
Modeling Niemann–Pick disease type C in a human haploid cell line allows for patient variant characterization and clinical interpretation
- Steven Erwood1,2,
- Reid A. Brewer1,
- Teija M.I. Bily1,
- Eleonora Maino1,2,
- Liangchi Zhou1,
- Ronald D. Cohn1,2,3 and
- Evgueni A. Ivakine1
- 1Program in Genetics and Genome Biology, The Hospital for Sick Children Research Institute, Toronto, Ontario, M5G 0A4, Canada;
- 2Department of Molecular Genetics, University of Toronto, Toronto, Ontario, M5S 1A8, Canada;
- 3Department of Pediatrics, University of Toronto and The Hospital for Sick Children, Toronto, Ontario, M5G 1X8, Canada
Abstract
The accurate clinical interpretation of human sequence variation is foundational to personalized medicine. This remains a pressing challenge, however, as genome sequencing becomes routine and new functionally undefined variants rapidly accumulate. Here, we describe a platform for the rapid generation, characterization, and interpretation of genomic variants in haploid cells focusing on Niemann–Pick disease type C (NPC) as an example. NPC is a fatal neurodegenerative disorder characterized by a lysosomal accumulation of unesterified cholesterol and glycolipids. In 95% of cases, NPC is caused by mutations in the NPC1 gene, for which more than 200 unique disease-causing variants have been reported to date. Furthermore, the majority of patients with NPC are compound heterozygotes that often carry at least one private mutation, presenting a challenge for the characterization and classification of individual variants. Here, we have developed the first haploid cell model of NPC. This haploid cell model recapitulates the primary biochemical and molecular phenotypes typically found in patient-derived fibroblasts, illustrating its utility in modeling NPC. Additionally, we show the power of CRISPR/Cas9-mediated base editing in quickly and efficiently generating haploid cell models of individual patient variants in NPC. These models provide a platform for understanding the disease mechanisms underlying individual NPC1 variants while allowing for definitive clinical variant interpretation for NPC.
Niemann–Pick disease type C (NPC) is a rare autosomal recessive lysosomal storage disorder affecting one in 90,000 individuals (Vanier 2010; Wassif et al. 2016). In 95% of cases, NPC is caused by mutations in the gene NPC1, which is required for the proper transport of sterols from the lysosome to other subcellular compartments (Vanier 2010). Although NPC is a clinically heterogeneous disorder with symptoms ranging from hepatosplenomegaly to ataxia and seizures, the disease is defined by fatally progressive neurodegeneration (Vanier 2010; Patterson et al. 2013). These symptoms are caused by the intracellular accumulation of cholesterol and glycolipids within late endosomes and lysosomes (Ory 2000; Wojtanik and Liscum 2003). This accumulation is easily visualized in patient-derived fibroblasts using a fluorescent dye called filipin, which is used as a primary assay in the diagnosis of NPC (McKay Bounford and Gissen 2014).
More than 200 disease-causing mutations have been identified in NPC1 that define a heterogeneous mutational spectrum that includes missense and nonsense mutations, small duplication, deletion and insertion mutations, and splice-site mutations (Millat et al. 2001; Tarugi et al. 2002; Park et al. 2003; Scott and Ioannou 2004; Fernandez-Valero et al. 2005). The primary source material used to understand NPC pathology in humans is patient-derived fibroblasts (Greer et al. 1999; Millat et al. 2001; Yamamoto et al. 2004; Gelsthorpe et al. 2008; Zampieri et al. 2012; Rauniyar et al. 2015). The majority of patients with NPC, however, present as compound heterozygotes that often harbor at least one private mutation. This presents a challenge in understanding the molecular mechanisms of disease underlying individual NPC1 variants, leaving most documented mutations as variants of uncertain significance. Further complicating variant interpretation, it has been shown that variant pathogenicity is contingent on level of expression. Specifically, certain variants that are pathogenic at physiologically relevant expression levels can rescue disease phenotypes when artificially overexpressed (Gelsthorpe et al. 2008; Zampieri et al. 2012).
