
Human long intrinsically disordered protein regions are frequent targets of positive selection
- Arina Afanasyeva1,2,3,4,6,
- Mathias Bockwoldt5,6,
- Christopher R. Cooney1,
- Ines Heiland5 and
- Toni I. Gossmann1
- 1Department of Animal and Plant Sciences, University of Sheffield, Sheffield S102TN, United Kingdom;
- 2Institute of Nanobiotechnologies, Peter the Great St. Petersburg Polytechnic University, Saint-Petersburg 195251, Russia;
- 3Petersburg Nuclear Physics Institute, B.P. Konstantinov NRC Kurchatov Institute, Gatchina, Leningrad District 188300, Russia;
- 4National Institutes of Biomedical Innovation, Health and Nutrition, Ibaraki City, Osaka 567-0085, Japan;
- 5Department of Arctic and Marine Biology, UiT The Arctic University of Norway, 9037 Tromsø, Norway
-
↵6 These authors contributed equally to this work.
Abstract
Intrinsically disordered regions occur frequently in proteins and are characterized by a lack of a well-defined three-dimensional structure. Although these regions do not show a higher order of structural organization, they are known to be functionally important. Disordered regions are rapidly evolving, largely attributed to relaxed purifying selection and an increased role of genetic drift. It has also been suggested that positive selection might contribute to their rapid diversification. However, for our own species, it is currently unknown whether positive selection has played a role during the evolution of these protein regions. Here, we address this question by investigating the evolutionary pattern of more than 6600 human proteins with intrinsically disordered regions and their ordered counterparts. Our comparative approach with data from more than 90 mammalian genomes uses a priori knowledge of disordered protein regions, and we show that this increases the power to detect positive selection by an order of magnitude. We can confirm that human intrinsically disordered regions evolve more rapidly, not only within humans but also across the entire mammalian phylogeny. They have, however, experienced substantial evolutionary constraint, hinting at their fundamental functional importance. We find compelling evidence that disordered protein regions are frequent targets of positive selection and estimate that the relative rate of adaptive substitutions differs fourfold between disordered and ordered protein regions in humans. Our results suggest that disordered protein regions are important targets of genetic innovation and that the contribution of positive selection in these regions is more pronounced than in other protein parts.
There is substantial experimental evidence that proteins or protein regions may be deprived of a specific three-dimensional structure (Daughdrill et al. 2005; Oldfield and Dunker 2014) and instead exist as intrinsically disordered proteins or protein parts (IDPs). The lack of a particular conformation may be an advantage where structural flexibility is required, for example for multifunctional proteins or for functional flexibility and regulation (Tompa et al. 2005; Oldfield et al. 2008; Hsu et al. 2013). In some cases, IDP regions can adopt a specific three-dimensional structure when environmental conditions change, for example, as entropic bristles (Santner et al. 2012), entropic springs (Smagghe et al. 2010), and entropic clocks (Zandany et al. 2015), or when binding to protein partners (Daughdrill et al. 1997). Experimental evidence suggests that real time state transition between ordered and disordered states can occur (Mohan et al. 2006), illustrating that protein structures are not static arrangements, as they are generally perceived (Ahrens et al. 2017). Protein disorder may also provide a genome-wide mechanism of adaptation to environmental conditions and lifestyle; e.g., host-changing parasites have a higher level of predicted disorder compared to obligate intra-cellular parasites and endosymbionts (Pancsa and Tompa 2012). Other complex organismal roles for disordered regions have been suggested, such as tissue-specific alternative splicing of disordered regions that may alter protein functions and thus change protein–protein interaction networks by recruiting new interaction partners (Buljan et al. 2013). These examples clearly demonstrate that IDPs are a heterogeneous group of protein regions that increase the functional plasticity of proteins and the flexibility of intermolecular interactions in the cell (Buljan et al. 2012; Mosca et al. 2012).
