
Experimental and pan-cancer genome analyses reveal widespread contribution of acrylamide exposure to carcinogenesis in humans
- Maria Zhivagui1,
- Alvin W.T. Ng2,3,4,
- Maude Ardin1,
- Mona I. Churchwell5,
- Manuraj Pandey1,
- Claire Renard1,
- Stephanie Villar1,
- Vincent Cahais6,
- Alexis Robitaille7,
- Liacine Bouaoun8,
- Adriana Heguy9,
- Kathryn Z. Guyton10,
- Martha R. Stampfer11,
- James McKay12,
- Monica Hollstein1,13,14,
- Magali Olivier1,
- Steven G. Rozen2,3,4,
- Frederick A. Beland5,
- Michael Korenjak1 and
- Jiri Zavadil1
- 1Molecular Mechanisms and Biomarkers Group, International Agency for Research on Cancer, Lyon 69008, France;
- 2Centre for Computational Biology, Duke–NUS Medical School, Singapore 169857, Singapore;
- 3Program in Cancer and Stem Cell Biology, Duke–NUS Medical School, 169857, Singapore;
- 4NUS Graduate School for Integrative Sciences and Engineering, Singapore 117456, Singapore;
- 5Division of Biochemical Toxicology, National Center for Toxicological Research, Jefferson, Arkansas 72079, USA;
- 6Epigenetics Group, International Agency for Research on Cancer, Lyon 69008, France;
- 7Infections and Cancer Biology Group, International Agency for Research on Cancer, Lyon 69008, France;
- 8Environment and Radiation Section, International Agency for Research on Cancer, Lyon 69008, France;
- 9Department of Pathology and Genome Technology Center, New York University, Langone Medical Center, New York, New York 10016, USA;
- 10IARC Monographs Group, International Agency for Research on Cancer, Lyon 69008, France;
- 11Biological Systems and Engineering Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA;
- 12Genetic Cancer Susceptibility Group, International Agency for Research on Cancer, Lyon 69008, France;
- 13Deutsches Krebsforschungszentrum, 69120 Heidelberg, Germany;
- 14Faculty of Medicine and Health, University of Leeds, LIGHT Laboratories, Leeds LS2 9JT, United Kingdom
Abstract
Humans are frequently exposed to acrylamide, a probable human carcinogen found in commonplace sources such as most heated starchy foods or tobacco smoke. Prior evidence has shown that acrylamide causes cancer in rodents, yet epidemiological studies conducted to date are limited and, thus far, have yielded inconclusive data on association of human cancers with acrylamide exposure. In this study, we experimentally identify a novel and unique mutational signature imprinted by acrylamide through the effects of its reactive metabolite glycidamide. We next show that the glycidamide mutational signature is found in a full one-third of approximately 1600 tumor genomes corresponding to 19 human tumor types from 14 organs. The highest enrichment of the glycidamide signature was observed in the cancers of the lung (88% of the interrogated tumors), liver (73%), kidney (>70%), bile duct (57%), cervix (50%), and, to a lesser extent, additional cancer types. Overall, our study reveals an unexpectedly extensive contribution of acrylamide-associated mutagenesis to human cancers.
Cancer can be caused by lifestyle factors, environmental or occupational exposures involving chemicals, their complex mixtures, and physical and biological agents. Many human carcinogens show shared key characteristics (Smith et al. 2016), and different carcinogens may have a spectrum of such characteristics and operate through distinct mechanisms to produce genetic alterations. Recognizable somatic alteration patterns characterize carcinogens that are mutagenic. Single-base substitution (SBS) mutational signatures can be expressed in simple mathematical terms that enable them to be extracted from thousands of cancer genomes (Alexandrov et al. 2013a, 2018). Several of the identified mutational signatures have been attributed to specific external exposures or endogenous factors through epidemiological and/or experimental studies (Alexandrov et al. 2018). The majority of the signatures remain of unknown origin, and additional, yet unrecognized, signatures are likely to be extracted from rapidly accumulating cancer genome data. Well-controlled experimental exposure systems can help identify the causes of the orphan mutational signatures and define new carcinogen-generated patterns (for review, see Hollstein et al. 2017; Zhivagui et al. 2017).
Various diet-related and iatrogenic exposures contribute to human cancer burden, involving, for instance, food contaminants (aflatoxin B1 [AFB1]) or alternative medicines (aristolochic acid [AA]) with well-documented mutagenic properties; AFB1 induces predominantly C:G > A:T and AA generates T:A > A:T transversions. These characteristic mutations, arising in preferred sequence contexts, allowed unequivocal association of exposure to AFB1 or AA with specific subtypes of hepatobiliary or urological cancers (Poon et al. 2013; Meier et al. 2014; Scelo et al. 2014; Jelaković et al. 2015; Hoang et al. 2016; Chawanthayatham et al. 2017; Huang et al. 2017; Ng et al. 2017; Zhang et al. 2017).
Among dietary compounds with carcinogenic potential, acrylamide (ACR) is of interest because of its ubiquitous presence. Important sources of exposure to ACR include tobacco smoke (Mojska et al. 2016), coffee (Takatsuki et al. 2003), and a spectrum of occupational settings (IARC 1994). ACR forms in carbohydrate-rich foods (e.g., potatoes and cereals) heated at high temperatures, because of Maillard reactions involving reducing sugars and the amino acid asparagine (Tareke et al. 2002). There is sufficient evidence that ACR is carcinogenic in rodents (Beland et al. 2013, 2015), and it was classified by the International Agency for Research on Cancer (IARC) as a probable carcinogen (Group 2A) (IARC 1994). The associations of dietary ACR exposure with renal, endometrial, and ovarian cancers have been explored in epidemiological studies (Hogervorst et al. 2008; Virk-Baker et al. 2014; Pelucchi et al. 2015). However, accurate ACR exposure assessment by questionnaires has been difficult, whereas more direct measures of molecular markers, such as hemoglobin adduct levels, may not yield conclusive findings on past exposures (Olesen et al. 2008; Wilson et al. 2009; Xie et al. 2013; Obón-Santacana et al. 2016a,b,c). Thus, innovative well-controlled exposure model systems can improve our understanding of the ACR exposure–associated effects and risk.