The advent of CRISPR/Cas9-based genome editing has allowed for modifications to genomes with a precision and efficiency unparalleled by previous technologies (Mali et al. 2013a). In brief, CRISPR/Cas9-based genome editing relies on a guide RNA programmable bacterial endonuclease, Cas9, to induce a targeted DNA double-stranded break (DSB). In the absence of a repair template, this break is predominantly repaired by nonhomologous end joining (NHEJ), which is stochastic and leads to small insertions or deletions (Jinek et al. 2012; Cho et al. 2013; Mali et al. 2013b). Typically, even when a repair template is exogenously supplied, NHEJ is responsible for the majority of genome editing outcomes with CRISPR/Cas9, making the establishment of models with specifically designed modifications inefficient. Recently, this challenge has been addressed with the introduction of CRISPR/Cas9-mediated base editing, which uses a nucleobase deaminase enzyme fused to a catalytically impaired Cas9 enzyme capable of inducing only single-stranded breaks (Rees and Liu 2018). These nucleobase deaminase enzymes, APOBEC1 and TadA for cytosine and adenine base editing, respectively, operate on single-stranded DNA (ssDNA), exclusively (Komor et al. 2016; Gaudelli et al. 2017). Similar to traditional CRISPR/Cas9-based genome editing, this fusion protein can be targeted to a guide RNA–specified genomic locus. When the guide RNA binds to its target sequence, the complementary strand is displaced, becoming available for modification by the deaminase enzyme (Nishimasu et al. 2014). In practice, only a portion of the displaced “R loop” is prone to deamination with the current generation of CRISPR/Cas9 base editors, corresponding to an ∼5-bp editing window located 13–17 bp upstream of the protospacer-adjacent motif sequence (PAM) (Komor et al. 2016; Gaudelli et al. 2017; Rees et al. 2017). Deamination of cytosine produces uridine, which base pairs as thymidine, whereas deamination of adenosine produces inosine, which has base-pairing preferences equivalent to guanosine (Yasui et al. 2008). The single-stranded nick produced on the unedited strand by the Cas9 enzyme then induces endogenous DNA repair pathways that will use the edited strand as a template, effectuating either a C•G-to-T•A or an A•T-to-G•C base pair transition.
Here, we aimed to show that by CRISPR/Cas9-mediated NPC1 gene editing, the HAP1 cell line, a human near-haploid cell line, can serve as an effective model of NPC. By doing so, we present a highly efficient approach to resolve the clinical interpretations of NPC1 variants that extends to those both seen and not yet seen in the clinic.
Results
Generation and characterization of an NPC1-deficient near-haploid cell line
As NPC is an autosomal recessive disorder, cellular disease modeling requires the disruption of each allele present in the target cell type. This presents a challenge given the diploid or often aneuploid nature of typical human cell lines, especially if uniform allele disruption is desired. HAP1 cells, however, are a near-haploid human cell line containing a single copy of each chromosome, with the exception of a heterozygous fragment of Chromosome 15, making them an excellent system for loss-of-function disease modeling (Carette et al. 2011). We used CRISPR/Cas9-mediated gene targeting to generate NPC1-deficient HAP1 cells. To disrupt NPC1 expression, we selected several single guide RNAs (sgRNAs) with minimal computationally predicted off-target activity that target exon 21 of the NPC1 locus. These sgRNAs were cloned into plasmids, allowing coexpression with Streptococcus pyogenes Cas9 (SpCas9), and tested for editing efficiency. The two most highly active sgRNAs were transfected separately into wild-type HAP1 cells (Fig. 1A; Supplemental Table S1). Following 72 h of antibiotic selection to enrich for successfully transfected cells, isogenic clones were isolated by limited dilution and screened by Sanger sequencing for locus disruption. Out of 15 clones screened, six isogenic cell clones were identified with unique mutations in the targeted locus, and three clones were carried forward for further characterization. A 28-bp deletion was detected in the first clone (NCBI reference sequence: NG_012795.1 NPC1: g.54927_54954del), resulting in a frame-shift and the formation of a premature stop codon six amino acids downstream from the deletion site (Fig. 1B). The second clone (NG_012795.1 NPC1: g.54902insA) harbored an insertion of an adenine nucleotide at the predicted DSB site, resulting in a frameshift and the formation of a premature stop codon four amino acids downstream from the insertion (Fig. 1C). The third clone (NG_012795.1 NPC1: g.54899_54904del) contained an in-frame 6-bp deletion (Fig. 1D). To assess whether these mutations were sufficient to disrupt NPC1 expression, we performed a western blot for NPC1 protein. Both clones with frameshift mutations showed a complete absence of NPC1 protein, whereas the third clone showed residual protein expression and appeared to run as a doublet, with a second band at a slightly lower molecular weight (Fig. 1E). For the first two clones, this indicates that both frameshift mutations are sufficient in generating null NPC1 alleles. In the third clone, the doublet staining of NPC1 likely indicates a heterogeneously glycosylated protein product, a phenomenon that has been previously reported for a variety of NPC variants (Watari et al. 1999; Zampieri et al. 2012; Nakasone et al. 2014), and the reduced expression indicates that perturbations to the NPC1 locus, in spite of a preserved reading frame, can negatively influence protein expression. Our three cell clones, with their disrupted NPC1 protein expression, represent the first haploid cell models of NPC.
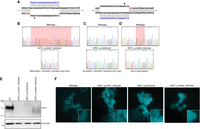
Generation and characterization of three haploid models of Niemann–Pick disease type C (NPC). (A) Diagrams illustrating the two targeted sites in NPC1. Arrowheads indicate the predicted DSB site. (B–D) Sequencing chromatographs showing wild-type NPC1 (top) and the specific disruption in each isogenic edited cell clone (bottom). (B,D) Red highlighted region indicates the locations of the deletions in edited clones. (C) Red highlighted region indicates the position of the insertion in the edited clone. (E) Western blot analysis from total protein lysate from wild-type HAP1 cells and the three edited cell clones illustrating absent or reduced NPC1 protein expression. Actin beta was used as a loading control. (F) Filipin staining reveals deposits of intracellular cholesterol in edited cells that are absent in wild-type cells. White dashed-bordered box has been enlarged twofold and inset at bottom right. Scale bars, 6.3 μm.