Due to the functional complexity of these protein regions, much research has been conducted to characterize IDPs and explore the underlying evolutionary mechanisms (Brown et al. 2002; Chen et al. 2006a,b; Bellay et al. 2011; Szalkowski and Anisimova 2011; Zea et al. 2013). The molecular rate at which proteins evolve at the DNA level is an important quantity in evolutionary biology and population genetics to determine the selective forces that have shaped protein composition and is generally dominated by selective constraint across many taxa (Gossmann et al. 2014). This is most prominently illustrated by the fact that, in protein coding regions, amino acid changing substitutions occur much less frequently than synonymous changes, i.e., mutations that do not change the amino acid but the underlying codon. Consequently, the rate ratio of these two types of substitutions is often found to be ≪1, illustrating evolutionary constraint at the amino acid level between species. There is, however, compelling evidence that substantial rate heterogeneity in the protein molecular rate exists across the genome and within proteins (Echave et al. 2016). Variation in the rate at which proteins evolve is, but not exclusively, attributed to functional importance, genomic context (e.g., the recombination environment or local effective population size), and structural features. For example, functionally active parts of proteins are more susceptible to selective pressure, and active sites of enzymes evolve significantly slower than other parts of the protein (Dean et al. 2002). On the other hand, parts exposed to the solvent protein surface evolve much faster (Franzosa and Xia 2009). IDPs generally tend to evolve more rapidly, largely attributed to relaxed purifying selection due to the lack of structural constraint (Brown et al. 2011), although Ahrens et al. (2016) found that sites that were predicted to be disordered and to have secondary structure were evolving at a lower rate than sites that were predicted to be ordered and to have secondary structure. Other important evolutionary determinants of the rate at which disordered regions evolve are synonymous constraint elements (Macossay-Castillo et al. 2014) and gene age, as evolutionarily young proteins tend to be enriched in disordered regions (Wilson et al. 2017).
Although our knowledge of the functional complexity and associated molecular pattern of IDPs is increasing, little is known about the role of positive selection in these important protein regions, in particular, in our own species. Rapid evolution of IDPs might suggest that positive selection contributed to the evolution of these protein regions. Disentangling the combined effects of relaxed purifying selection, genetic drift, and positive selection is crucial to identifying the underlying selective forces for the observed rapid evolution. Currently, evidence for positive selection in IDPs stems from comparative work in yeast species (Nilsson et al. 2011) and protein secondary structural elements in six Drosophila species (Ridout et al. 2010). However, the latter study finds evidence for an enrichment of positively selected residues in coiled-coil regions, but not in β-turns—regions that are also associated with protein disorder. Substitution-based tests of positive selection with few species are limited in power (Anisimova et al. 2001) and possibly prone to alignment quality issues (Markova-Raina and Petrov 2011); thus, it remains unclear if certain gene categories have experienced more positive selection than others. In humans, the selective effects of deleterious, but not advantageous, mutations (e.g., within a population genetic framework) in IDP regions have been investigated (Khan et al. 2015). Mutations in these regions experience less selective constraint than other structural protein elements, consistent with the observation that IDPs are rapidly evolving between species. The role of positive selection remained unexplored within this data set to circumvent potential alignment issues (Khan et al. 2015).
Here, we analyze human proteins with long intrinsically disordered regions to disentangle the evolutionary forces that have acted on these protein regions, with emphasis on the rate of positive selection.
Results
To analyze the evolutionary features of human proteins with long IDPs, we apply a comparative phylogenetic approach using publicly available genome data from human and other mammalian genomes. After rigorous alignment preprocessing, we focused on 6663 proteins with high-quality alignments (see Supplemental Methods).
Proteins with disordered regions are functionally associated with intermolecular binding
The vast majority of disordered regions in our protein data set correspond to small fractions of the entire protein (average disordered content is <15% and <100 amino acids) (Supplemental Fig. S1). Generally, these proteins tend to be involved in protein and DNA/RNA binding (Gene Ontology [GO] enrichment analysis) (Supplemental Table S1). Amino acid residues in disordered regions tend to occur predominantly at the surface of proteins (SASA scores) (Supplemental Table S2). Disordered regions are enriched in post-translational modification sites as well as regions and motifs (annotated sequence stretches of biological importance) in comparison to their ordered counterparts (Supplemental Table S3). Disease-associated SNPs tend to occur less frequently in disordered regions, with an exception for musculoskeletal disease-associated SNPs (Supplemental Table S4). This is in keeping with other research (Uversky et al. 2009; Marsh and Teichmann 2011; Gao and Xu 2012; Peng et al. 2015).