Oxidation of ACR by cytochrome P450 produces the highly reactive electrophilic epoxide glycidamide (GA) (Segerbäck et al. 1995; Sumner et al. 1999; Ghanayem et al. 2005). The Hras mutation loads in neoplasms of mice exposed to ACR or GA were higher upon exposure to GA (Von Tungeln et al. 2012), and more mutations in the cII reporter gene of Big Blue mouse embryonic fibroblasts were obtained by GA treatment in comparison to ACR (Besaratinia and Pfeifer 2003, 2004). In vivo and in vitro reporter gene mutagenesis studies showed an increased association of ACR and GA exposure with T:A > C:G transitions and T:A > A:T and C:G > G:C transversions (Besaratinia and Pfeifer 2003, 2004; Von Tungeln et al. 2009, 2012; Ishii et al. 2015; Manjanatha et al. 2015). In addition, GA exposure induces C:G > A:T transversions (Besaratinia and Pfeifer 2004). However, these ACR- and GA-specific patterns were based on limited mutation counts and do not allow translating adequately the reported mutation types into genome-wide patterns.
Massively parallel sequencing allows studying a large number of mutations in a single sample, thus significantly enhancing the power of mutation analysis in experimental models. Analogously to human cancer genome projects, genome-scale mutational signatures can be extracted from highly controlled carcinogen exposure experiments using mammalian cells and animal models, in combination with advanced computational methods (Olivier et al. 2014; Nik-Zainal et al. 2015; Huang et al. 2017). By integrating massively parallel sequencing and DNA adduct analysis in a mammalian cell clonal expansion model (Olivier et al. 2014; Nik-Zainal et al. 2015; Huskova et al. 2017) and by computational interrogation of the Pan-Cancer Analysis of Whole Genomes (PCAWG) data, we aimed to systematically investigate the mutational signatures of ACR and GA and to determine the contribution of ACR/GA to human carcinogenesis.
Results
Human TP53 mutations generated by ACR or GA treatment
Primary Hupki MEF cultures from three different embryos (Prim_1, Prim_2, and Prim_3) exposed to ACR or GA at the predetermined cytotoxic and genotoxic conditions yielded multiple immortalized clones (Methods) (Supplemental Fig. S1) suitable for massively parallel sequencing (Olivier et al. 2014). Sanger sequencing of TP53 in the clones derived from ACR exposure (ACR clones) and GA exposure (GA clones) and spontaneous immortalization (Spont), showed that ACR clones obtained from the Prim_2 MEFs showed loss of heterozygosity in the TP53 codon 72 involving a loss of the proline allele (ACR_1 clone), and also loss of the arginine allele resulting in a hemizygous ACR_2 clone (Table 1). No TP53 mutations were observed in the Spont clones. The detection of TP53 mutations in three out of seven ACR clones and in one out of five GA clones (Table 1) provided a sound rationale for extended sequencing at the exome scale.
Summary of cell lines, treatment conditions, and TP53 mutation status
Analysis of mutation spectra
Whole-exome sequencing (WES) of all Spont as well as exposed clones revealed that the total number of acquired SBS did not differ markedly between the ACR and Spont clones. The Spont clones harbored on average 190 (median = 151, range = 141–277) SBSs, whereas the ACR clones had on average 208 (median = 173, range = 151–262) SBSs. In contrast, the total number of SBSs was considerably increased in the GA clones, with an average of 485 SBSs (median = 448, range = 370–592) (Supplemental Tables S1, S2). This finding reveals stronger mutagenic properties of GA in the MEFs.
Principal component analysis (PCA) performed on the resulting SBS spectra unambiguously separated the GA clones from other experimental conditions (Fig. 1A). The ACR-exposed samples showed a diffuse pattern across the six SBS classes, whereas the Spont clones showed an enrichment of C:G > G:C SBS in the 5′-GCC-3′ context, also present across the exposed cultures (Supplemental Fig. S2). This background mutation type appears related to the culture conditions used for the MEF immortalization assay, and its consistent formation has been observed previously (Olivier et al. 2014; Nik-Zainal et al. 2015). No significant transcription strand bias (TSB) was observed for any mutation class in the Spont or ACR clones (Supplemental Fig. S3). In the clones derived from the GA-treated primary MEF cultures, we observed an enrichment of T:A > A:T and C:G > A:T transversions and T:A > C:G transitions (Supplemental Fig. S2B), marked by significant TSB (Supplemental Fig. S3). The GA-associated clones showed lower numbers (25 per clone) of small insertions/deletions (indels) in comparison to the ACR (44 per clone) or Spont clones (39 per clone) (see Supplemental Tables S1, S3). Thus, higher SBS counts owing to GA treatment may selectively promote the senescence bypass and the selection, with a decreased functional contribution of indels, whereas an inverse scenario is plausible for the Spont and ACR clones, consistent with a previous report based on the Big Blue mouse embryonic fibroblasts and cII transgene (Besaratinia and Pfeifer 2005).