A hallmark biochemical feature of NPC pathology is the accumulation of unesterified cholesterol and glycolipids within late endosomes and lysosomes. Presently, the demonstration of defective intracellular cholesterol transport and homeostasis is considered the most definitive functional diagnostic test for NPC (McKay Bounford and Gissen 2014). This defect is readily visualized in NPC patient fibroblasts by staining with the fluorescent compound filipin, which stains unesterified cholesterol deposits (Supplemental Fig. S1). We assessed whether the NPC1-deficient HAP1 cells displayed a similar biochemical phenotype using filipin staining. All three cell clones display distinct foci of intracellular filipin staining that are absent in unedited HAP1 cells, indicative of impaired trafficking of unesterified cholesterol in the NPC1-deficient HAP1 cells (Fig. 1F). The development of disease-relevant pathology in NPC1-deficient HAP1 cell clones shows the potential of these cells in understanding disease mechanisms in NPC.
Modeling NPC1 variants in a haploid cell model using CRISPR/Cas9-mediated base editing
After showing the effectiveness of HAP1 cells in recapitulating a primary cellular phenotype of NPC, we sought to investigate the feasibility of modeling individual NPC1 variants using HAP1 cells. To do this, we elected to use CRISPR/Cas9-mediated base editing. CRISPR/Cas9-mediated base editing technologies are capable of targeted single-nucleotide transitions within a designated editing-window upstream of the PAM sequence (Komor et al. 2016; Gaudelli et al. 2017). We selected mutations that span the NPC1 protein and that are representative of the NPC1 mutation spectrum, including missense, nonsense, and synonymous mutations and a splice site mutation. We selected variants that have or have not been previously documented in clinical databases, as well as variants of both known and unknown pathogenicity. The 19 variants modeled are documented in Table 1. Although we focused on the C-terminal luminal loop domain, spanning residues 855–1098 of NPC1, where 45% of NPC patient mutations occur (Greer et al. 1999; Li et al. 2017), we selected sgRNAs to establish at least one mutation in each of five functional protein domains (Davies and Ioannou 2000). These sgRNA cassettes were cloned into a U6-driven expression vector and individually cotransfected into wild-type HAP1 cells alongside either a Cas9 cytosine or adenine base editor plasmid (Koblan et al. 2018; Nishimasu et al. 2018; Zafra et al. 2018; Huang et al. 2019). Following antibiotic selection, transfected cells were subject to limited dilution to isolate isogenic clones. In each case, editing was apparent in a bulk population, ranging from 10% to 48% (Supplemental Fig. S2). When clones were individually screened by Sanger sequencing by analyzing the genomic sequence ranging from at least 100 bp both upstream of and downstream from the sgRNA binding site, between 8% and 60% of isogenic clones were positively edited (Table 1). Our system for model generation resulted in clonal isolation in just >2 wk with an average frequency of positive clone-selection of ∼27% (Table 1; Fig. 2A). The editing in all but two of the 19 variants isolated was contained to a single codon. During the generation of the NPC1 p.R1077X variant, the editing window contained a second cytosine adjacent to the targeted cytosine, and in our screened clones, we were only able to identify clones in which both bases were edited. The secondary mutation, however, is a silent mutation in which the adjacent histidine codon has been changed from TAC to TAT, resulting in an NPC1 p.Y1076=/R1077X cell line (hereafter referred to as NPC1 p.R1077X) (Supplemental Fig. S3). Similarly, when isolating the NPC1 p.I1061T variant, an adjacent adenine two bases upstream was uniformly targeted in all edited clones, resulting in a secondary silent mutation and an NPC1 p.L1060=/I1061T cell line (hereafter referred to as NPC1 p.I1061T). For the rest of the NPC1 variants, however, multiple clones were isolated with editing contained to a single codon (Table 1; Supplemental Fig. S3). These data show that both CRISPR/Cas9-mediated cytosine and adenine base editing are highly efficient in HAP1 cell model generation, providing a viable solution to the documented poor efficiency of introducing single-nucleotide variants (SNVs) by typical CRISPR/Cas9-based homology-directed repair, which is particularly inefficient in HAP1 cells (Findlay et al. 2018).
