even phospholipids (Wang and Chang 2011; Lin et al. 2017) and regulate complex regulatory networks composed of DNA, RNA, and proteins. The complexity of these networks allow alterations in lncRNA expression levels to affect a broad spectrum of genes via their multiple partners and orchestrate profound phenotypic changes (Wang and Chang 2011; Long et al. 2017). While the modular nature of lncRNAs is widely accepted, its regulatory principles remain largely unknown after >6 yr from the publication of an influential review (Guttman and Rinn 2012).
The full repertoire of ncRNAs and a mechanistic understanding of their functional involvement in the regulation of cellular processes, and by extension in the onset and progression of human disease, remain largely unknown (Kapranov et al. 2007; Cech and Steitz 2014; Ling et al. 2015), as is the molecular and structural basis for their function. We analyze together the short miRNAs and the long lncRNAs, as similar structural principles can be applied to both categories. We propose that two classes of functional elements can be identified in ncRNAs: first, the “interactor elements” (IEs), necessary for direct physical interaction with various partners through base complementarity (with other nucleic acids) and sequence-specific recognition by RNA-binding proteins (RBPs) (Table 1); and, second, the “structural elements” (SEs), governing the emergence of secondary and/or tertiary 3D ncRNA structures, that direct their functional interactions with other cellular partners (Table 1). A noncoding transcript can have multiple IEs and multiple SEs, located either separately or overlapping within the RNA sequence. IEs might be structured as well. For instance, many RBPs bind to double-strand RNA or to secondary structures such as a hairpin, a bulged nucleotide, or even more complex 3D architecture, and these structures generally influence binding affinity and specificity (Helder et al. 2016). IEs and SEs are components of more complex and structured segments of RNA named “domains,” containing only IEs or only SEs or combinations of both (Fig. 1). We anticipate that most of ncRNAs have both IE and SE, one exception being the mature miRNAs that, due to their short size, lack SEs. By analogy with the “universal grammar” (Hauser et al. 2002; Chomsky 2017; Yang et al. 2017; Box 1), we envision that the patterns of nucleotides that we name “elements” are important for the structure (SEs) and function (IEs) of ncRNA, like patterns of letters and words are important for the structure of a language. Such patterns are structured in a genomic syntax composed of ncRNA interactor networks (NINs), which can be targeted in new therapeutic applications (Fig. 2 and Box 1).

Examples of interactor elements (IEs) and structural elements (SEs) in ncRNAs. (A) The IEs from the miR-15a/miR-16 cluster target the proapoptotic oncogene BCL2. When this miRNA cluster is down-regulated in human cancer cells, the BCL2 protein is overexpressed and can be targeted by the antiapoptotic small molecule venetoclax (Croce and Reed 2016). (B) The processing of another member of the miR-16 family, miR-195, is regulated by a direct interaction with the ultraconserved ncRNA uc.283. When this lncRNA is overexpressed in human cancer due to promoter hypomethylation, this interaction prevents DROSHA cropping of the mir-195 primary transcript, leading to down-regulation of the mature miR-195, a new mechanism of tumor-suppressor microRNA inactivation (Liz et al. 2014). (C) LINC01139 (LINK-A) is the first lncRNA known to interact with lipids, specifically with PIP3, facilitating AKT activation and consequent resistance to AKT inhibitors (Lin et al. 2017). (PH-domain) Pleckstrin homology domain. (D) CCAT2 harbors a conserved SE, within which a SNP alters the secondary structure of the lncRNA so that the CCAT2 alleles bind to the CFIm splicing complex with distinct affinities. The cancer risk G allele induces the oncogenic GAC glutaminase-C isoform that causes colorectal cancer progression (Redis et al. 2016).
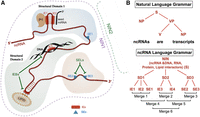
The analogy between the natural language grammar and the ncRNA structure grammar. (A) The elements of the ncRNA language grammar. A schematic view of IEs, SEs, structural domains of a ncRNA, and the noncoding RNA interactor network (NIN) composed by the ncRNA, interactor RNAs (such as miRNAs), interactor proteins (such as P1), interactor DNA elements, and interactor lipids (such as PIP3). Each ncRNA can contain multiple structural domains (here we show three for simplicity): one or multiple IEs and/or one or multiple SEs. These elements can be targeted by IE-blockers (IEBs), which release a specific interactor molecule (either DNAs, RNAs, proteins or lipids), and SE-lockers (SELs), which lock the lncRNA structure in a specific conformation favoring specific interactions with multiple molecules. A new type of therapy based on the correction of various interconnected genetic alterations that occur in the complex NINs by targeting the IEs can be envisaged, because these are docking sites for multiple types of molecules (DNA, RNA, proteins, and lipids) and/or the SEs, that directly affects the conformation of a lncRNA and indirectly the functional interactions with interactor molecules. (B) A comparison between the natural language and ncRNA language grammars. The various elements of the ncRNA language grammar are assembled under a “merge” (combine) function: The IEs and SEs from a lncRNA are combined during evolution by multiple rounds of merge (here, steps 1–6). (S) Subject; (NP) noun phrase; (VP) verb phrase; (V) verb.
