
Sex-biased microRNA expression in mammals and birds reveals underlying regulatory mechanisms and a role in dosage compensation
- Maria Warnefors1,
- Katharina Mössinger1,
- Jean Halbert2,
- Tania Studer1,
- John L. VandeBerg3,
- Isa Lindgren4,
- Amir Fallahshahroudi4,
- Per Jensen4 and
- Henrik Kaessmann1
- 1Center for Molecular Biology of Heidelberg University (ZMBH), DKFZ-ZMBH Alliance, D-69120 Heidelberg, Germany;
- 2Ludwig Institute for Cancer Research, University of Lausanne, CH-1066 Lausanne, Switzerland;
- 3South Texas Diabetes and Obesity Institute, School of Medicine, The University of Texas Rio Grande Valley, Brownsville, Texas 78520, USA;
- 4Department of Physics, Chemistry and Biology, Linköping University, SE-581 83 Linköping, Sweden
Abstract
Sexual dimorphism depends on sex-biased gene expression, but the contributions of microRNAs (miRNAs) have not been globally assessed. We therefore produced an extensive small RNA sequencing data set to analyze male and female miRNA expression profiles in mouse, opossum, and chicken. Our analyses uncovered numerous cases of somatic sex-biased miRNA expression, with the largest proportion found in the mouse heart and liver. Sex-biased expression is explained by miRNA-specific regulation, including sex-biased chromatin accessibility at promoters, rather than piggybacking of intronic miRNAs on sex-biased protein-coding genes. In mouse, but not opossum and chicken, sex bias is coordinated across tissues such that autosomal testis-biased miRNAs tend to be somatically male-biased, whereas autosomal ovary-biased miRNAs are female-biased, possibly due to broad hormonal control. In chicken, which has a Z/W sex chromosome system, expression output of genes on the Z Chromosome is expected to be male-biased, since there is no global dosage compensation mechanism that restores expression in ZW females after almost all genes on the W Chromosome decayed. Nevertheless, we found that the dominant liver miRNA, miR-122-5p, is Z-linked but expressed in an unbiased manner, due to the unusual retention of a W-linked copy. Another Z-linked miRNA, the male-biased miR-2954-3p, shows conserved preference for dosage-sensitive genes on the Z Chromosome, based on computational and experimental data from chicken and zebra finch, and acts to equalize male-to-female expression ratios of its targets. Unexpectedly, our findings thus establish miRNA regulation as a novel gene-specific dosage compensation mechanism.
Phenotypic variation between males and females of the same species, also known as sexual dimorphism, is thought to arise largely as a consequence of sex-biased gene expression (Grath and Parsch 2016; Mank 2017). Studies addressing the genetic underpinning of sexual dimorphism from the perspective of protein-coding genes have revealed thousands of sex-biased genes in mammals and birds (Yang et al. 2006; Itoh et al. 2010; Harrison et al. 2015; Melé et al. 2015). Less attention has been given to the potential impact of sex-biased microRNAs (miRNAs)—short regulatory RNAs (∼21 nt) that enable expression fine-tuning of target mRNAs through transcript degradation or translational inhibition (Jonas and Izaurralde 2015). Because each miRNA can potentially target many hundred mRNAs (Baek et al. 2008; Selbach et al. 2008), it has been proposed that miRNAs might have widespread effects that contribute to shaping male and female transcriptomes, both in healthy and diseased tissues (Morgan and Bale 2012; Dai and Ahmed 2014; Sharma and Eghbali 2014).
In some cases, sex-biased miRNA expression might arise as a consequence of differential gene dosage (number of gene copies) between the sexes. In most mammals, females have two X Chromosomes, whereas males have one X and one Y Chromosome (Cortez et al. 2014; Graves 2016), resulting in sex-specific gene dosage of X-linked and Y-linked genes. For the majority of genes, the effect on expression is mitigated through a dosage compensation mechanism in which one of the X Chromosomes is inactivated, although a fraction of genes are able to escape inactivation, thereby becoming more highly expressed in females (Deng et al. 2014). Even so, the X Chromosome is generally associated with male-biased expression and is enriched for young, testis-expressed miRNAs (Guo et al. 2009; Meunier et al. 2013). Contrary to mammals, birds do not have global dosage compensation and they therefore display more pervasive transcriptomic differences between the sexes: The bird Z Chromosome is present in two copies in males, whereas females carry one Z and one W Chromosome, and Z-linked protein-coding genes are, on average, expressed at 1.5-fold higher levels in males (Ellegren et al. 2007; Itoh et al. 2007; Mank and Ellegren 2009; Julien et al. 2012). Consistent with incomplete dosage compensation, the Z-linked miRNA miR-2954-3p was found to be male-biased in birds (Zhao et al. 2010; Luo et al. 2012), and studies in zebra finch suggested a role for the miRNA in sex-specific song response (Gunaratne et al. 2011; Lin et al. 2014). The role of miR-2954-3p in non-singing species, such as chicken, has not been extensively explored, although its strongly male-biased expression throughout chicken development hints at a contribution of the miRNA toward the establishment of sex identity in individual cells, especially as this feature is not dependent on gonadal hormones in chicken (Zhao et al. 2010).