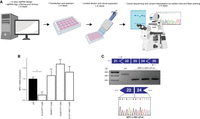
NPC1 expression in NPC variant cell models. (A) Schematic overview of the process of cell model generation and characterization. (B) Expression of NPC1 mRNA is significantly decreased in the NPC1 p.R1077X cell model (n = 3, P = 0.002 by two-tailed t-test) but is unchanged in all other variants assayed. (C, top) Diagram illustrating PCR assay used to analyze splicing. (Middle) PCR amplification results in a shorter amplicon in NPC1 c.3591+2T>C cells compared with the wild type. (Bottom) Sequencing chromatogram from NPC1 c.3591+2T>C cells showing exclusion of exon 23.
Summary of NPC1 mutations modeled
Haploid cell models of NPC1 variants allow for variant characterization and clinical interpretation
The majority of NPC patients are compound heterozygotes and often carry at least one private mutation (Park et al. 2003; Fernandez-Valero et al. 2005). As a consequence, it remains challenging to attribute a specific molecular mechanism of disease to an individual NPC1 variant. By using our haploid models, we sought to characterize the 19 aforementioned NPC1 variants. First, we assayed the expression of NPC1 mRNA in four of the cell models—NPC1 p.D945N, NPC1 p.R1077X, NPC1 p.D1097N and NPC1 c.3591+2T>C—in which variant interpretation has been previously documented. Although there was a trend toward increased NPC1 mRNA expression in NPC1 p.D945N, NPC1 p.D1097N, and NPC1 c.3591+2T>C compared with the wild type, no measurement reached significance (Fig. 2B). This aligns with previously reported data from NPC patient fibroblasts, in which select missense mutations and in-frame deletion mutations have been shown to result in modestly increased NPC1 mRNA expression (Yamamoto et al. 2004; Gelsthorpe et al. 2008). In the NPC1 p.R1077X cell model, however, there was a significant reduction in NPC1 mRNA expression (25.3% ± 14%, P = 0.002, n = 3) (Fig. 2B). We suspect the reduction in NPC1 mRNA expression is the result of nonsense mediated decay owing to the introduced premature stop codon, as previously reported for other nonsense variants in NPC and a variety of other genetic disorders (Frischmeyer and Dietz 1999; Macias-Vidal et al. 2009). The modeled splice-site mutation, NPC1 c.3591+2T>C, is predicted to disrupt canonical splicing of the NPC1 transcript. To assess splicing, we designed a cDNA-based PCR assay that amplified a region between exons 21 and 25 (Fig. 2C). In our assay, amplification of the NPC1 c.3591+2T>C splice-site mutation model resulted in a band ∼100 bp shorter than the wild-type amplicon (Fig. 2C). By Sanger sequencing, we confirmed that the shorter amplicon was indeed the result of exon 23 (114 bp in length) exclusion (Fig. 2C).
Next, by analyzing NPC1 protein expression via western blot, we found that mutant NPC1 expression varied in apparent molecular weight and level of expression. Six of the 19 variants—NPC1 p.R4H, NPC1 p.F402P, NPC1 p.Y634=, NPC1 p. I635T, NPC1 p.V1158=, and NPC1 p.M1159T—ran with an equivalent molecular weight to wild-type NPC1 protein (Fig. 3). Each of these variants had similar NPC1 expression compared with the wild type, with exception of NPC1 p.F402P, which had a moderate reduction in protein expression. Seven of the 19 variants—NPC1 p.E406G, NPC1 p.Y634C, NPC1 p.G765K, NPC1 p. L1060P, NPC1 p.I1061T, NPC1 p.D1097N, and NPC1 c.3591+2T>C—ran as a single band at a lower molecular weight than wild-type NPC1 protein (Fig. 3) and showed reduced expression compared with the wild type. Of note, the reduction in protein expression found in the NPC1 c.3591+2T>C implies that despite the exon 23 exclusion leaving the open reading frame intact, the protein is likely being targeted for degradation. Four of the 19 variants—NPC1 p.F402S, NPC1 p.L785S, NPC1 p.D898N, and NPC1 p.D945N—showed a reduction in total protein and ran as two bands, one equivalent to the wild type and the other equivalent to that found in the lower-molecular-weight mutants (Fig. 3). The distinct molecular weights found in a subset of the mutant variants modeled is consistent with findings in patient-derived fibroblasts and have been attributed to endoplasmic reticulum–associated protein degradation and heterogeneous glycosylation (Watari et al. 1999; Yamamoto et al. 2000; Millat et al. 2001; Gelsthorpe et al. 2008; Zampieri et al. 2012; Nakasone et al. 2014). The NPC1 p.R1077X model, consistent with the reduced mRNA expression, showed a complete absence of NPC1 protein (Fig. 3). In preliminary assays, the NPC1 p.Y1267C model also displayed a total absence of NPC1 protein (Fig. 3). The amino acid change for this variant, however, occurs within immunogen sequence of the primary C-terminal antibody. Upon further analysis with an N-terminal antibody, we found that the NPC1 p.Y1267C protein ran similarly to the wild type (Supplemental Fig. S4). Having observed different levels of expression and migration patterns in our models, we assayed cholesterol homeostasis in each of the cell lines (Fig. 4; Supplemental Fig. S5). With the exception of NPC1 p.L785S, all variants that ran either as a single lower-molecular-weight band or a doublet showed defective cholesterol trafficking indicated by the distinct foci of cholesterol deposits revealed by filipin staining. Despite a marked reduction in NPC1 protein, the NPC1 p.L785S model appeared largely indistinguishable from wild-type HAP1 cells by filipin staining, with only a minor subset (∼5%) of cells showing lysosomal cholesterol accumulation (Fig. 4). It is likely that this variant represents what has been well documented in a minority of NPC patients, a variant biochemical phenotype. In these biochemical variants, filipin staining of patient-derived fibroblasts is less definitive (Vanier et al. 1991). Six of the 19 variants—the two variants harboring silent mutations, NPC1 p.Y634= and NPC1 p.V1158=, and the missense mutants NPC1 p.R4H, NPC1 p.I635T, NPC1 p.M1159T, and NPC1 p.Y1267C—appeared comparable to wild type (Supplemental Fig. S5).