Examples of experimentally supported interactor elements and structural elements in noncoding RNAs
The ‘universal grammar’ of the ncRNA structure
In analogy with linguistics, we consider the two elements of ncRNAs, IEs and SEs, as analogous to two types of letters, such as consonants and vowels. Just like words are composed of both, structural domains contain both IEs and SEs in various combinations. By not considering SEs in short RNAs such as precursor miRNAs and the interactor mRNAs or long ncRNAs could explain why target prediction programs, developed by many scientists to be based mostly on sequence complementarity, never performed well enough. Diverse combinations of multiple structural domains compose full-length ncRNAs in a similar way that phrases are composed of different words (see the definition of a phrase and the difference with sentence at https://www.eurocentres.com/blog/clause-phrase-sentence-learn-the-difference/). The domains do not have equal influence on the final structure, as those containing more SEs than IEs are expected to participate mostly in the secondary and 3D structure of a ncRNA, like different words have different grammar roles: Without a verb and a subject, no sentence can be assembled, while adjectives, for example, are not mandatory for the sentence structure. The IEs are important for the context of ncRNA interactor networks (NINs). Just like the universal grammar (UG) concept (Chomsky 2005, 2017; Dabrowska 2015) that considers the initial state of language development to be determined by “the genetic endowment,” the ncRNA language assembles according to rules transmitted hereditarily. For example, the initial mutations identified in the essential tumor-suppressor miR-15a and miR-16-1 for the development of CLL were germline, distributed in a family with multiple cases of cancer (Calin et al. 2005), and located within an IE for the splicing factor SRSF3 (Auyeung et al. 2013). As the number of confirmed ncRNAs of any types (including short or long) already is much larger than protein-coding genes (Ezkurdia et al. 2014), we anticipate that the “sentences” (i.e., NINs) will contain more noncoding than coding words. Protein-coding genes are usually more conserved phylogenetically and, therefore, can be considered “archaic” words (archaism); many ncRNAs are recently evolved (as pyknon transcripts are primate- and/or human-specific), and therefore, these could be considered the “neologisms” of the language. These different parts compose a ncRNA UG essential for the construction of the ncRNA functional language. The composition of structural domains from already constructed IEs and SEs can be called a “merge” function, a fundamental operation of structure building in human language (Fig. 2B; Yang et al. 2017). A “merge” (combine) function can be extraordinary conserved through hundreds of millions of years of evolution: The oncogenic THORLNC ncRNA contains an ultraconserved region responsible for the interaction with the IGF2BP1, and this interaction is conserved from zebrafish to humans (Hosono et al. 2017), meaning that the IE and the necessary SE are merged (combined) in both homologous transcripts, although zebrafish is thought to have arisen ∼340 Myr ago from a common ancestor with humans (Howe et al. 2013). By “merge” operations, the various elements can be combined during evolution of species in homologous transcripts that are analogous to redundant language phrases and sentences that have similar meanings (contribution to the same NINs). An example is provided by uc.339. The miRNA binding elements for miR-339-3p, miR-663b-3p, and miR-95-5p represent IEs, whose disruption prevents the interaction between uc.339 and the three miRNAs, thereby affecting the expression levels of CCNE2. Even if these IEs are intact, a mutation in the TREs modifies the secondary structure of the uc.339 in such a way that the miRNAs cannot interact with their target, even if the IEs are left intact. Therefore, TREs act as bona fide SEs. Let us assume that a hypothetical RBP (RBP1) recognizes the complex of uc.339 bound to miR-663b-3p. In this case, the combination of IE, SE, and miR-663b-3p bound to the IE would constitute a structural domain (SD1), whose integrity is necessary for RBP1's function. Now, let us assume that another hypothetical RBP (RBP2) recognizes another structural domain (SD2) composed of the IE necessary to interact with miR-95-5p, the TRE (SE) that affects this interaction and the miR-95-5p itself. In this case, a protein working only if RBP1 and RBP2 are properly bound to their structural domains could accurately function only by recognizing NIN1 (composed of SD1 + SD2). Similarly, in linguistics a sentence “S” (NIN1) is composed of phrases (SD), written with words (IE and SE) (Fig. 2B).
The IEs in ncRNAs
IEs are generally short and include stretches of functional nucleotides
Many well-characterized IEs are located in miRNAs, which follow the paradigm of target recognition through sequence complementarity (base-pairing) to other RNAs (e.g., mRNAs or lncRNAs) or DNAs (e.g., promoters). The archetypal IE is the “seed” sequence, a conserved heptamer including the nucleotides at positions 2–8 at the 5′-end of miRNAs, which provides most of the binding energy between the miRNA and its target (Fig. 1A). Only the “seed” region is perfectly complementary to the target miRNA's response element on messenger RNAs or lncRNAs, while the remaining base pairs do not necessarily match perfectly. The principles underlying the interaction between the seed regions of miRNAs and their target mRNAs are well covered in other reviews (Bartel 2004, 2009, 2018). Other types of miRNA interactions through specific elements outside the “seed region” have been described, and their functional role investigated as well (Tay et al. 2008).
Single-nucleotide mismatches due to single-nucleotide polymorphisms (SNP) located on either the target miRNA response element (Nicoloso et al. 2010; Saridaki et al. 2014) or on miRNA IEs (Króliczewski et al. 2018) suffice to affect the function of the IE and can have clear biological consequences, such as increased risk for cancer. This is the case for the insulin-like growth factor-1 receptor (IGF1R) that promotes cancer cell growth and survival and is essential for malignant transformation by many classical cancer-related genes, such as TP53 and BRCA1 (Suleymanova et al. 2017; Worrall et al. 2017). The SNP rs28674628, located in the IGF1R 3′ UTR, associates with increased cancer risk and earlier age at diagnosis of breast cancer in Jewish Ashkenazi carriers of the 185delAG BRCA1 mutation (Gilam et al. 2013). Functional studies demonstrated that IGF1R expression is directly down-regulated by miR-515-5p, whose “seed” IE recognizes the sequence contained within the rs28674628 but only when the common A allele is present; the A-to-G single-nucleotide substitution enables IGF1R mRNA to escape this negative control, with resultant changes in age at diagnosis in the 185delAG BRCA1-mutation carriers (Gilam et al. 2013).