Alternatively, sex-biased miRNA expression can be achieved through various types of sex-biased regulation. In the perhaps simplest scenario, miRNAs that are located in the introns of protein-coding genes (Baskerville and Bartel 2005) could potentially become sex-biased indirectly if they are coexpressed with a sex-biased host (Marco 2014). Many intronic miRNAs, as well as those that are intergenic, are nevertheless transcribed from their own promoters (Ozsolak et al. 2008) and would require targeted sex-biased regulation, for example by hormones, to become differentially expressed between males and females. In Drosophila, hormonal regulation by ecdysone promotes sex-biased expression of the miRNA locus let-7-C, which was found to contribute to sexual identity (Fagegaltier et al. 2014). Concordant with this finding, the expression of many human miRNAs, including members of the let-7 family, is regulated by sex hormones (Yang and Wang 2011; Klinge 2012). Other hormones can also be secreted in a sex-biased manner, such as growth hormone, which contributes to sexual dimorphism of the mammalian liver (Waxman and O'Connor 2006), including differential expression of miRNAs (Cheung et al. 2009). Although some miRNAs are primary targets of nuclear hormone receptors (Bhat-Nakshatri et al. 2009), sex-biased regulation is in other instances mediated through hormonally regulated transcription factors (Castellano et al. 2009). Finally, the expression of key factors involved in the processing of primary miRNA transcripts into mature miRNAs (Ha and Kim 2014) has also been found to be sensitive to sex hormones (Bhat-Nakshatri et al. 2009; Nothnick et al. 2010), suggesting an additional layer of post-transcriptional, sex-biased regulation.
Taken together, previous studies suggest that sex-biased miRNA expression might be widespread and have potentially profound functional implications. Even so, previous efforts to identify sex-biased miRNAs in mammals and birds were restricted to a single species and typically also to a single tissue, and many had limited resolution, because they were based on microarray technology and/or lacked biological replicates (Mishima et al. 2008; Bannister et al. 2009; Cheung et al. 2009; Ciaudo et al. 2009; Chiang et al. 2010; Luo et al. 2012; Mujahid et al. 2013; Murphy et al. 2014; Ziats and Rennert 2014; Kwekel et al. 2015, 2017; Link et al. 2017). To provide a more complete view of mammalian and avian sex-biased miRNAs, we have conducted the first comprehensive survey of male and female miRNA transcriptomes based on small RNA sequencing of somatic and gonadal tissues from mouse, opossum, and chicken. Our results highlight the diverse mechanisms underlying sex-biased miRNA expression across tissues and species and establish a role for miR-2954-3p regulation in gene-specific dosage compensation in birds.
Results
Identification of miRNAs with sex-biased expression in mouse, opossum, and chicken
To identify miRNAs with sex-biased expression in mouse, gray short-tailed opossum, and chicken (red jungle fowl), we prepared and sequenced a total of 72 small RNA libraries corresponding to brain, heart, liver, and gonad (ovary and testis) samples from three male and three female adult individuals per species (Methods). To mitigate the extensive sequencing biases that have been identified for standard library preparation methods (Sorefan et al. 2012; Baran-Gale et al. 2015), we chose to prepare the libraries with the NEXTflex Small RNA-seq Kit (Methods), which improves the ligation step through the use of partially degenerate adapters (Baran-Gale et al. 2015). We found that this protocol allowed substantially broader coverage of expressed miRNAs compared to standard methods (Supplemental Fig. S1). After adapter trimming and filtering based on quality scores and read length, each library was represented by an average of 19.4 million high-quality reads with a size of 15–28 nt. Novel miRNA precursors were predicted for each library using miRDeep2 (Friedländer et al. 2012), with a cutoff score of 5, corresponding to a signal-to-noise ratio of at least 10:1 in all tissues except chicken testis (Methods). Only precursors that were identified in at least three of six tissue replicates (male or female) and that did not overlap with other noncoding RNAs (ncRNAs) were kept for further analysis (Methods). Genomic coordinates of all known and putative novel miRNA loci investigated in this study are provided in Supplemental Table S1. All novel miRNAs loci have furthermore been submitted to miRBase (Kozomara and Griffiths-Jones 2014) and their assigned miRBase IDs are listed in Supplemental Table S2. After an additional filtering step performed by miRBase, 40 mouse, 220 opossum, and 135 chicken miRNA precursors were added to the database.