NPC1 expression varies across haploid cell models of NPC. Expression of NPC1 protein was measured via western blot for all NPC1 variants modeled. Actin beta was used as a loading control.
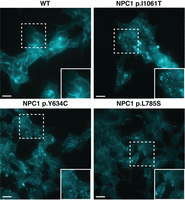
Filipin staining of haploid models of NPC1 variants. Wild-type (WT) HAP1 cells (top left) displayed no defect in cholesterol trafficking. Pathogenic variants, NPC1 p.I1061T and NPC1 p.Y634C (top right and bottom left, respectively), show distinct foci of filipin staining indicative of a defect in cholesterol trafficking. NPC1 p.L785S (bottom right) was found to show a biochemical variant phenotype, with less definitive filipin staining. White dashed-bordered box has been enlarged twofold and inset at bottom right. Scale bars, 13 μm.
Taken together, these data provide strong functional evidence for the clinical pathogenicity of each variant modeled, summarized in Table 1. Critically, our data resolve the clinical interpretation of three variants presently documented as variants of uncertain significance—NPC1 p.Y634C, NPC1 p.D898N, and NPC1 p.D945N, indicating that each variant is pathogenic (Landrum et al. 2016). For the remaining variants, we have either confirmed existing clinical interpretations or made the first interpretation of clinical significance. Together, these data show the utility of our haploid models of NPC1 variants in the delineation of disease mechanisms and the interpretation of clinical variants.
Discussion
By using CRISPR/Cas9-based genome editing, we have developed the first haploid cell model of NPC. Our model recapitulates the primary biochemical and diagnostic phenotype found in patient-derived fibroblasts. Although other human cells lines deficient in NPC1 expression have been reported (Rodriguez-Pascau et al. 2012; Du et al. 2017; Tharkeshwar et al. 2017; Zhao and Ridgway 2017), these have all been diploid or aneuploid, without established uniform allelic disruption, and NPC1 deficiency confirmed by immunoblotting only. By using CRISPR/Cas9-mediated gene editing, we obtained two unique NPC1-null cell models with indels resulting in verified coding frameshifts, and one NPC1-deficient cell model containing a 6-bp in-frame deletion. Similar to NPC patient–derived fibroblasts, these models display a distinct defect in cholesterol trafficking, resulting in the accumulation of unesterified cholesterol. To date, greater than 200 disease-causing mutations in NPC1 have been reported. Accordingly, it is common for NPC patients to be compound heterozygotes, often harboring at least one private mutation. The precise nature of a given mutation in NPC1 can vary widely, including both missense and nonsense mutations, splice-site mutations, small duplication mutations, and indel mutations (Millat et al. 2001; Tarugi et al. 2002; Park et al. 2003; Fernandez-Valero et al. 2005). Despite the vast diversity found in the NPC1 mutation spectrum, most research detailing the molecular mechanisms of NPC have been focused on one mutation, NPC1 p.I1061T (Gelsthorpe et al. 2008; Rauniyar et al. 2015; Schultz et al. 2018). As this allele accounts for between 15% and 20% of all NPC disease alleles (Davies and Ioannou 2000; Millat et al. 2001; Park et al. 2003), it is readily available in homozygous patient fibroblasts, a unique occurrence given the well-documented heterogeneity of NPC1 mutations. A detailed understanding of the majority of NPC1 variants using patient-derived fibroblasts, however, remains challenging as they are rarely found in isolation.
Here, we have developed a platform that allows for the rapid generation and analysis of NPC1 variants. Specifically, our approach allows for the isolation and expansion of mutant cell clones in <14 d. Once established, this system requires only the ordering and cloning of a single pair of oligos encoding a unique sgRNA for each new mutation of interest. By using CRISPR/Cas9-mediated base editing, we modeled 19 unique NPC1 variants, showing the utility of the system in terms of variant characterization and clinical interpretation and expanding our understanding of the genotype–phenotype relation in NPC1. The 19 modeled variants show varied outcomes of NPC1 mutations at the level of mRNA and protein, suggesting different mechanisms of pathogenicity. As such, our results emphasize the need for a mutation-by-mutation analysis of the NPC1 gene, which could lead to further insights into basic NPC1 function and help identify unique therapeutic avenues.