IEs within lncRNAs are more diverse compared with those in miRNAs and more poorly understood in part because they are embedded within larger RNA structures. Some IEs contain specific base sequences: For example, a G-rich motif in the lncRNA braveheart long non-coding RNA (Bvht) specifies the interaction with the CNBP/ZNF9 zinc-finger transcription factor and dramatically affects cardiomyocyte differentiation. This sequence is located in a motif named AGIL (from 5′-asymetric G-rich internal loop), and the deletion of 11 nt from this motif out of the ∼590 nt of lncRNA sequence has a major phenotypic impact: the failure of the transition from nascent mesoderm to the cardiac progenitor state (Xue et al. 2016). IEs within lncRNAs are shaped by evolution: Some IEs are conserved over hundreds of millions of years from rodents to humans, as is the case of the ultraconserved IE from uc.63 with the oncogenic miR-155 (Calin et al. 2007) or the miR-7 binding site within OIP5-AS1 (known also as Cyrano) (Ulitsky and Bartel 2013). Others are present only in primate-specific transcripts, as in the case of the IE sequence located in the pyknon motif from the N-BLR lncRNA interacting with miR-200 family members (see Box 2; Rigoutsos et al. 2017).
Inter- and intra-genomic conservation of IEs and SEs
The IEs and SEs of ncRNAs are codified at the DNA level as short nucleotide motifs embedded in the 3 billion-nucleotide-long human genome. According to the levels of conservation, such motifs can be divided in two categories, both important for biological processes and involved in diseases such as cancer. First are the highly conserved motifs, with the most extreme conservation being reported for the ultraconserved regions, pieces of DNA fully conserved for ∼300 Myr of evolution from rodents to humans (Bejerano et al. 2004). These regions are often transcribed as long ncRNAs (Calin et al. 2007; Ferdin et al. 2013; Hosono et al. 2017), and the ultraconservation at the genomic level can therefore dictate the function of ncRNAs due to the presence of very conserved IEs, SEs, and RNA domains, as in the case of the conserved interaction of THORLNC with the insulin like growth factor 2 mRNA binding protein 1 IGF2BP1. At the opposite spectrum of conservation are the extremely short primate- and/or human-specific motifs named pyknons: 16-nt-long sequences that exhibit exceptional intra-genomic conservation, each being repeated in at least 40 places of the human genome (Rigoutsos et al. 2006). At least in a specific instance, the pyknon located in the N-BLR transcript harbors IEs for miR-200c and miR-141, and this interaction is important for the epithelial-to-mesenchymal transition and colorectal cancer metastases (Rigoutsos et al. 2017). Although the pyknons are located in about 225,000 places within the human genome, other more abundant genetic elements influence the structure of ncRNAs, and these are the SNPs. The examples of SNPs influencing the function of ncRNAs through the structural changes in ncRNAs are increasing at a rapid pace (Castellanos-Rubio et al. 2016; Redis et al. 2016; Bal et al. 2017; Shah et al. 2018).
Multiple IEs can function together to modulate the biological function of a ncRNA
Different types of IEs located within the same transcript can have complementary functional effects. One example is the RNA decoy function of miR-328 that modulates the poly(rC) binding protein PCBP2 (known also al hnRNP E2) during regulation of mRNA translation in leukemic blasts (Eiring et al. 2010). The precursor miR-328 harbors three C-rich elements that resemble the inhibitory PCBP2 binding site in the intercistronic mRNA region of the transcription factor CEBPA. Mechanistically, pre-miR-328 directly interacts with PCBP2 protein through these motifs located in an IE and prevents its binding to the CEBPA mRNA region, thus restoring CEBPA expression that, in turn, directly enhances miR-328 transcription. This interaction is important for the restoration of myeloid maturation in chronic myeloid leukemia progenitor cells. Furthermore, miR-328 also impairs clonogenicity of the same progenitors through a canonical miRNA pathway that involves the interaction between its seed region from a distinct IE in mature miRNA with the 3′ UTR of PIM1 kinase mRNA, proving that two distinct IEs can, together, balance the function of the pre-miR-328 (Eiring et al. 2010).
IEs regulate miRNA processing through RNA–RNA interactions
Specific sequences within distinct RNAs are necessary for RNA–RNA interactions. An example is the regulation of pri-miRNA processing by a lncRNA transcribed from an ultraconserved region: The uc.283 + A controls the processing of pri-mir-195 (Liz et al. 2014) into the highly conserved miR-195, a member of the tumor-suppressor miR-15/16 family. This interaction requires imperfect complementarity between two IEs: 15 nt within the lower stem region of the pri-mir-195 transcript (first IE), and 12 nt from the ultraconserved sequence (100% conserved among human, mice, and rats) of the uc.283 + A transcript (second IE) (Fig. 1B). The interaction precludes cleavage of pri-mir-195 by the nuclear exonuclease DROSHA. Mutations of either of these IEs in either of two distinct RNA partners disrupt pri-miR-195 regulation and function both in vivo and in vitro (Liz et al. 2014).