Ordered and disordered protein parts differ in their evolutionary rates due to genetic drift and differences in purifying selection
We subdivided our protein alignments into disordered and remaining parts of the protein (referred to hereafter as ordered
protein regions) to separately estimate the molecular evolutionary rates in coding DNA (ω = dN/dS) on a gene-by-gene basis. We found that ω ratios are significantly higher for disordered regions (Fig. 1) in comparison with their ordered counterparts (Wilcoxon signed-rank test, paired, P < 2.2 × 10−16). The difference in ω is largely driven by differences in the substitution rates at nonsynonymous sites (dN, Wilcoxon signed-rank test, P < 2.2 × 10−16), as the difference in substitution rates at synonymous sites (dS) between disordered and ordered regions is less prominent (Wilcoxon signed-rank test, P < 4 × 10−4). Furthermore, dS, a proxy for local mutation rate, is significantly lower in disordered regions (median 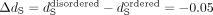 ). This could indicate slight differences in the local mutation rate or differences in selection on synonymous sites between
ordered and disordered sites, although these effects are currently difficult to disentangle (Smith et al. 2018). We estimated the proportion of substitutions fixed by genetic drift (i.e., dN/dS = 1) in ordered and disordered regions along with the intensity of purifying selection for nonneutral sites (Nearly Neutral
Model sites model as implemented in PAML). The proportion of neutrally evolving sites is significantly different between ordered
and disordered regions (Wilcoxon signed-rank test, P < 2.2 × 10−16, with genes with evidence for positive selection removed from the sample) (Fig. 2). We also found that ω for nonneutral sites is higher in disordered regions (median 0.078 compared to 0.041).
). This could indicate slight differences in the local mutation rate or differences in selection on synonymous sites between
ordered and disordered sites, although these effects are currently difficult to disentangle (Smith et al. 2018). We estimated the proportion of substitutions fixed by genetic drift (i.e., dN/dS = 1) in ordered and disordered regions along with the intensity of purifying selection for nonneutral sites (Nearly Neutral
Model sites model as implemented in PAML). The proportion of neutrally evolving sites is significantly different between ordered
and disordered regions (Wilcoxon signed-rank test, P < 2.2 × 10−16, with genes with evidence for positive selection removed from the sample) (Fig. 2). We also found that ω for nonneutral sites is higher in disordered regions (median 0.078 compared to 0.041).
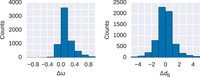
Histograms of paired differences in ω (ω = dN/dS) and dS values in proteins with disordered and ordered protein regions. Shown are the pairwise differences in disordered minus ordered protein regions (Δω and ΔdS) of the same protein. Substitution rates for nonsynonymous (dN) and synonymous (dS) sites are obtained from a one-ratio model. ω values are significantly different for ordered and disordered regions (Wilcoxon signed-rank test, paired, P < 2.2 × 10−16); the difference dS has a greater P-value (P < 4 × 10−4).
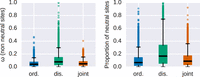
Estimates of sequence evolution in a nearly neutral model. Distributions of ω = dN/dS values (left panel) and the proportion of the neutrally evolved sites (right panel) for ordered (blue), disordered (green), and ordered and disordered protein regions jointly analyzed (orange).