CRISPR/Cas9-mediated base editing in HAP1 cells is so efficient that, by screening only a moderate number of clones, one can isolate multiple positive colonies. Recently, two studies have documented off-target editing events as a result of cytosine base editors (Jin et al. 2019; Zuo et al. 2019). It was shown that these off-target events were independent of sgRNA sequence and were enriched in highly transcribed regions. Accordingly, we performed an analysis on three independent cell clones for each variant of interest, minimizing the potential that any observed phenotype to be off-target dependent. Furthermore, we performed RNA sequencing on the parental HAP1 line and nine edited cell clones representing the three biological replicates from three of our modeled variants, NPC1 p.R1077X, NPC1 p.D1097N, and NPC1 c.3591+2T>C. By using these sequencing reads, we performed variant analysis evaluating the distribution of mutations throughout the cell clones. Although we found between nine to 20 mutations in each clone that were not present in the parental cell line, no single mutation was shared by multiple cell clones indicating that the observed cholesterol accumulation phenotype is independent of potential off-target base editing in the transcriptome (Supplemental Fig. S6A). Additionally, all mutations found were outside of the NPC1 gene with exception of the desired on-target mutation. Furthermore, of the mutations found there was no apparent bias toward C-to-T or G-to-A substitutions in clones generated using cytosine base editing and no apparent bias toward A-to-G or T-to-C substitutions in clones generated using adenine base editing (Supplemental Fig. S6B). This indicates the mutations observed are unlikely true off-targets but are likely a result of random mutation events.
The American College of Medical Genetics guidelines classify the results of functional assays as strong evidence for or against variant pathogenicity (Richards et al. 2015). We have shown that NPC1 perturbation leads to a readily visualized defect in cholesterol trafficking in HAP1 cells. This makes haploid models of NPC1 variants an ideal system for clinical variant interpretation. We showed this by confirming the clinical interpretation of four documented NPC1 variants, resolving the clinical interpretation of three NPC1 variants of uncertain significance and establishing the clinical interpretation of 12 presently undocumented NPC1 variants.
Given the overt phenotype induced by NPC1 perturbation, it can be envisioned that, if appropriately scaled, a CRISPR/Cas9-mediated base editing screen using HAP1 cells could serve as an effective platform for the clinical interpretation of a multitude of NPC variants. Presently, there are 1839 nonsynonymous NPC1 mutations documented in the gnomAD database (Lek et al. 2016), 56% of which, are the result of transitional nucleotide substitutions. In the present study, we showed efficient targeting using multiple engineered SpCas9 variants (Table 1). Indeed, with the expanding list of available Cas9 enzymes, each with a unique PAM consensus sequence (Hu et al. 2018; Yang et al. 2018; Hua et al. 2019), it is possible that the vast majority of documented NPC1 variants could be modeled using CRISPR/Cas9-mediated base editing. Furthermore, base editing systems are not strictly limited to transitional nucleotide substitutions, which further expands their utility in variant interpretation. Although occurring in a less predictable manner, the targeted activation-induced deaminase (AID)-mediated mutagenesis system allows for all nucleotide substitutions of a cytosine (Ma et al. 2016), and the CRISPR-X system allows for all substitutions of both cytosines and guanines (Hess et al. 2016; Ma et al. 2016). More recently still, it was shown that adenine base editors are capable of cytosine deamination, ultimately resulting in both cytosine-to-guanine or cytosine-to-thymine substitutions (Kim et al. 2019).
Presently, our approach is limited by the editing window of current base editing technologies, which is ∼5 bp wide (Komor et al. 2016, 2017; Rees et al. 2017). Although the percentage of total modelable mutations are limited by the currently permissible editing, efforts are underway to engineer enzymes with a much narrower editing window (Tan et al. 2019). We noted at least one editing event with nucleotide substitutions occurring well outside of the predicted editing window for both SpCas9 cytosine and SpCas9 adenine base editors (Supplemental Fig. S3). Although these events were in the minority, they suggest the boundaries of the SpCas9 base editing window are not absolute. Accordingly, it may be possible to design enzymes with a shifted editing window, further expanding the catalog of targetable mutations.
In this study, by using CRISPR/Cas9-mediated gene editing, we have generated and characterized three models of NPC1 deficiency, which represent the first human haploid cell models of NPC. We showed that these models effectively capture the principle diagnostic readout of NPC. The sheer number of NPC1 variants, combined with most patients being compound heterozygotes, presents a challenge to understanding the disease mechanisms underlying individual patient variants. To overcome this challenge, we sought to model unique NPC1 variants in HAP1 cells. We showed that this is readily achievable using CRISPR/Cas9-mediated base editing. Finally, we showed that our haploid models of NPC1 variants allow for efficient variant characterization and clinical interpretation. Although we have focused on NPC, it is worth of note that the largest class of known pathogenic mutations in humans are point mutations (Landrum et al. 2014, 2016). Given the ease and efficiency of our approach, one can envision applying this strategy to a variety of genetic disorders for which a suitable cellular phenotype exists, thus providing a platform for the establishment of detailed genotype–phenotype correlations.