The interaction of IEs with proteins can regulate the production of small RNAs
Long ncRNAs such as pri-miRNAs rely on IEs for processing. The murine knockout model of Dleu2, the primary noncoding transcript in which the sequence of miR-15/16 is embedded (Bullrich et al. 2001), showed a more aggressive chronic lymphocytic leukemia (CLL) phenotype compared with the miR-15/16 knockout model, suggesting that Dleu2 participates in the development of CLL, the most common form of leukemia (Klein et al. 2010). Reduced miR-16 expression associated with CLL is typically due to deletions involving both miR-15a and miR-16-1 (Calin et al. 2002; Calin and Croce 2006). However, there are cases of CLL that retain high levels of pri-miRNAs in malignant leukemic cells while carrying a C-to-T germline mutation near the 3′ region of the miR-16-1 hairpin (Calin et al. 2005). This mutation corresponds to the first C in the “CNNC” motif, an IE for the splicing factor SRSF3 (named also SRp20) binding to pre-miR-16-1 (Auyeung et al. 2013). When the mutation occurs, the interaction between the IE and SRSF3 protein is disrupted, pri-miRNA processing is impaired, and the expression of mature miR-16 is reduced, leading to CLL. SRSF3 is also involved in mRNA export from the nucleus and translation (Corbo et al. 2013). Further supporting the importance of this interaction, the SRSF3-binding motif (CNNC) was found downstream from most pre-miRNA hairpins in bilaterian animals. In fact, a miR-16 mutation in the same IE was also found in the mouse synthenic region of New Zealand black mice, a strain that naturally develops CLL at older ages but not in other strains, including its nearest neighbor strain, New Zealand white, which has no higher risk for CLL (Raveche et al. 2007).
IEs might even interact with lipids
Recent findings expanded the interactor capacities of lncRNAs through the potential identification of lipid-binding lncRNAs in cancer cells (Lin et al. 2017). Phosphatidylinositol (3,4,5)-trisphosphate (PtdIns(3,4,5)P3), abbreviated to PIP3, functions to activate downstream signaling partners, the most notable being the protein kinase AKT (Lien et al. 2017). LINC01139 (also known as LINK-A, the lincRNA for kinase activation) directly interacts with the AKT pleckstrin homology domain and PIP3 at the single-nucleotide level, facilitating AKT–PIP3 interaction and subsequent enzymatic activation. Genomic deletions of the LINC01139 PIP3-binding motif sensitized breast cancer cells to AKT inhibitors, while LINC01139-dependent AKT hyperactivation led to resistance to AKT inhibitors and increased tumorigenesis (Fig. 1C; Lin et al. 2017). It is possible that this IE is in fact a combined IE-SE as this element might have also a defined structure that facilitates the interaction.
IEs signal the interaction of miRNAs with receptors
It was reported that extracellular secreted miRNAs can act as paracrine agonists of Toll-like receptors (TLRs) in immune cells, triggering a TLR-mediated prometastatic inflammatory response that leads to tumor growth and metastasis (Fabbri et al. 2012) and increased resistance to cisplatin (Challagundla et al. 2015). Specific sequences were found to be relevant to this process: GU-rich motifs at positions 18–21 (GUUG for miR-21 and GGUU for miR-29a) seem to be important for the activation of the downstream signaling of human TLR8 (or its murine ortholog TLR7) upon miRNA binding. Tumor-associated macrophages (TAMs) at the tumor interface express TLR8 in the intracellular endosomal compartment, and the cancer cell–derived miR-21 and miR-29a (shuttled from the cancer cells to TAMs as cargo of extracellular vesicles) are able to bind to TLR8 and activate it in a MYD88-dependent manner. The induced NF-kB signaling leads to increased secretion of IL6 and TNF by the TAMs, promoting cancer growth and metastasis (Fabbri et al. 2012). By performing a systematic point mutagenesis of the miRNA GU region, the nucleotide in position 20 was identified as critical for the TLR8 binding activity of both miR-21 and miR-29a, whereas substituting G with U in position 18 of miR-21 increased the activation of TLR8.
Some IEs serve to ‘signal’ the intracellular location of ncRNAs
Some miRNAs are confined to specific subcellular compartments; for example, miR-29b (but not its close homolog miR-29a) is enriched in the nucleus and contains a particular hexanucleotide terminal motif (5′-AGUGUU) that increases its stability (Hwang et al. 2007). The motif is unique and not found at the 3′-end of other mammalian miRNAs. This observation suggests that there may be a correlation between different miRNA functions and their intracellular localization and that there might be mechanisms controlling the intracellular distribution of some miRNAs. The presence of mature miRNAs (not only the primary miRNA transcripts) in the nucleus explains how specific miRNAs can regulate transcription of some genes at the DNA level. For example, miR-551b-3p binds a complementary sequence on the STAT3 promoter, recruiting RNA polymerase II and the TWIST1 transcription factor to activate STAT3 transcription, therefore directly up-regulating STAT3 expression. This interaction promotes resistance to apoptosis and increases the survival and proliferation of cancer cells in vitro and in vivo (Chaluvally-Raghavan et al. 2016).
Low-specificity versus high-specificity IEs
Most lncRNAs interact with proteins located in the same subcellular compartment (Chen et al. 2016; Carlevaro-Fita and Johnson 2019). Generally, the same lncRNA was reported to interact with various proteins in different cell types; likewise, the same protein was identified to interact with several lncRNAs. Some of these interactions are ubiquitous (e.g., the interaction between the PRC2 complex, specifically the EZH2 enzyme, and several lncRNAs) (Cifuentes-Rojas et al. 2014), while others are highly specific for some lncRNAs (e.g., the LINC01139 interaction with the PIP3, or the interaction of CCAT2 with the Cleavage Factor I (CFIm) complex) (Redis et al. 2016; Lin et al. 2017). We separate interactions that occur through “high-specificity IEs” (highly specific for a protein or family of proteins) and interactions through “low-specificity IEs,” which interact with lower specificity, perhaps through a common RNA secondary or 3D structure.