The role of positive selection is substantially more pronounced in disordered regions
To investigate whether elevated ω ratios are additionally caused by the effect of positive selection (e.g., a few sites for which dN/dS > 1), we conducted site-specific dN/dS analyses (Wong et al. 2004; Yang et al. 2005) using a gene-by-gene approach. First, we jointly analyzed ordered and disordered regions and found that 363 proteins (5.4%) showed evidence for positive selection based on a likelihood ratio test (FDR = 0.1) (Table 1). Second, we conducted the same analysis separately for ordered and disordered regions. We found evidence of positive selection in the disordered regions of 377 genes and in the ordered regions of 252 genes (significantly different, Fisher's exact test [2 df], P < 0.001, FDR = 0.1) (Supplemental Table S5). There were 240 genes (64%) with evidence for positive selection from the disordered set that were not identified as positively selected when the protein regions were jointly analyzed (in contrast to ≈11% for the ordered region). Moreover, considering that disordered regions are generally shorter than their ordered counterparts, this difference is likely to be an underestimate, as the power to detect positive selection decreases with shorter protein length (Anisimova et al. 2001). We therefore repeated our analysis by using the same number of sites for ordered and disordered regions by down-sampling to the number of sites of the shorter region. Using this approach, we found a roughly 10-fold (38 versus 363 genes) difference in the number of positively selected genes in ordered regions compared to disordered regions (Fisher's exact test [2 df], P ≪ 0.001) (Table 1). We tested whether alignment scores before applying our alignment-processing pipeline are significantly different between the identified gene sets and find no evidence for that (Supplemental Fig. S2). Taken together, this illustrates that positive selection has played an intensified role in disordered regions and suggests that an a priori distinction of disordered sites substantially increases the power to detect adaptive processes. There is also no evidence that alignment quality issues have artificially created a signature of positive selection.
Number of genes with evidence of positive selection in ordered, disordered, and joint estimates
The detected level of positive selection is likely to be an underestimate
We investigated whether there is sufficient power and accuracy to detect positive selection for the estimated parameter range. For this, we conducted extensive sequence evolution simulations for proteins with a signature of positive selection, as well as 250 randomly chosen proteins from the remaining set. On average, more than 90% of the simulated data sets for which we simulated a signature of positive selection were identified as such and less than 10% were significant when no positive selection was simulated (Supplemental Fig. S3). We observed a slight increase in the detection of positive selection in the simulated data sets for disordered regions where only the corresponding ordered region was initially identified as positively selected. As we do not generally see this phenomenon for disordered regions, it suggests that, for these proteins, the disordered region might be contributing to the signal of positive selection. Possible reasons include a misclassification of ordered and disordered boundaries, heterogeneity of the lengths of ordered and disordered regions across species, and heterogeneity of positively selected sites within the disordered regions. As PAML does not consider gapped positions, we investigated the impact of indels on our alignment-processing pipeline. We additionally simulated protein sequences composed of disordered and ordered regions under varying indel rates, to simulate the alignment difficulties usually observed for disordered regions. Applying the site test to the processed alignments in comparison to the true simulated alignment, we find that, with an increasing indel rate, the power to detect positive selection decreases (Supplemental Fig. S4). The false positive rate stays low and remains on a comparable level when the test is applied on the true alignment. Taken together, these simulations suggest that our alignment pipeline does not artificially create signatures of positive selection but, on the contrary, is rather conservative, and that the true rate of positive selection in disordered regions is likely to be even more pronounced than reported here.
Biological implications of disordered regions with positively selected target sites
We were interested in the functional associations and interactions of the proteins for which the corresponding genes showed evidence of positive selection. We performed a protein network analysis using STRING (Szklarczyk et al. 2017) for protein products of the genes that were uniquely identified to have evolved under positive selection in disordered and ordered regions (322 and 197 genes, respectively). We find that protein interaction networks for the two sets are significantly enriched in interactions but differ drastically in their layout (Supplemental Fig. S5). While the proteins from the ordered set show small-sized clusters with less than eight interaction partners, we find one large-size cluster connecting more than 50 proteins from the disordered set that includes important proteins such as breast cancer type 1 susceptibility protein BRCA1, the telomere binding protein TERF1, involved in telomere length homeostasis, and the NAD-dependent de-acetylase SIRT1 that is an important drug target in aging research (Hubbard et al. 2013). Differences in maximum cluster size are also observed when differences in the protein numbers are accounted for (Supplemental Fig. S6). The interaction network from the disordered gene set showed significant functional enrichment in RNA binding and nucleic acid binding, while the ordered gene set was associated with immune response and T-cell activation (Supplemental Table S6). This suggests an important, and to our knowledge hitherto unattributed, role of positive selection of disordered regions in transcriptional and/or translational regulation and may be driven by co-evolutionary mechanisms in regulatory arms races. Rapid evolution of immune defense genes has been reported in many taxa (Mondragón-Palomino et al. 2002; Viljakainen et al. 2009; Bonneaud et al. 2011; McTaggart et al. 2012) and is often associated with adaptive immune response.