Methods
Cell culture and transfection
HAP1 cells were obtained from Horizon Genomics. HAP1 cells were cultured in Iscove's Modified Dulbecco's Medium (Gibco) supplemented with 10% fetal bovine serum, 1% L-glutamine, and 1% Pen-Strep (Gibco). Patient-derived fibroblasts were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum, 1% L-glutamine, and 1% Pen-Strep (Gibco). We seeded 500,000 HAP1 cells for transfection in 2 mL of media in six-well plates using Lipofectamine 3000 (Thermo Fisher Scientific). Cells were transfected with 1250 ng of a Cas9 base editor expression vector and with 1250 ng of a sgRNA expression vector containing a puromycin-resistance gene. The sgRNA expression vector was generated as follows: A puromycin-resistance gene was PCR amplified from the pSpCas9(BB)-2A-Puro (PX459) V2.0 plasmid, which was a gift from Feng Zhang (Addgene plasmid 62988), appending an AgeI cut site to the 5′ end of the amplicon. This amplicon was inserted into the pX600-AAV-CMV::NLS-SaCas9-NLS-3xHA-bGHpA plasmid, which was a gift from Feng Zhang (Addgene plasmid 61592), replacing the SaCas9 coding region and resulting in a CMV-driven puromycin-resistance gene. By using In-Fusion cloning (ClonTech), this puromycin-expression cassette was inserted into an EcoRI linearized BPK1520 plasmid, which was a gift from Keith Joung (Addgene plasmid 65777). The pLenti-FNLS-P2A-Puro plasmid used for a portion of the cytosine base editing experiments was a gift from Lukas Dow (Addgene plasmid 110841). The pSI-Target-AID-NG plasmid used for the remaining cytosine base editing experiments was a gift from Osamu Nureki (Addgene plasmid 119861). The pCMV_ABEmax_P2A_GFP, VRQR-ABEmax, NG-ABEmax plasmids used for adenine base editing experiments were gifts from David Liu (Addgene plasmids 112101, 119811, 124163). To enrich for transfected cells, 24-h post-transfection cells were subjected to 0.8 µg/mL of puromycin for 72 h. Transfected cells were expanded for genomic DNA isolation and limited dilution as previously described (Essletzbichler et al. 2014).
Genomic DNA isolation and PCR
Genomic DNA was isolated using the DNeasy blood and tissue kit (Qiagen) according to the manufacturer's protocol. PCR was performed using DreamTaq polymerase (Thermo Fisher Scientific) according to the manufacturer's protocol.
Estimation of genome editing
PCR amplification from the genomic DNA of a bulk population of edited cells centered on the predicted editing site was performed. These amplicons were PCR purified using a QIAquick PCR purification kit following the manufacturer's protocol (Qiagen) and Sanger sequenced using the forward primer. To test guide efficiency, the Sanger sequencing files from unedited and edited cells were used as an input into the online sequence trace decomposition software, TIDE (Brinkman et al. 2014). To ascertain base editing percentage, the Sanger sequencing AB1 files were input into the online base editing analysis software, editR (Kluesner et al. 2018).
Protein isolation and western blot analysis
HAP1 cells were trypsinized from their well, pelleted, and washed three times with 1× PBS (Gibco). Protein was isolated from HAP1 cells by resuspending in 150 µL of a one-to-one solution of RIPA homogenizing buffer (50 mM Tris HCl at pH 7.4, 150 nM NaCl, 1-mM EDTA) and RIPA double-detergent buffer (2% deoxycholate, 2% NP-40, 2% Triton X-100 in RIPA homogenizing buffer) supplemented with a protease-inhibitor cocktail (Roche). Cells were subsequently incubated on ice for 30 min. Cells were then centrifuged at 12,000g for 15 min at 4°C, and the supernatant was collected and stored at −80°C. Whole-protein concentration was measured using a Pierce BCA protein assay kit according to the manufacturer's protocol (Thermo Fisher Scientific). SDS-Page separation was completed by running 2 μg of total protein on a NuPAGE 3%–8% Tris-acetate gel (Thermo Fisher Scientific). Next, proteins were transferred to a nitrocellulose membrane using the iBlot 2 transfer apparatus (Thermo Fisher Scientific). A 5% milk solution in 1× TBST was used for blocking for 1 h at room temperature. The membrane was then incubated with the NPC1 primary antibody (Abcam ab106534 in main body figures or Novus Biologicals H00004864-M02 in Supplemental Fig. S4) overnight at 4°C. Primary antibody solution was removed, and the membrane was washed three times with 1× TBST. This was followed by a 1-h incubation at room temperature with horseradish peroxidase conjugated goat anti-rabbit IgG (Abcam: ab6721). After three washes with 1× TBST, signal detection was achieved using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) according to the manufacturer's protocol.