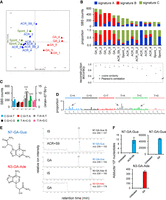
Analysis of the mutation patterns derived from experimental exome sequencing data. (A) Principle component analysis (PCA) of WES data. PCA was computed using as input the mutation count matrix of the clones that immortalized spontaneously (Spont) or were derived from exposure to acrylamide (ACR) or glycidamide (GA). Each sample is plotted considering the value of the first and second principal components (Dim1 and Dim2). The percentage of variance explained by each component is indicated within brackets on each axis. Spont and ACR- and GA-exposed samples are represented by differently colored symbols. (B) Mutational signatures (sig A, sig B, and sig C), identified by NMF, and their contribution to each sample (x-axis), assigned either by absolute SBS counts or by proportion (bar graphs). The reconstruction accuracy of the identified mutational signatures in individual samples is shown in the bottom dot plot (y-axis value of 1 = 100% accuracy). (C) Transcription strand bias analysis for the six mutation types in GA-exposed clones. For each mutation type, the number of mutations occurring on the transcribed (T) and nontranscribed (N) strand is shown on the y-axis. (***) P < 10−8, (*) P < 10−2. (D) Extraction of GA signature, with arrows pointing at the enriched SBS classes. The contribution of signature 17 (T:A > G:C in 5′-NTT-3′ context), present in all clones, was decreased by performing NMF on human-TP53 knock-in (Hupki) MEF samples pooled with primary tumor samples with high levels of signature 17 (see Methods and Supplemental Methods). (E) DNA adducts analysis as determined by LC-MS/MS. (F) Levels of N7-GA-Gua adduct in ACR + S9- and GA-treated cells and N3-GA-Ade DNA adduct level in GA-treated cells compared with untreated cells yielding no adducts. The data are presented as the number of adducts in 108 nucleotides in replicated experiments (n ≥ 2).
Variant allele frequency (VAF) analysis performed for GA clones detected a large proportion of acquired mutations manifesting at VAF between 25% and 75% (Supplemental Fig. S4C). Upon grouping of substitutions into bins of high (67%–100%), medium (34%–66%), and low (0%–33%) VAF, the predominant GA-specific mutation types (T:A > A:T, T:A > C:G, and C:G > A:T) started manifesting at high VAF and became increasingly enriched in the medium and low VAF intervals. The background 5′-N[T>G]T-3′ SBS, corresponding to COSMIC signature 17 arising in cultured mouse cells including MEFs (Behjati et al. 2014; Nik-Zainal et al. 2015; Milholland et al. 2017), displayed minor, although not statistically significant, lower-VAF enrichment (P = 0.25, assessed by χ2 test) (Supplemental Fig. S5). These observations suggest early effects of the GA exposure, reproducible contribution of the induced mutations to senescence bypass, and their clonal propagation during the immortalization stage.
Mutational signature of GA
Three distinct mutational signatures were extracted from all MEF clones, termed signatures A, B, and C. Signatures A and C were enriched in the Spont and ACR clones, whereas the more robust signature B was selectively enriched in the GA clones (Fig. 1B; Supplemental Fig. S6). The TSB analysis in the GA clones revealed significant enrichment of the prominent mutation types C:G > A:T, T:A > A:T, and T:A > C:G (using the pyrimidine-based mutation class convention) on the transcribed strand (P < 0.05, χ2 test), consistent with the less efficient transcription-coupled nucleotide excision repair because of adduct formation on purines (Fig. 1C; Supplemental Fig. S3). In signature C and to a lesser extent in signatures A and B, we observed an admixture of a pattern identical to the COSMIC signature 17 (T:A > G:C in the 5′-NTT-3′ trinucleotide context), present in human cancers (notably esophageal and gastric adenocarcinomas) but also seen in AFB1-driven mouse liver cancers (Huang et al. 2017), in murine small cell lung carcinoma initiated by loss of Trp53 and Rb1 (McFadden et al. 2014), and in primary MEF-derived clones (Olivier et al. 2014; Nik-Zainal et al. 2015). This signature has been linked to cell culture conditions (Behjati et al. 2014; Milholland et al. 2017) and may be linked to oxidative stress effects on the free dGTP pool (Tomkova et al. 2018). To further refine the putative GA mutational signature from signature B, we used extended-input nonnegative matrix factorization (NMF) by combining the MEF clone data with signature 17–rich esophageal adenocarcinoma data from the International Cancer Genome Consortium (ICGC) ESAD-UK study (Secrier et al. 2016), as well as with The Cancer Genome Atlas (TCGA) esophageal adenocarcinoma (ESCA) and gastric carcinoma (STAD) samples enriched for or lacking signature 17 (see Methods) (Supplemental Methods; Supplemental Figs. S6, S7). This considerably reduced (average = 47%, median = 48%) the signature 17–specific T > G peaks in signature B associated with GA treatment and resulted in a cleaner pattern (Fig. 1D; Supplemental Fig. S6). The refined GA signature retains the strand-biased enrichment of the T:A > A:T transversions and T:A > C:G transitions in the 5′-CTG-3′ and 5′-CTT-3′ trinucleotide contexts, as well as the C:G > A:T component (Fig. 1D; Supplemental Fig. S8A; Supplemental Table S4).