Molecular dynamics of positively selected target sites in IL21
As we applied a site-specific test of positive selection, it is possible to specifically pinpoint amino acid residues that potentially have evolved under positive selection. Unfortunately, three-dimensional structural information for IDPs is difficult to obtain and, hence, substantially underrepresented in respective databases. A systematic study of important residues in these regions on the three-dimensional features is therefore very limited. Out of 7652 residues in IDPs that we classified to be on the surface and, hence, accessible to interaction partners (Supplemental Table S2), 83 residues (1.08%) show evidence for positive selection. In stark contrast, none of the residues identified to be on the surface in the ordered regions show evidence for positive selection, suggesting that the disordered state, but not the ligand accessibility, lead to increased molecular rates. An exception suitable for a more detailed study of three-dimensional properties of positively selected sites in IDPs is one of our candidate genes under positive selection, interleukin 21, a protein that plays a fundamental role in the innate and adaptive immune responses (Yi et al. 2010; Ju et al. 2016). Based on a partly resolved NMR structure of the disordered region, we pinpointed three residues under positive selection (Fig. 3A). This region has been shown to be a part of a helix C motif, which exists in both ordered and disordered states in different conformers and is involved in receptor binding (Bondensgaard et al. 2007). As disordered regions exist as ensembles of structures rather than as a static snapshot, we conducted a molecular dynamic (MD) analysis by simulating molecular movements of the disordered region based on the resolved NMR structure (PDB: 2OQP) for 200 nanoseconds (nsec) in explicit water solvent (Fig. 3B). This allows us to investigate the effect of the three identified residues on the structural flexibility within this protein region (Rajasekaran et al. 2011; Papaioannou et al. 2015). All three residues (S81, G85, R91) are located in a highly flexible region according to B-factor values, suggesting that these sites contribute to the structural flexibility in the disordered state (Radivojac et al. 2004). Comparing these positions across species (Supplemental Fig. S7), we find at least three different variant types at each site and with the exception of Tarsius syrichta (I91), none of these variants is a protein order-promoting amino acid (Oldfield and Dunker 2014). The MD simulation also reveals three neighboring residues of high flexibility at positions 61–64 outside of the annotated disordered region (Fig. 3B). The initial study by Bondensgaard et al. (2007) reported a segment from position 57 to 84 as regions of two interchangeable conformers (i.e., disordered region), adding to the notion that precise boundaries of disordered regions are difficult to capture with different methods.
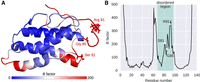
Three-dimensional features of positively selected sites in the disordered region of human interleukin 21. (A) Cartoon of the NMR structure of human interleukin 21 (PDB Code: 2OQP) including its disordered region indicating B-factor scores from the molecular dynamics analysis. Three residues have been identified as positively selected in a PAML branch-site test (Ser81, Gly85, and Arg91) in the disordered region. (B) Molecular dynamics analysis; shown are the B-factors of all residues. Here, B-factors reflect the fluctuation of single amino acids (Cα atom) about their average positions during the MD simulation. The predicted disordered region by MobiDB is indicated in green as well as the three identified residues under positive selection (S81, G85, and R91).