These variations of interactor partners, specificity, and strength of interaction might be at the basis of ncRNA functional versatility. An example of a high-specificity interaction is the one between Bvht and the CNBP/ZNF9 (a zinc-finger transcription factor that bind single stranded G-rich sequences) and occurs probably through a very short 11-nt-long asymmetric G-rich internal loop, with functional consequences on cardiomyocyte differentiation (Xue et al. 2016). An example of low-specificity interaction is represented instead by the Polycomb repressive complex-2 (PRC2) interaction with multiple lncRNA (Guil et al. 2012). PRC2 is a histone methyltransferase required for epigenetic silencing during cellular development and cancer (Davidovich and Cech 2015; Spitale et al. 2015). Mammalian PRC2 binds thousands of RNAs in vitro and in vivo, including multiple lncRNAs from animals (HOTAIR, GAS5, etc.) and plants (COLDAIR). Out of >2300 nt of HOTAIR, no more than 300 nt is required to bind its target PRC2 (Tsai et al. 2010), while for the Drosophila roX1 RNA, chromatin binding activity was attributed to only three small finger-like elements out of 4832 nt of RNA (Ilik et al. 2013). Yet, well-defined PRC2-binding motifs within target RNAs have been elusive, and the PRC2 RNA-binding subunits contain no known RNA-binding motifs, complicating the functional studies (Davidovich et al. 2015).
An intriguing point, which is still underexplored in the field of ncRNA interaction, is to what extent the stoichiometry affects the relationship between ncRNAs and their binding/interacting molecular partners (Thomson and Dinger 2016). Epigenetic modifications (such as promoter CpG island hypermethylation) of host genes for ncRNA, such as the gene TUSC3 for circ104555, are common in cancer cells and can modify the expression of the circRNA, leading to a modulation of the expression of miRNAs harboring an IE with the specific circRNA (Ferreira et al. 2018). In addition to the amount of interactor molecules, their location in specific cellular compartments (nucleus, cytoplasm, or mitochondria) (Carlevaro-Fita and Johnson 2019) and their stability, as well as the number of IEs and their high or low interaction specificity, are all factors influencing the interaction stoichiometry.
Identification of protein-binding sites (IEs) within lncRNAs
Chromatin remodeling enzymes such as PRC1, PRC2, and EZH2 (Margueron and Reinberg 2011) and many other proteins associate with ncRNAs, but it is unclear where and how they interact with them. For example, the class of RBP is composed by a mixture of proteins directly binding to RNAs and proteins binding to these RBPs instead of RNAs. Consequently, it is not known whether these ncRNA:RBP complexes interact directly with specific fragments (IEs) or conserved structural domains (SEs) specifically, perhaps by recognizing conserved folded structures, or whether they bind to the full RNA without specificity for a given region (Margueron and Reinberg 2011; Davidovich et al. 2013). RNA secondary structure provides a better understanding of how RBPs and epigenetic enzymes interact with lncRNAs. Advances in UV crosslinking and immunoprecipitation (CLIP) methods (Lee and Ule 2018), as well as several databases, provide faster and more efficient methods to identify proteins likely to be associated with a given RNA in cells (McHugh et al. 2015). For example, the server RAPID (Bellucci et al. 2011) predicts RNA interaction propensities for known RBPs. Protein partners of a given ncRNA can be identified within the ENCODE enhanced CLIP database (eCLIP) as well. In a recent study (A. Jones, G Pisgnano, G Varani et al, in prep.), we showed that most changes in in cell SHAPE (the Selective 2′ Hydroxyl Acylation analyzed by Primer Extension method) reactivity profile, compared with in vitro SHAPE, occurred at or near binding sites of proteins identified by eCLIP. By defining the secondary structure and independently folded domains, it is easier to address the question of where these enzyme complexes and proteins bind to ncRNA.
SEs in ncRNAs
Functional SNPs provide evidence that SEs are important in gene regulation
Although SEs in ncRNAs are poorly defined and even less studied, there is growing evidence supporting their functional importance (Table 1; Blythe et al. 2016; Zampetaki et al. 2018). For instance, because the ncRNA XIST has a conserved A-repeat element, it adopts in vivo an inter-repeat structure essential for mediating its gene silencing function (Pintacuda et al. 2017). It was recently reported that the cancer-risk-associated rs6983267 SNP located at the 8q24 amplicon induces changes in the secondary structure of the overlapping lncRNA CCAT2 that leads to allele-specific reprogramming of cellular energy metabolism (Redis et al. 2016). Secondary structure predictions using the RNAfold webserver anticipated major local structural changes induced near the putative upstream binding sequence of NUDT21 (Cleavage Factor I subunit, CFIM25) (Masamha et al. 2014), by a single-nucleotide variation between the G and T alleles. Such changes may plausibly translate into distinct secondary and perhaps tertiary folds and could explain binding of the G and T alleles with different affinities. In this model, formation of the RNA:protein complex is controlled by the rs6983267 SNP through its effect on the secondary structure of CCAT2. The consequence of the allele-specific interaction between CCAT2, the splicing complex CFIM, and the metabolic enzyme glutaminase (GLS) pre-mRNA appears to be the preferential splicing to the GAC isoform (Redis et al. 2016), the more catalytically active of the two GLS isoforms (termed KGA and GAC, respectively) and therefore more effective at replenishing intermediates of the TCA cycle (Fig. 1D; Cassago et al. 2012; Le et al. 2012). No other lncRNA has been so far proven to interact with two subunits (CFIM25 and CFIM68) of this complex, providing an example of a high-specificity interaction: The G and the T alleles bind to the NUDT21(CFIm25) and CPSF6 (CFIm68) subunits of the CFIm cleavage factor with different affinities, due to the presence of a high-specificity ID containing the rs6983267 site. The CCAT2–CFIm–GLS regulation axis is altered in about two-thirds of colorectal cancers (Redis et al. 2016).