Quantitative DNA adduct analysis supports the GA mutational signature
Following metabolic activation, ACR induces GA-DNA adducts at the N7 and N3 positions of guanine and adenine, respectively. Analysis using liquid chromatography–tandem mass spectrometry (LC-MS/MS) revealed the absence of these adducts in the untreated samples, as well as in MEFs exposed to ACR in the absence of S9 fraction (with levels below the limit of detection [LOD]). This suggests a lack of Cyp2e1 activity normally required for the metabolism of ACR to GA in the MEFs. Upon addition of human S9 fraction, N7-(2-carbamoyl-2-hydroxyethyl)-guanine (N7-GA-Gua) levels increased to 11 adducts/108 nucleotides (twice the LOD levels), suggesting limited metabolic activation of ACR despite the enzymatic activity of the S9 fraction (Fig. 1E,F). In contrast, cells exposed to GA showed high DNA adduct levels, with N7-GA-Gua and N3-(2-carbamoyl-2-hydroxyethyl)-adenine (N3-GA-Ade) observed at 49,000 adducts/108 nucleotides and 350 adducts/108 nucleotides, respectively, after subtracting the trace amount of contamination from the internal standard (Fig. 1E,F). These observed DNA adducts provide a possible mechanistic basis for the mutation types, the TSB, and the mutational signature arising upon treatment with GA, the reactive metabolite of ACR.
Comparison of the GA signature with PCAWG mutational signatures
We next performed cosine similarity comparison of the putative GA signature with the recently updated PCAWG SBS mutational signatures (Alexandrov et al. 2018) and with known T:A > A:T-rich experimental signatures (Fig. 2A; Supplemental Figs. S7, S9). The highest cosine similarity value (84%) corresponded to PCAWG SBS25 (Fig. 2A). However, unlike the GA signature, neither SBS25 nor any other signatures show TSB for the three mutation classes (C:G > A:T, T:A > A:T, and T:A > C:G). Thus, the mutation patterns with a three-class strand bias generated by the GA treatment render the resulting mutational signature unique and novel.

Comparison of GA signature to known signatures. (A) Cosine similarity matrix comparing GA mutational signature with the human PCAWG data (SBS3, -4, -5, -8, -22, -25, -35, -39, and -40) and other A > T-rich mutational signatures from experimental exposure assays using specific carcinogens (7,12-dimethylbenz[a]anthracene [DMBA], urethane, and aristolochic acid [AA]). (B) Comparison of PCAWG SBS4 with two experimentally derived signatures: B[a]P_exp = benzo[a]pyrene mutational signature extracted from HMECs; GA_exp = GA mutational signature extracted from MEF cells. Cosine similarity between the T > N (adenine) components of SBS4 and GA signature is shown on the right. (C) Transcription strand bias analysis for the six mutation types underlying the signatures in panel B. For each mutation type (using the pyrimidine convention), the number of mutations occurring on the transcribed (T) and nontranscribed (N) strand is shown on the left y-axis. The significance is expressed as –log10(P-value) indicated on the right y-axis. (***) P < 10−8, (**) P < 10−4, (*) P < 10−2.
GA signature in the human pan-cancer genomes
The initial visual comparison with PCAWG signatures indicated similarity between the GA signature and signature SBS4 of tobacco smoking (Supplemental Fig. S8; Alexandrov et al. 2018), in keeping with the established presence of ACR in tobacco smoke. This was further corroborated by the cosine similarity of 94% between the adenine (T > N) components of SBS4 and the GA signature (Fig. 2B). We thus hypothesized that SBS4 reflects the coexposure to benzo[a]pyrene (B[a]P; generating the predominant, strand-biased C > N/guanine mutations) and to GA (generating strand-biased T > N/adenine mutations) (Fig. 2B,C; Supplemental Fig. S8). To obtain experimental evidence, we modeled a B[a]P mutational signature by whole-genome sequencing (WGS) of cell clones derived from B[a]P-exposed normal human mammary epithelial cells (HMECs) (Stampfer and Bartley 1985, 1988). This yielded a robust pattern characterized by predominant strand-biased guanine (mainly C:G > A:T) mutation levels and negligibly mutated adenines (T > N) (Fig. 2B; Supplemental Figs. S8, S10; Supplemental Table S4). Next, we interrogated the PCAWG data for the presence of the experimentally defined, 192-class (strand-biased) GA and B[a]P signatures in 1584 tumors of 19 cancer types from 14 organ sites (Fig. 3; Supplemental Table S5). The stringency of the process was controlled by determining the P-value and the false-discovery rate (FDR) for the signature presence test and the reconstruction accuracy (Supplemental Table S6) and by modeling false-positive rates (FPRs) and FDRs of the experimental signature detection using 2000 synthetic tumors as described in the Methods and in Supplemental Tables S7 through S10. In the subset of PCAWG-7 cancers known to carry SBS4 signature (adenocarcinomas and squamous cell carcinomas of the lung, hepatocellular carcinomas of the liver and head, and neck squamous cell carcinomas), we compared the GA and B[a]P signatures to estimated levels of SBS4 and found that in the lung and head and neck cancers, a combination of the GA and B[a]P signatures accounted for very similar numbers of mutations as SBS4, suggesting that SBS4 represents combined and highly correlated exposure to GA and B[a]P (Fig. 3A). In contrast, we found more variability in the assignment of mutation numbers to GA and B[a]P versus SBS4 in liver cancers (Fig. 3), which may reflect a weaker relationship between GA and B[a]P exposure because of generally more complex exposure history in the liver. Successful reconstruction of SBS4 by the experimental 192-class (strand-biased) GA and B[a]P signatures in the lung and liver human tumors enabled correct assignment of the GA signature in a subset of 24 lung adenocarcinomas, 42 lung SCCs, and 239 liver tumors with a subset of 184 GA-positive HCCs lacking the B[a]P signature mutations (Fig. 3B; Supplemental Table S11). Moreover, we identified the GA signature in additional 15 cancer types without SBS4, including clear cell renal cell carcinoma (78 GA-positive of 111 analyzed tumors), papillary renal cell carcinoma (26 GA-positive out of 32), biliary adenocarcinoma (20 GA-positive out of 35), colorectal adenocarcinomas (24 GA-positive out of 60), stomach adenocarcinoma (17 GA-positive out of 75), bladder transitional cell carcinoma (six GA-positive out of 23), and uterine adenocarcinoma (10 GA-positive out of 51) (Fig. 3B,C). The signature assignments results for the 537 individual GA-positive PCAWG tumors are summarized by cancer type in the Supplemental Table S11.