The distribution of fitness effects differ between ordered and disordered protein regions in humans
To get a better understanding of the role of genetic drift and the purifying selective forces currently acting in IDP regions, we used polymorphism data from the 1000 Genomes Project (The 1000 Genomes Project Consortium 2015). The rate ratio of nonsynonymous to synonymous diversity (πN/πS) in coding regions can be regarded as a rough indicator of the effectiveness of negative selection on amino acid-changing mutations, with larger values indicating less effective purifying selection (Chen et al. 2017). Mean πN/πS is increased for disordered regions compared to their ordered counterparts (0.277 versus 0.17, Wilcoxon signed-rank test, P < 3.9 × 10−18) (Fig. 4A). We inferred the distribution of fitness effects (DFE) of new amino acid-changing mutations by applying a method that uses the frequency distribution of mutations at nonsynonymous sites relative to synonymous sites as neutral reference (Keightley and Eyre-Walker 2007). The DFE in ordered and disordered regions differs significantly, with roughly 23% of nonsynonymous mutations being effectively neutral in disordered regions compared to 12% for ordered regions. In contrast, the proportion of mutations with strong selective effects (i.e., Nes > 100) is reduced for disordered regions relative to their ordered counterparts (48% versus 63%). Taken together, these results are in full agreement with the substitution rate analyses, hinting at a prominent role of genetic drift and reduced purifying selection in disordered regions. When conducting a joint analysis of ordered and disordered regions, results are very similar to ordered regions. Taking into account that the majority of sites lie in ordered protein regions, indicating that the selective effects in disordered regions are somewhat obscured by the vast number of mutations in the ordered protein parts.
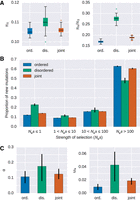
Evidence for differences in the selective effects in disordered and ordered protein regions in humans. (A) Nucleotide diversity at synonymous sites (πS, left panel) and the ratio of nucleotide diversity at nonsynonymous sites over synonymous sites (πN/πS, right panel) for ordered, disordered, and jointly obtained protein regions in humans. (B) The distribution of fitness effects of nonsynonymous mutations estimated separately for ordered and disordered protein regions, as well as when jointly estimated. Error bars represent the standard error. Nes denotes the effective population size (Ne) scaled strength of selection (s). (C) Estimates of the role of positive selection for the analyzed protein set. The proportion of nonsynonymous substitutions that can be attributed to positive selection (α) and the adaptive divergence relative to the synonymous divergence (ωa) estimated separately for ordered and disordered protein regions, as well as when jointly estimated. All pairwise comparisons are significantly different (Wilcoxon signed-rank test, paired, P < 3 × 10−12).
Adaptive amino acid changes have substantially contributed to the evolution of disordered protein regions in recent human evolution
To infer the role of positive selection in IDP regions in recent human and ape evolution, we compared the ratio of nonsynonymous
to synonymous sites between intra-species polymorphisms and inter-species divergence (McDonald-Kreitman test [MK test]) (McDonald and Kreitman 1991). This mathematical contrast can be used to obtain the proportion of fixed substitutions that were driven by positive selection
(α) and the rate of adaptive substitutions relative to the synonymous divergence (ωa) (Gossmann et al. 2010). Since the DFEs for ordered and disordered regions differ (Fig. 4B), we applied a derivate of the MK test that corrects for the effect of slightly deleterious mutations based on the DFE (Eyre-Walker and Keightley 2009). We find that disordered regions show a higher proportion of adaptive mutations (α = 17% versus 11% for ordered regions) (Fig. 4C). Since there is strong evidence that the proportion of effectively neutral nonsynonymous mutations and substitutions appear
to be different (Figs. 2 and 4B) between ordered and disordered regions, a contrast of ωa between the two regions will reveal the contribution of positive selection in absolute terms independent of fixation events
of neutral and slightly deleterious mutations contributing to the nonsynonymous divergence. We observe a significant difference
in ωa values (≈fourfold, 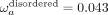 versus
versus 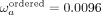 ) (Fig. 4C), suggesting that the rate of adaptive substitutions relative to the synonymous rate is elevated in disordered regions.
As expected, we also find reduced rates of adaptive evolution when the MK test is conducted jointly on ordered and disordered
regions. This illustrates that the role of positive selection in IDP regions is difficult to capture when not taken into account
a priori.
) (Fig. 4C), suggesting that the rate of adaptive substitutions relative to the synonymous rate is elevated in disordered regions.
As expected, we also find reduced rates of adaptive evolution when the MK test is conducted jointly on ordered and disordered
regions. This illustrates that the role of positive selection in IDP regions is difficult to capture when not taken into account
a priori.