Filipin staining
Coverslips were incubated for 1 h at 37°C in 1 mL of 1:30 solution of collagen I rat protein (Thermo Fisher Scientific) to 1× PBS. The collagen solution was then aspirated, and 250,000 HAP1 cells were seeded onto the coverslips in 12-well plate 24 h before staining. Cells were washed three times with 1× PBS and then fixed in 4% paraformaldehyde in 1× PBS for 30 min at room temperature. After fixation, cells were washed three times in 1× PBS and then stained with 50 μg/mL of filipin III (Sigma-Aldrich) for 1 h at room temperature. Coverslips were then washed three times in 1× PBS and mounted onto a slide using ProLong Gold Antifade Mountant (Thermo Fisher Scientific). Cells were visualized using the Zeiss Axiovert 200M epifluorescent microscope, and images were captured with a Hamamatsu C4742-80-12AG camera.
RNA isolation and quantitative PCR
Cells were harvested, washed three times with 1× PBS, pelleted, and then resuspended in TRIzol reagent (Thermo Fisher Scientific). Isolation of mRNA was isolated following the manufacturer's protocol. Next, 500 ng of mRNA was reverse transcribed using SuperScript III reverse transcriptase (Thermo Fisher Scientific) following the manufacturer's protocol. Quantitative PCR using fast SYBR Green master mix (Qiagen) on a StepOnePlus real-time PCR (Applied Biosystems) was performed. NPC1 expression was analyzed by amplification using a forward primer spanning the junction of exon 12 and 13 and using a reverse primer specific to exon 13. Primers against endogenous GAPDH were used as an internal control. ΔΔCt was analyzed to assess fold changes between edited and unedited samples.
RNA sequencing and off-target analysis
RNA sequencing was performed by the Centre for Applied Genomics in Toronto using the Illumina HiSeq 2500 system, producing 120-bp paired-end reads. Raw transcript reads were aligned to the GRCh38 human genome using HISAT2 (Kim et al. 2015). The Picard program (v2.21.1; http://broadinstitute.github.io/picard) was used to mark duplicative and sort reads. The Genome Analysis Toolkit (GATK; v4.1.3.0) (Poplin et al. 2018) was used to split the reads that contained Ns in their CIGAR string (McKenna et al. 2010). Variant calling was conducted with both FreeBayes (v1.3.1) and LoFreq (v2.1.3) independently (Garrison and Marth 2012; Wilm et al. 2012). Only variants called by both software programs were considered true variants. Any variants with a read depth of less than four, or an allele frequency less than 0.4, were filtered from the final list of unique mutant variants. The exon coordinates of GRCh38 version 86 were retrieved from the Ensembl database (Hunt et al. 2018), and any called variants falling outside of these coordinates were excluded. The final list of variants was manually inspected using the Integrative Genomics Viewer (IGV) software (Thorvaldsdottir et al. 2013).
Statistical analyses
All graphs were plotted as the mean with error bars indicating standard error. Differences between groups was assessed by two-tailed Student's t-test. P-values < 0.05 were considered statistically significant.
Data access
All sgRNA and primer sequences are available in Supplemental Table S2. The sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA580451.
Acknowledgments
We thank Niemann–Pick Canada and the Marcogliese Family Foundation for their support and commitment to our research into Niemann–Pick disease type C. We thank Ebony Thompson, Sonia Evagelou, and Kyle Lindsay for their technical assistance. Figure 2A was created with Biorender.com. This work was supported by the Rare Disease Foundation and the BC Children's Hospital Foundation (2304 to S.E.), The Hospital for Sick Children (Restracomp scholarship to S.E.), Niemann Pick Canada, and the Marcogliese Family Foundation.
Author contributions: Conceptualization was by S.E., E.A.I., and R.D.C. Methodology was by S.E., R.A.B., T.M.I.B., E.M., L.Z., E.A.I., and R.D.C. Formal analysis was by S.E., R.A.B., T.M.I.B., E.A.I., and R.D.C. Investigation was by S.E., R.A.B., T.M.I.B., E.M., E.A.I., and R.D.C. Resources were by E.A.I. and R.D.C. Data curation was by S.E., R.A.B., T.M.I.B., E.A.I., and R.D.C. Writing, original draft preparation, was by S.E. Writing, review and editing, was by S.E., R.A.B., T.M.I.B., E.M., L.Z., E.A.I., and R.D.C. Supervision was by E.A.I. and R.D.C. Project administration was by E.A.I. and R.D.C. Funding acquisition was by S.E., E.A.I., and R.D.C.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.250720.119.
- Received March 20, 2019.
- Accepted November 1, 2019.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