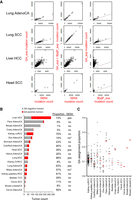
Identification of experimental GA signature in the human cancer PCAWG data sets. (A) Scatter plots of the experimental GA_exp and B[a]P_exp mutational signature assignments by mSigAct show reconstruction of tobacco-smoking signature SBS4 assignments in cancer types with SBS4 present. (Lung.AdenoCA) Lung adenocarcinoma, (Lung.SCC) lung squamous cell carcinoma, (Liver.HCC) liver hepatocellular carcinoma (Head.SCC) head squamous cell carcinoma. The combination of GA_exp and B[a]P_exp mutation counts reconstructed SBS4 mutation counts in Lung.AdenoCA and Lung.SCC and, to an extent, in Head.SCC. In liver HCCs, GA counts alone partially reconstructed SBS4 mutation counts and indicate GA_exp-positive and B[a]P_exp-negative tumors (third row, right scatter plot). The lines in GA versus B[a]P scatter plots have a slope of 0.3, reflecting the 3:1 ratio of B[a]P:GA mutation counts that reconstruct SBS4. (B) Summary of GA mutation assignment analysis of 1584 individual tumors of 19 cancer types from the PCAWG data sets. Assignments were performed using mSigAct (positivity was determined by the signature.presence.test tool at FDR < 0.05) with the PCAWG annotations of signature present in each subtype, in addition to the GA and B[a]P signatures. The tumor types manifesting or lacking SBS4 signature of tobacco smoking are labeled accordingly in the column SBS4. Asterisk denotes borderline SBS4 presence in PCAWG Billiary.AdenoCA (two of 173, 1.16%) and Eso.AdenoCA (two of 347, 0.06%). Proportion indicates percentage of GA-positive tumors within each listed cancer type. (C) The dot plot shows the proportion of mutations assigned to GA signature among other identified signatures (see Supplemental Material) in individual tumors of cancer types not showing the direct effects of tobacco smoking (i.e., lacking signature SBS4). Red horizontal lines denote median values (y-axis, 1 = 100%).
Discussion
ACR and GA exposures induce an almost identical set of tumors in both mice and rats, providing a substantial argument for a GA-mediated tumorigenic effect of ACR (Beland et al. 2015). This is supported by further mechanistic studies showing that lung tissue from mice exposed to ACR and GA displays comparable DNA adduct patterns, as well as similar mutation frequencies in the cII transgene (Manjanatha et al. 2015). Similar observations were made in the context of in vitro mutagenicity of ACR in human and mouse cells, suggesting the key role for the epoxide metabolite GA to form premutagenic DNA adducts (Besaratinia and Pfeifer 2004). Thus, in keeping with the established ACR/GA carcinogenicity in rodents (IARC 1994; Olstørn et al. 2007; Von Tungeln et al. 2012; Beland et al. 2015), our findings provide new information on the characteristic mutagenic effects of GA and their contribution to tumor development.
The observation that ACR itself is not efficiently metabolized by MEFs is consistent with similar differences reported by previous animal carcinogenicity studies. In neonatal B6C3F1 mice, GA, but not ACR, induces hepatocellular carcinomas, likely because of the inability of neonatal mice to efficiently metabolize ACR (Von Tungeln et al. 2012). Moreover, unlike ACR, GA induces tumors in the small intestine in a dose-dependent manner upon perinatal exposure (Olstørn et al. 2007). Similar differences between GA and ACR mutagenicity, possibly because of limited metabolization of ACR, were observed in vitro (Besaratinia and Pfeifer 2004). We addressed the lack of ACR activation by the addition of human S9 fraction, yet the assessment of DNA adducts suggested limited metabolic activation of ACR with adduct levels substantially lower compared with the direct GA exposure. This may explain the mutagenicity differences observed between GA and ACR. A consistent minor contribution of the GA mutational signature was detected in the majority of ACR clones, whereas it was mostly absent in the Spont clones, suggesting subtle metabolic activation of ACR in the MEFs resulting in low levels of GA. However, a robust mutational signature in the experimental setting was generated exclusively by exposing the cells directly to GA.
Single reporter gene studies had previously linked ACR and GA exposure to multiple different mutation types. Thanks to the larger number of mutations obtained by exome sequencing, we were able to attribute to the GA exposure a particular mutational signature characterized by three strand-biased mutation classes (C:G > A:T, T:A > A:T, and T:A > C:G). The identification of the N7-GA-Gua and N3-GA-Ade DNA adducts originating from the metabolic conversion of ACR (Segerbäck et al. 1995; da Costa et al. 2003; Besaratinia and Pfeifer 2005), underlines the relationship between DNA adduct profiles and the mutational signature of GA. N3-GA-Ade and N7-GA-Gua are depurinating adducts resulting in apurinic/apyrimidinic sites. During replication, these lead to misincorporation of deoxyadenine, leading to the respective T:A > A:T and C:G > A:T transversions observed in the GA signature. The T:A > C:G transitions enriched in the GA signature correspond to the miscoding N1-GA-Ade adduct, the most commonly identified adenine adduct in vitro (Randall et al. 1987; da Costa et al. 2003; Besaratinia and Pfeifer 2005; Ishii et al. 2015). The levels of the guanine adduct were especially high in the GA-exposed MEF cells, whereas the associated C:G > A:T transversions in the resulting postsenescence clones were less represented. This could reflect differences in DNA repair efficiency concerning the individual guanine and adenine adduct species or the fact that the resulting clones are derived from single cells that selectively immortalized but do not accurately represent the bulk exposed primary cell population in which the GA-DNA adduct levels were measured after exposure. It is also plausible that the excessive and possibly highly cytotoxic N7-GA-Gua adduct burden leads to negative selection of a large number of affected cells.