Discussion
Here, we investigated the evolutionary pressures that may have acted on proteins containing long IDP regions in humans. Our approach differs from previous analyses of the evolutionary rate analysis of these regions. First, in our comparative analysis, we used sequence data from more than 90 species, many more species than in any other previous study on this topic. In doing so, we were able to exclude low-quality sequences by excluding sequences of questionable alignability but still were able to conduct our test statistics with a sufficient number of orthologs in high-quality alignments. Second, we accounted for the genomic context and differences in the amino acid composition of disordered regions by conducting our test statistics separately for ordered and disordered regions of the same protein in a pairwise manner. Third, unlike previous approaches, we have combined inter-specific and intra-specific data for humans and other ape species to conduct a derivative of the McDonald-Kreitman test to investigate whether positive selection has played a role in the evolution of disordered regions in the human lineage.
We show that the molecular evolutionary rates, as measured by the nonsynonymous to synonymous rate ratio in coding regions, are elevated in disordered regions compared to their ordered counterparts and identify three main contributors for these elevated rates: (1) relaxed purifying selection; (2) intensified genetic drift; and (3) positive selection. This is in agreement with studies from other taxa such as yeast species and Drosophila and suggests general features of the evolvability of IDP regions. The lack of three-dimensional constraint may explain the fast evolution of these protein regions. However, it has been shown that there are amino acid residues that maintain the intrinsic disorder and that these residues are under stronger evolutionary constraint than in ordered regions (Ahrens et al. 2016). It is therefore important to note that the vast majority of IDP regions experienced substantial purifying selection during their evolution (Fig. 2), hinting at their important functional role despite their relaxed structural features for certain residues. The difference in the selective effects between ordered and disordered regions may be attributed to a net shift of strongly deleterious to slightly deleterious and effectively neutral mutations (Fig. 4B). Hence, the selective effects on mutations in IDP regions are more dependent on fluctuations of the effective population size (Charlesworth 2009)—which may explain rapid evolution and higher molecular diversification due to periodic episodes of random genetic drift (Nabholz et al. 2013).
A major outcome of our study is that positive selection has played a pronounced role in certain proteins with disordered regions across the entire mammalian tree, as we find evidence for positive selection in 377 IDP regions. To our knowledge, this is the first time that the increased role of positive selection in these regions has been attributed in mammals. We show that there is limited power to detect adaptive processes if intrinsic features of disorder are not taken into account a priori. This may explain why the role of adaptive substitutions has not been emphasized thus far. We also identify, based on an extended version of the McDonald-Kreitman test that takes into account the distribution of fitness effects of new mutations, that there is a fourfold increase in the rate of adaptive substitutions relative to the rate of synonymous substitutions (ωa) in intrinsically disordered regions. It is important to note that ωa, but not α, should be used to compare the adaptive rates in these regions. If ordered and disordered regions would experience the same amount of adaptive substitutions, we would expect α to be lower in disordered regions; as there are more neutral fixations, this reduces the relative proportion of adaptive substitutions.
It has recently been suggested that younger proteins tend to be enriched in disordered regions and over evolutionary time become more ordered (Wilson et al. 2017). If the loss of intrinsic disorder is potentially associated with a selective advantage (e.g., gain of protein domain function), then positive selection could be a general mechanism explaining the evolvability of these IDPs. However, it is difficult to determine the turnover of intrinsic disordered regions across various species from our data set and consequently whether our set of positively selected genes between species supports such a model of adaptive losses. Evidence for repeated events of positive selection across the entire mammalian tree may suggest repeated functional diversification within the mammalian lineage due to multiple independent losses of intrinsic disorder. It is well possible that we underestimate the true role of positive selection in IDP regions. Our estimates suggest that there might be up to a 10-fold difference in the amount of adaptive changes between disordered and ordered protein parts when sequence lengths are accounted for. This is due to technical limitations of the applied test statistic, with short sequence length and high quality alignments that are difficult to obtain for some IDPs due to increased rate of fixed indels (Khan et al. 2015). Hence, disordered residues in regions of ambiguous alignability are not included in this study and their evolutionary patterns remain unexplored. However, we were able to elucidate the role of positive selection in IDPs because we included structural information a priori, and it is likely that our estimates of the amount of positive selection in these regions are conservative. Taken together, our results suggest that IDP regions are important targets of genetic innovation and that the contribution of positive selection in these regions is more pronounced than in other parts of proteins.