Further supporting the importance of SNPs in the function of lncRNAs, it was reported that lnc13, harboring a celiac disease–associated haplotype block, forms a complex with the RBP HNRNPD and the histone deacetylase HDAC1 that represses the promoters of proinflammatory genes (Castellanos-Rubio et al. 2016). Of note, the lnc13 interaction with HNRNPD depends on the genotype of the SNP rs917997: This interaction is stronger with the “wild-type” CC genotype than with the disease-associated genotype TT, and the lncRNA structure was predicted to be different based on the SNP genotype (Castellanos-Rubio et al. 2016). Furthermore, in basal cell carcinomas, mutations in an enhancer RNA impaired its activity and reduced the host actin-related gene ACTRT1 expression, ultimately leading to aberrant activation of Hedgehog signaling (Bal et al. 2017).
SEs entrap miRNAs within lncRNA 3-dimensional structures
Recently, a new type of RNA:RNA interaction was described involving a T-UCR. The transcript of uc.339 harbors three IEs for miR-339-3p, miR-663b-3p, and miR-95-5p; of note, the uc.339 interaction with these miRNAs leads to up-regulation of their common target CCNE2, the cyclin E2 that plays a role in cell cycle G1/S transition and promotes lung carcinogenesis (Vannini et al. 2017), overriding a canonical competing endogenous RNA (ceRNA)-type of interaction (Poliseno et al. 2010; Cesana et al. 2011) in which overexpression of the lncRNA or of the miRNAs down-regulates the interacting counterpart. Conversely, while up-regulation of uc.339 reduces the levels of miR-339, miR-663b, and miR-95, modulation (both up- and down-) of these three miRNAs does not significantly affect the expression levels of the uc.339 transcript. This system introduces “entrapping,” distinct from ceRNA, because the structural context is important: The interaction between uc.339 and the three miRNAs is modulated by distinct 4-nt sequences in the lncRNA transcript, located outside of the IEs and called trapping related elements (TREs). These sequences represent a particular type of SE. The disruption of the TREs impairs the binding of miR-339, miR-663b, and miR-95 to uc.339 (Vannini et al. 2017). TREs are likely to be present in other lncRNAs and mRNA. For example, the levels of the SNH6-003 lncRNA that interacts with miR-26a/b did not change when Huh7 cells were treated with either miR-26a or miR-26b mimics or inhibitors (Cao et al. 2017). Therefore, their existence needs to be taken into consideration in every RNA:RNA interaction prediction, because mutations or SNPs in TREs might modulate intermolecular interactions even when the RNA:RNA match (the IE) is intact.
RNA structure is highly hierarchical
Studies of RNA structure are facilitated by its highly hierarchical nature: Secondary structure forms first, creating local stem–loops and helices that fold locally and coalesce into higher-ordered structural domains. Tertiary and higher-order interactions generally form only after the secondary structure is established, with only rare cases of secondary structure rearrangements induced by tertiary structure formation (Wu and Tinoco 1998). This hierarchical folding generates architectures composed of structured domains connected to each other perhaps flexibly, which interact with specific protein complexes, other RNAs, or lipids and bring them in proximity to each other and with target chromatin (Guttman and Rinn 2012). In fact, primary-sequence elements that influence the processing of the primary miRNAs (in fact long ncRNAs) have started to be identified (Chang et al. 2015; Roden et al. 2017). Thus, it is reasonable to hypothesize that lncRNAs contain highly structured regions or actual RNA domains that we might call “structural domains,” that are evolutionarily conserved at the secondary and perhaps tertiary structure and would control function (Ulitsky and Bartel 2013). Such domains might harbor one or multiple SEs or both SEs and IEs (Fig. 2A). RBPs can interact with an IE or with a SE or can even recognize a structural domain or a NIN. These individual structural domains, each composed of SEs and IEs, can be assembled in various configurations according to a ncRNA universal grammar (Fig. 2B and Box 1).