The established animal models (Beland et al. 2013, 2015) of ACR- and GA-mediated tumorigenesis provide a suitable starting point for a comparison of the mutational signatures obtained from the mouse and in vitro. Next, genome-scale sequencing of human tumors and adduct analysis of normal tissues collected in well-designed molecular epidemiological studies focusing on ACR intake are warranted to provide further evidence that the GA signature mutations identified in various cancer types indeed correlate with the exposure to ACR.
The GA signature has not been identified among the currently known computationally extracted PCAWG signatures (Fig. 2A; Alexandrov et al. 2018). Here we show that a new pattern can be identified in a large subset of pan-cancer tumors when experimentally modeled signatures are combined with sophisticated computational signature reconstruction methods while considering the extended features, such as TSB supported by premutagenic adduct analysis. Such integrated approaches can thus lead to future identification of yet unrecognized carcinogen signatures that may be eluding the solely computation-based analyses of the pan-cancer data.
The quest for understanding the contribution of ACR to cancer development is reflected by recent accumulation of mechanistic data on the compound's mutagenicity and carcinogenicity in experimental models. The possibly carcinogenic effects of ACR in humans were recommended for re-evaluation by the Advisory Group to the Monographs Program of the International Agency for Research on Cancer (Straif et al. 2014). Our findings related to the reconstruction of signature SBS4 by the experimental signatures of GA and B[a]P, together with the detection of the GA signature in lung and liver cancer, are relevant given the established high content of ACR in tobacco smoke. Compared with the GA effects, experimental B[a]P exposure generates very few T > N (adenine) mutations. However, we cannot exclude a possibility that in the human tissues directly exposed to tobacco smoke the adenine residues can be targeted by carcinogens such as B[a]P derivatives or nitrosamines.
A subset of 184 liver tumor samples identified in this study harbored the GA signature but no features of the B[a]P signature or SBS4 (Fig. 3B; Supplemental Material). Furthermore, we found 217 GA-positive, SBS4-negative tumors of additional 15 cancer types (Fig. 3B,C). The numerous GA-positive, SBS4-negative tumors are of particular interest as they likely reflect dietary and/or occupational exposures to ACR unrelated to tobacco smoking. Overall, our findings offer new insights into the thus-far tenuous association of ACR with human carcinogenesis.
Methods
Source and authentication of primary cells
Primary human-p53 knock-in (Hupki) MEFs were isolated from 13.5-d-old Trp53tm/Holl mouse embryos from the Central Animal Laboratory of the Deutsches Krebsforschungszentrum as described previously (Liu et al. 2004). The mice had been tested for specific pathogen-free (SPF) status. The derived primary cells were genotyped for the human TP53 codon 72 polymorphism (Table 1) to authenticate the embryo of origin. Cells from three different embryos (E210, E213, and E214) were used for the exposure experiments (Table 1). All subsequent cell cultures were routinely tested at all stages for the absence of mycoplasma.
Cell culture, exposure, and immortalization
The primary MEF cells were expanded in advanced DMEM supplemented with 15% fetal calf serum, 1% penicillin/streptomycin, 1% pyruvate, 1% glutamine, and 0.1% β-mercaptoethanol. The cells were then seeded in six-well plates and, at passage 2, were exposed for 24 h to 5 mM ACR (A4058, Sigma-Aldrich) in the presence of 2% human S9 fraction (Life Technologies) complemented with NADPH (Sigma-Aldrich) or the absence of S9 to 10 mM ACR or 3 mM GA (04704, Sigma-Aldrich), or to vehicle (PBS). Exposed and untreated control primary cells were cultured until they bypassed senescence and immortalized clonal cell populations could be isolated (Todaro and Green 1963). The HMEC cultures used in this study for WGS were generated from primary HMECs (passage 4) exposed to B[a]P and propagated in M87A medium to passage 13, as described previously (Stampfer and Bartley 1985, 1988; Garbe et al. 2009; Severson et al. 2014).
MTT assay for cell metabolic activity and viability
Cells were seeded in 96-well plates and treated as indicated. Cell viability was measured 48 h after treatment cessation using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega). Plates were incubated for 4 h at 37°C, and absorbance was measured at 492 nm using the Apollo 11 LB913 plate reader. The MTT assay was performed in triplicate for each experimental condition.
Phospho-H2AFX immunofluorescence
Immunofluorescence staining of phosphorylated histone H2AFX (γH2AFX) was performed using phospho-histone H2A.X (Ser139) (20E3) Rabbit monoclonal antibody (9718, Cell Signaling Technology). Briefly, primary MEFs were seeded on coverslips in 12-well plates and, the following day, treated as indicated in duplicate for 24 h. Four hours after treatment cessation, the cells were fixed with 4% formaldehyde for 15 min at room temperature. Following blocking in 5% normal goat serum (31872, Life Technologies) for 60 min, they were incubated with the γH2AFX-antibody (1:500 in 1% BSA) overnight at 4°C. Subsequent incubation with a fluorochrome-conjugated secondary antibody (4412, Cell Signaling Technology) was performed for 60 min at room temperature. Coverslips were mounted in Vectashield mounting medium with DAPI (Eurobio). Immunofluorescence images were captured using a Nikon Eclipse Ti.