Methods
Protein annotations of disordered regions in human proteins and multiple sequence alignments
We obtained information of long intrinsically disordered regions for human proteins from MobiDB v2.2 (Di Domenico et al. 2012). To conduct a phylogenetically based analysis, we performed multiple sequence alignments using a customized automated pipeline (Supplemental Methods; Supplemental Fig. S8). As a phylogenetic framework, we used the near-complete species-level mammalian consensus tree assembled by Bininda-Emonds et al. (2007) and updated by Rolland et al. (2014) and pruned the complete tree to leave only those species corresponding to samples in our genomic data set (Supplemental Fig. S9). This resulted in 6663 human proteins with disordered regions and their corresponding orthologs in other mammalian species. Details are described in the Supplemental Methods.
Phylogenetic models for site-specific analyses and site annotation
The ratio of nonsynonymous to synonymous substitutions (i.e., ω = dN/dS) can be interpreted as a measurement of selective pressure that has acted during the evolution of a protein. Here, we use site-specific dN/dS models for which we assume that there is variation of selective pressures between different types of sites within a protein but not between species. Since these models are computationally very expensive, we randomly down-sampled the number of species in cases when there were too many (Supplemental Fig. S10). We conducted sequence simulations using the INDELible package (Fletcher and Yang 2009). Functional associations and structural data were obtained from UniProt and PDB Details, and based on the relative solvent-accessible surface area (SASA), we predicted whether a protein site is buried or more likely to be positioned at the surface of the protein. Details are described in the Supplemental Methods.
Molecular dynamics analysis
Molecular dynamics simulations were performed using a standard protocol for pmemd simulations included in the AMBER 14 software package (Salomon-Ferrer et al. 2013). A high-resolution three-dimensional structure of human interleukin 21 (IL21) resolved by heteronuclear NMR spectroscopy (PDB code: 2OQP) was used (Bondensgaard et al. 2007).
Polymorphism statistics, DFE, and McDonald-Kreitman type test of positive selection
We obtained coding gene information for 46 unrelated Yorubian individuals from the 1000 Genomes Project (The 1000 Genomes Project Consortium 2015) and excluded genes on the X Chromosome as well as genes that could not clearly be assigned to the respective MobiDB database entry. Divergence data for the respective gene was obtained by randomly obtaining the ortholog from a closest related non-ape species we had in our between-species data set. We used DFE-alpha (Keightley and Eyre-Walker 2007; Eyre-Walker and Keightley 2009) to estimate the distribution of fitness effects of new nonsynonymous mutations (Eyre-Walker and Keightley 2007) along with the proportion of substitutions attributed to positive selection and the relative rate of adaptive substitutions to synonymous divergence (ωa) (Gossmann et al. 2010) for ordered and disordered regions as well as jointly for both together. Details are described in the Supplemental Methods.
Software availability
Customized scripts for the alignment processing pipeline are available in Supplemental Material and at https://www.github.com/tonig-evo/3D_gaps.
Acknowledgments
We thank Tobias Warnecke for helpful comments on an earlier version of this manuscript, and we also thank three anonymous reviewers for comments that have helped to improve the quality of this manuscript. The computations were partially performed on resources provided by UNINETT Sigma2–the National Infrastructure for High Performance Computing and Data Storage in Norway. Financial support for the work came from a FEBS Short-Term Fellowship to A.A., a UiT BFE mobility grant to M.B., and a Leverhulme Early Career Fellowship Grant (ECF-2015-453) and Natural Environment Research Council grant (NE/N013832/1) to T.I.G.
Author contributions: T.I.G. designed the study; A.A., M.B., and T.I.G. conducted the research; C.R.C. contributed to the research; A.A. and T.I.G. wrote the draft of the manuscript; and all authors contributed to editing of the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.232645.117.
- Received November 21, 2017.
- Accepted June 1, 2018.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