Simple sequence motifs often suffice to confer specific recognition features to a ncRNA; for example, N6-methyladenosine alters RNA structure to regulate binding to low-complexity regions in RBPs (Liu et al. 2017b). However, other functional properties of the ncRNA rely on secondary and 3D structure. RNAs belonging to different functional classes possess different degrees of secondary structure, as remarked by high-throughput technologies that couple chemical modification with deep sequencing (Berkhout and van Wamel 2000; Lu et al. 2011, 2016; Chu et al. 2015; Spitale et al. 2015). Recent studies suggest that the extent of base-pairing in lncRNAs is comparable to that of the ribosome (Batey et al. 2004; Yonath 2005), and reported the observation of distinct structural domains arranged in a manner that approaches the complexity of RNA enzymes (Novikova et al. 2012; Somarowthu et al. 2015; Hawkes et al. 2016; Liu et al. 2017a). However, relatively few lncRNAs have been characterized at the molecular level, besides the longtime-known cases of H19 (Hurst and Smith 1999; Juan et al. 2000) and Xist (Wutz et al. 2002; Pintacuda et al. 2017) and other more recently characterized examples, such as HOTAIR (Somarowthu et al. 2015), COOLAIR (Hawkes et al. 2016), or lincRNA-p21 (Chillon and Pyle 2016) and MALAT1 (Brown et al. 2014). Of note, this conclusion does not collide with the functional implications of structures and their evolutionary significance (Rivas et al. 2017) because, in cells, RBPs such as hnRNPs (Dreyfuss et al. 1993) might keep these RNAs unfolded. Nevertheless, although no correlation can be found between large stretches of predicted secondary structure in a particular ncRNA and its evolutionary conservation (Ulitsky et al. 2011; Ulitsky and Bartel 2013; Ulitsky 2016), RNA structure is likely to be more conserved than sequence. The frequently intimidating size of lncRNAs (several in the range of tens of kilobases) makes the combinatorial problem of finding the true folded structure difficult because of sampling (search space is vast), convergence (whether the absolute minimum in the free energy of folding is found), and flexibility (multiple structures might be equally consistent with the experimental data, perhaps because of conformational heterogeneity in the transcript). In addition, folding in cells and in vitro might very well differ because of kinetic constraints on RNA cotranscriptional folding and the presence of RBPs (Leamy et al. 2016). Advances in techniques such as SHAPE (Mustoe et al. 2018) and dimethyl sulfate (DMS) mapping (Novikova et al. 2012; Kwok et al. 2013), psoralen-cross linking (Lu et al. 2016), and high-throughput ligation followed by deep sequencing (Ramani et al. 2015) provide increasingly rapid access to RNA secondary structure (Carlson et al. 2018). In contrast, RNA 3D organization remains very challenging to study and requires methods that are highly specialized and technically demanding and provide very low throughput, such as small angle X-ray scattering (SAXS) (Rambo and Tainer 2013; Bai et al. 2014) or x-ray crystallography and NMR. More specific information can be found in several recent publications (Lin et al. 2019; Qian et al. 2019).
The NIN and the functional roles of IEs and SEs
RNA–RNA interactions through IEs are now widely recognized and conceptually assembled under the nonsynonymous concepts of “ceRNA” and “miRNA sponges,” discussed in several reviews (Tay et al. 2014; Thomson and Dinger 2016). We propose here a general concept of interactor ncRNAs, that we name the noncoding RNA interactor network (NIN), based on the new paradigm that a ncRNA can interact not only with RNAs (as exemplified by ceRNAs or sponges) but also with DNAs, proteins, and lipids, adding complexity and functional versatility to the regulatory networks. Each NIN is composed of one or more ncRNAs, each having IEs and SEs that participate in direct physical interactions (through IEs) or modulate these interactions (through SEs) with multiple partners (Fig. 2A). Further increasing the intricacy of NINs is the finding that some of the lncRNAs code for proteins that also have functional effects. For example, the protein encoded by the WRAP53 gene is overexpressed in a variety of cancers and promotes cellular transformation, while its down-regulation activates the proapoptotic mitochondrial pathway (Mahmoudi et al. 2011). The lncRNA WRAP53 is also a natural TP53 antisense transcript, functionally archetypal of another class of regulatory lncRNAs. WRAP53 transcript regulates endogenous TP53 mRNA levels and increases translation of the TP53 protein by targeting the 5′ untranslated region of TP53 mRNA, thus sensitizing the cells to TP53-dependent apoptosis (Mahmoudi et al. 2009).
Examples of NINs continuously expand (Marín-Béjar et al. 2017). In addition to the miR-328-PCBP2-mRNA of CEBP alpha-3′ UTR of PIM1 kinase mRNA (Eiring et al. 2010) network presented above (through two different IEs: a repeat-rich IE for PCBP2 protein and an antisense complementary IE used for PIM1 recognition), another recent example is provided by the lncRNA BCAR4 (Xing et al. 2014), which promotes cancer cell metastasis by coordinating a noncanonical Hedgehog signal transduction pathway. At the molecular level, BCAR4 is part of a protein complex containing two RBPs (SNIP1 and PPP1R10 [known also as PNUTS]), one kinase (CIT), and one transcription factor (GLI2) coordinating transcription in response to signaling activation. BCAR4 contains at least two IEs, one at position 235–288 for the interaction with SNIP1 and a second at position 991–1044 for the interaction with PPP1R10. In response to the CCL21 cytokine, BCAR4 binds to SNIP1 and PPP1R10 and releases the SNIP1's inhibition of EP300-dependent histone acetylation, subsequently enabling PPP1R10 recruitment by the lncRNA to bind to H3K18ac and relief of the inhibition of RNA Pol II by PP1 phosphatase activation. This mechanism activates a noncanonical Hedgehog/GLI2 transcriptional program that promotes cell migration (Xing et al. 2014).