DNA adduct analysis
GA-DNA adducts (N7-GA-Gua and N3-GA-Ade) were quantified by LC-MS/MS with stable isotope dilution as previously described (da Costa et al. 2003). The DNA was isolated from the cells using standard digestion with Proteinase K, followed by phenol-chloroform extraction and ethanol precipitation. The DNA was subsequently treated with RNase A and T1, extracted with phenol-chloroform, and reprecipitated with ethanol. N7-GA-Gua and N3-GA-Ade were released by neutral thermal hydrolysis for 15 min, using Eppendorf Thermomixer R (Eppendorf North America) set to 99°C. The samples were filtered through Amicon 3K molecular-weight cutoff filters (Merck Millipore) to separate the adducts from the intact DNA. The LC-MS/MS used for quantification consisted of an Acquity UPLC system (Waters) and a Xevo TQ-S triple quadrupole mass spectrometer (Waters). The following MRM transitions were monitored with a cone voltage of 50 V and a collision energy of 20 eV: N3-GA-Ade, m/z 223→178; [15N5]N3-GA-Ade (internal standard), m/z 228→183; N7-GA-Gua, m/z 239→152; and [15N5]N7-GA-Gua (internal standard), m/z 244→157 (da Costa et al. 2003).
TP53 genotyping
Exons 4 to 8 of the knocked-in human TP53 gene (NC_000017.11) were sequenced using standard protocols. Sanger sequencing of PCR products was performed at BIOfidal, using the Applied Biosystems 3730xl genetic analyzer. The amplicon and sequencing primers are listed in the Supplemental Methods. Sequences were analyzed using the CodonCode Aligner version 7.1 software.
Library preparation and WES
Refer to the online Supplemental Methods for details on the standard procedures for library preparation and WES, sequencing data preprocessing, read alignment, and the calling of the SBS and indel variants in the MEF and HMEC cell lines.
Bioinformatics and extraction of experimental mutational signatures
Refer to the Supplemental Methods for detailed information on PCA, assessment of sequencing-related artifacts and damage, and computation of the TSB and its significance. The TSB was considered statistically significant at P-value ≤ 0.05. To analyze the mutation spectra and treatment-specific mutational signatures, filtered mutations were classified into 96 types corresponding to the six possible base substitutions (C:G > A:T, C:G > G:C, C:G > T:A, T:A > A:T, T:A > C:G, T:A > G:C) and the 16 combinations of flanking nucleotides immediately 5′ and 3′ of the mutated base. Mutation patterns were then deconvolved into mutational signatures using NMF (Brunet et al. 2004; Alexandrov et al. 2013b) embedded in the MutSpec suite (Ardin et al. 2016). For details on estimates of the optimal number of signatures to extract, see the Supplemental Methods. The reconstruction error calculation evaluated the accuracy with which the deciphered mutational signatures describe the original mutation spectra of each sample by applying Pearson's correlation and cosine similarity.
The GA mutational signature was further polished by using an extended input including samples from ICGC (ESAD-UK study) with high level of signature 17 (>65% contribution as determined by independent NMF analysis), and with samples from the TCGA esophageal adenocarcinoma (ESCA) and gastric cancer (STAD) collection (exon data, to address comparable coverage of the genome). The samples used for this procedure are listed in the Supplemental Methods, and the results are summarized in Supplemental Figures S6 and S7.
Cosine similarity analysis was used to evaluate the concordance between the identified T:A > A:T-rich mutational signature of GA with the newly characterized SBS mutational signatures from the PCAWG (pan-cancer whole genome) data (Alexandrov et al. 2018). Cosine similarity values of more than 0.5 were found for PCAWG SBS3, SBS4, SBS5, SBS8, SBS22, SBS25, SBS35, SBS39, and SBS40 and the experimentally derived mutational signature of AA (Olivier et al. 2014; Ardin et al. 2016), 7,12-dimethylbenz[a]anthracene (DMBA) (McCreery et al. 2015; Nassar et al. 2015), and urethane (Westcott et al. 2014).
The experimental B[a]P signature was generated by WGS (using Illumina HiSeq X Ten by GENEWIZ) of finite lifespan poststasis clones derived from primary HMECs treated with B[a]P, as previously described (Stampfer and Bartley 1985, 1988; Severson et al. 2014). Following read alignment to NCBI GRCh38 genome build, mutations were called in the two poststasis samples with MuTect2 or Strelka2.8 using a primary HMEC sample as a comparison. Only mutations called by both algorithms were retained, and additional criteria were applied to filter out mutations with a match in public SNP databases (dbSNP150, and/or AF > 0.001 in either 1000 Genomes, gnomAD or NHLBI-ESP), with an allele frequency above zero in the primary sample, with coverage lower than 10 reads, or mutations overlapping tandem repeats. Finally, a cut-off was applied on VAF, and only mutations with a VAF equal or higher than 20% were retained, being 54,587 unique mutations. The NMF procedure to extract the experimental B[a]P signature used input extended with SBS data from the TCGA lung cancer collection (15 Lung.AdenoCA positive [>50%] for tobacco-smoking SBS4, 15 Lung.AdenoCA negative for SBS4, 15 Lung.SCC positive [>50%] for SBS4 and 15 Lung.SCC negative for SBS4). See the Supplemental Methods for sample details. The recovered signatures showed the strongest enrichment of the C > A-based signature B (Supplemental Fig. S10) in the B[a]P-treated HMEC clones. We next calculated the reconstruction error to evaluate the accuracy with which the extracted B[a]P_exp signature describes the original mutation spectra of each sample by applying Pearson's correlation and cosine similarity (Supplemental Fig. S10).