Exploiting ncRNA structures for therapeutic developments
The exploding awareness of the role of noncoding genes in normal human physiology and disease such as cancer (Ling et al. 2013; Shah et al. 2016) is prompting renewed interest in the discovery of both oligonucleotide analogs and more drug-like chemistries aiming at targeting RNA in a sequence and/or structure-specific manner (Matsui and Corey 2017). Exploiting RNA as a drug target has been strongly advocated in antiviral and antibacterial research for >25 yr (for review, see Gallego and Varani 2001; Thomas and Hergenrother 2008; Aboul-ela 2010; Guan and Disney 2012) but not yet reduced to practice. Small molecules or peptidic or antisense molecules targeting ncRNAs may provide an alternative approach to oligonucleotides (Gumireddy et al. 2008; Chirayil et al. 2009; Zhang et al. 2010; Velagapudi and Disney 2014; Shortridge et al. 2017; Monroig-Bosque et al. 2018), and several lines of evidence suggest that the challenges, while significant, are not insurmountable (Blount and Breaker 2006; Wilson 2014). RNA can be readily selected to recognize small molecules with greater sensitivity to fine details of the chemistry than antibodies (Warner et al. 2014). It can be envisioned that targeting the IEs directly (by sequence complementarity) or indirectly (by affecting the production of the ncRNA harboring the IEs) by using IE-blocker small molecules (IEBs), will have a double effect on the ncRNAs (long or short) that harbor the IEs and also on the interactor molecules (RNAs, DNAs, proteins, or lipids) by making them available to interact with other partners. The same is also true for targeting SEs by using SE-lockers (SELs), which lock the ncRNA structure in a stable conformation by locking the SEs. For instance, IEB can be considered an oligonucleotide antisense or a protein able to “hide” the IE from the interaction with its effector, therefore functioning as an “inverse agonist.” A SEL could be an antisense oligonucleotide or an integrating transposon that disrupts the SE sequence affecting the secondary and tertiary structure of the lncRNA. Therefore, IEBs and SELs include different types of molecules and should not be considered synonyms of “agonist/antagonist” or “ligand/inhibitors” sensu strictu. Changing the conformation of a lncRNA will indirectly disrupt the interaction through IE(s) and will change the interactor spectrum of proteins, RNAs, DNAs, or lipids as well and affect their poorly understood functions in perhaps unpredictable ways. Of note, by not inducing the degradation of the lncRNA, but only “freezing” the conformation, the SELs will not modify the abundance of the ncRNA but only affect downstream signaling pathways. The effects of SELs could be of higher functional magnitude compared with IEBs, because affecting the structure of a ncRNA would influence a wide spectrum of interactor molecules and not only those involved in a specific interaction (Fig. 2). However, at the moment, these are only hypotheses that await testing and validation in cellular and small animal models.
Concluding perspectives
The number of lncRNAs has long surpassed the number of protein-coding genes, and their functional versatility expands continuously. A simplifying framework, like “universal grammar” (Chomsky 2005; Dąbrowska 2015; Chomsky 2017) can help us think about how the functional language of ncRNAs is organized and frame the identification of new therapeutic targets within these functional transcripts. Potential therapeutic agents could interact with regions harboring IEs or SEs (the “letters”) by IEBs or the structural domains (the “words”) by SELs or affect globally the 3D structure of an ncRNA (the “phrase”) by SELs as well. The ultimate goal is to affect the function of an entire NIN (a “sentence” composed of various phrases producing a common “meaning”) that influences one or multiple cancer hallmarks (the “meanings”). For example, agents (small molecules, oligonucleotides, or peptides) that lock the ncRNA structure in a stable conformation by locking the SEs (the SELs) and indirectly disrupt the interaction through IE(s) with specific DNA elements, other oncogenic RNAs, or proteins would represent a new approach to the therapy of cancer or any disease involving alterations of ncRNAs (Fig. 2). Some advances have already been made: The MALAT1 triple-helix SE involved in stability was proven to be selectively targetable with small molecules (Donlic et al. 2018), but this field is just in its infancy. Building on the specific model based on the interaction of TERT RNA and telomerase (Zappulla and Cech 2004), in 2011, Wang and Chang proposed a general model of lncRNAs function as “molecular scaffold” (Wang and Chang 2011). While embracing the specific types of interactions anticipated by those two models, the paradigm we propose here essentially extends beyond these models by clearly distinguishing between sequences that interact with effectors (IEs) and sequences that affect the secondary/tertiary structure of the lncRNA (SEs). The field of ncRNAs has matured well beyond descriptive expression–phenotypic correlations toward the more laborious, time-consuming, but ultimately most rewarding stage of establishing structure–function–phenotype correlations. Understanding the structure–function relationship of ncRNAs using their domain architecture as an organizing principle will facilitate the sorting of the very large and growing class of ncRNAs within functional categories and provide a foundation for more comprehensive mechanistic studies, ultimately leading to their exploitation for clinical purposes. The ultimate goal is to affect the function of an entire NIN to modulate normal cellular states and pathological conditions.
Acknowledgments
Due to the vast expansion of the covered topic in the literature, we apologize to the colleagues whose work was not cited. We thank Chunlai Li and Mihnea Dragomir for the comments on the manuscript and Enrique Fuentes-Mattei for the drawings of the Figures 1 and 2. G.A.C. is the Felix L. Haas endowed professor in Basic Science. Work in G.A.C.’s laboratory is supported by National Institutes of Health (NIH/NCATS) grant UH3TR00943-01 through the NIH Common Fund, Office of Strategic Coordination (OSC), NCI grants 1R01 CA182905-01 and 1R01CA222007-01A1, an NIGMS 1R01GM122775-01 grant, a U54 grant #CA096297/CA096300–UPR/MDACC Partnership for Excellence in Cancer Research 2016 Pilot Project, a Team DOD (CA160445P1) grant, a Chronic Lymphocytic Leukemia Moonshot Flagship project, a Sister Institution Network Fund (SINF) 2017 grant, and the Estate of C.G. Johnson, Jr. The work at the University of Washington (G.V.) is supported by NIH NIGMS RO1 GM103834 and R35 GM126942. Research support for L.G.’s laboratory is received from the Swedish Research Council, Swedish Cancer Society, The Swedish Childhood Cancer Foundation, Crown Princess Margareta's Foundation for the Visually Impaired, SINF StraCan, King Gustaf V Jubilee Foundation, Stockholm Cancer Society, Stockholm County, and Karolinska Institute. M.F. is supported by the NIH/NCI grants R01CA215753 and R01CA219024 and the Pablove Foundation Accelerator Award.
Footnotes
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.247239.118.
-
Freely available online through the Genome Research Open Access option.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