Identification of the experimental signatures in PCAWG data
We used the mutational signature activity (mSigAct v0.10.R) software (Ng et al. 2017) to test for the presence of the experimental mutational signatures of GA and B[a]P in the human primary tumor data from PCAWG study. mSigAct conducts a statistical test for optimal reconstructions of the observed human tumor mutation spectrum with and without the GA mutational signature, in addition to a set of other mutational signatures from the PCAWG study. The 192-class strand-biased versions of the GA and B[a]P mutational signatures (Supplemental Fig. S8; Supplemental Table S4) were used to detect tumors with the experimentally defined signatures present, at high stringency achieved also by incorporating the same TSB information in the 192-class reconstructions of each tumor. To generate a 192-class reconstructed spectrum, the assignment of mutation counts for each 192-class signature is determined by mSigAct and multiplied with the 192-class versions of the PCAWG, GA, and B[a]P mutational signatures. The 192-class versions of each signature and spectrum is equivalent to the 96-class versions when the mutation counts on each strand are summed and then represented in the pyrimidine mutations (C > A, C > G, C > T, T > A, T > C, T > G). Specifically, B[a]P was added to cancer types with tobacco-smoking SBS4 signature previously found in the PCAWG signature set, and a combination of B[a]P and GA signatures was used in these cancers to reconstruct SBS4. For other signatures and cancers without evidence of SBS4 present, only GA was used to reconstruct the tumor spectra. This was followed by computing the likelihood ratio test between the original spectrum and the reconstructed tumor. A total of 1673 tumor samples from the PCAWG repository from 20 cancer subtypes were interrogated. We excluded hypermutated and recently identified AA signature–containing tumors (Ng et al. 2017) as the presence of strong T > A signature adversely affected the reconstruction process. A set of active mutational signatures were obtained from the PCAWG annotations of each cancer subtype, with flat signatures (SBS3, SBS8) removed to improve the sparsity of the mutation assignments. Final assignments of mutations to each mutational signature were performed by using the 96-class mutational signatures. Further fine-tuning was conducted using parameters for a negative binomial model, and the FDR was adjusted for mutational signature presence (FDR < 0.05).
The proportion matrices of the strand-biased and NMF versions of the experimental GA signature, the GA signature normalized to the human genome trinucleotide frequency to allow for human PCAWG data screening, and the strand-biased and NMF versions of the whole-genome B[a]P signature are available in Supplemental Table S4. The statistics underlying the assignment of GA_exp to PCAWG cancer data sets (P-values for “signature.presence.test” and cosine similarity between the reconstruction and spectra) are summarized in Supplemental Table S5.
FPR and FDR estimation for GA signature detection in synthetic tumors
To determine how often false positives arise when detecting the GA signature with mSigAct and to accurately estimate the FDR of the detection of GA signature, we performed a deeper validation analysis. We generated 2000 synthetic tumors with signatures from the PCAWG-7 data set and assignments sampled from the assignments to each signature in the PCAWG-7 data set, which represented the tumor types in which we found GA signature present, with similar signatures and mutation burdens associated with each signature. The synthetic tumors had the same frequency of observing a particular signature for a cancer type, similar to the PCAWG-7 tumors. One hundred tumors per 20 tumor types (included in the main analysis and listed in Supplemental Table S9) have been generated, with 1015 of the tumors harboring GA signature and 985 with GA signature absent. By using the synthetic tumor set and mSigAct to assign GA signature, we established the true-positive rates (TPRs), FPRs and FDRs (calculated by using the raw synthetic tumor counts and the formula FP/(TP + FP)). The results are shown as a short summary (Supplemental Table S7), raw tumor counts (Supplemental Table S8), per cancer type distribution (Supplemental Table S9) and a full listing of TPRs, FPRs, and FDRs (Supplemental Table S10).
Data access
Aligned WES reads from the primary MEF cells and clones arising from ACR- and GA-treated cultures and immortalized spontaneously, as well as Sanger sequencing files, have been submitted to the NCBI BioProject database (BioProject; https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA238303 (for the individual BioSample accession numbers, refer to Supplemental Tables S12, S13). The WES data reported here are a new extension of the BioProject PRJNA238303 dedicated to systematic identification of mutational signatures of carcinogenic agents (Olivier et al. 2014).
Acknowledgments
We thank the New York University Genome Technology Center, funded in part by the NIH/NCI Cancer Center support grant P30CA016087, and GENEWIZ, for expert assistance with Illumina sequencing. The study was supported by funding obtained from INCa-INSERM (Plan Cancer 2015 grant to J.Z.), NIH/NIEHS (1R03ES025023-01A1 grant to M.O.), and the Singapore National Medical Research Council (NMRC/CIRG/1422/2015 grant to S.G.R.) and the Singapore Ministry of Health via the Duke-NUS Signature Research Programmes to S.G.R. M.R.S. was supported by the U.S. Department of Energy under contract no. DE-AC02-05CH11231. M.P. was supported by the European Commission FP7 Marie Curie Actions-People-COFUND Fellowship. The views expressed in this article do not necessarily represent those of the U.S. Food and Drug Administration.
Author contributions: M.Z., M.K., and J.Z. drafted the manuscript and prepared figures. M.I.C. and F.A.B. performed DNA adduct analyses. M.Z., M.P., S.V., and M.K. performed laboratory studies under the supervision of M.R.S., J.M., A.H., M.H., and J.Z.; M.Z., A.W.T.N, M.A., C.R., V.C., A.R., L.B., M.O., and S.R.G. performed computational analyses and prepared relevant display items. M.Z., A.W.T.N., A.H., K.G., J.M., M.O., F.A.B., M.K., and J.Z. edited the manuscript, and M.Z., K.G., M.H., M.K., and J.Z. designed the study.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.242453.118.
-
Freely available online through the Genome Research Open Access option.
- Received July 31, 2018.
- Accepted February 1, 2019.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