Next, we estimated miRNA expression levels by remapping all reads to the respective genome using Bowtie (Langmead et al. 2009) and a sequence of decreasingly stringent settings (Methods) to allow for mismatches due to miRNA editing and 3′ modifications (Kawahara et al. 2007; Burroughs et al. 2010; Chiang et al. 2010; Warnefors et al. 2014). Because some miRNAs belong to multicopy families with high sequence similarity, it was not always possible to pinpoint the precise genomic locus from which a miRNA read was derived. We therefore grouped mature miRNAs into multimap groups (MMGs; Methods), which were thereafter treated as single expression units (Robert and Watson 2015). We chose a relatively conservative MMG cutoff of 5%, meaning that if a miRNA shared >5% of its total reads with another miRNA, these two miRNAs were assigned to the same MMG. Reads that mapped two MMGs (i.e., <5% cutoff) were divided equally between the MMGs. The 843 mouse, 893 opossum, and 564 chicken MMGs are listed in Supplemental Table S3. MMGs were considered expressed in a given tissue if they contributed at least 10 transcripts per million miRNA-mapped reads (TPM) in three of six replicates.
For each tissue and species, we then used DESeq2 (Love et al. 2014) to identify miRNAs with sex-biased expression at a global false discovery rate (FDR) of 5% (Methods). Differential expression between ovary and testis samples was analyzed and corrected separately due to the mixture of sex-related and tissue-related effects, which otherwise reduced the stringency of the significance test for the somatic samples (Methods). An overview of sex-biased miRNA expression per tissue is given in Figure 1, with complete details available in Supplemental Table S4. The most male-biased and most female-biased miRNA for each species and tissue are listed in Table 1. We did not identify any individual miRNAs that were sex-biased in a somatic tissue in more than one species (Discussion), although several were sex-biased in multiple tissues from a single species (Supplemental Table S4).
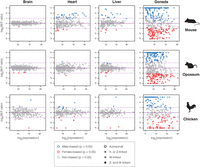
Sex-biased miRNA expression in mouse, opossum, and chicken. Male-to-female (M:F) ratios were calculated for three male and three female replicates per tissue and the significance assessed with DESeq2 (Love et al. 2014), with Benjamini-Hochberg correction for multiple tests (Benjamini and Hochberg 1995) performed separately for somatic and gonadal samples. For display purposes, the log2-transformed M:F ratios were capped at −3 and 3, with more extreme ratios replaced by these values. Each data point corresponds to a multimap group (MMG) comprised of one or more mature miRNAs (see main text), and MMGs were considered sex-linked if they included at least one sex-linked member. Expression values correspond to the mean normalized counts provided by DESeq2. The most sex-biased miRNAs per tissue are listed in Table 1. The Z-linked and W-linked miRNA miR-122-5p is visible as the rightmost data point in the chicken liver panel. All data shown in this figure are included in Supplemental Table S4.
The most sex-biased miRNAs per tissue
Independent transcription drives sex-biased miRNA expression in mouse tissues
We found evidence of sex-biased miRNA expression in all of the investigated somatic tissues (Fig. 1), with the largest proportion found in mouse heart (13%, 33 significant MMGs of 253 tested) and liver (8.6%; 19 of 222). These findings are consistent with the large numbers of protein-coding genes that are expressed in a sex-biased manner in various mouse tissues, notably with more pronounced differences in liver compared to brain (Yang et al. 2006). Although it is well established that many X-linked miRNAs are male-biased with high expression levels in the mouse and opossum testis (Song et al. 2009; Meunier et al. 2013), and this trend was visible also in our data (Fig. 1; Supplemental Table S4), we found that most sex-biased miRNAs in somatic tissues were autosomal (Fig. 1). The expression pattern of these miRNAs thus cannot be explained by escape from X inactivation or other effects immediately linked to the presence of sex chromosomes, but is instead likely achieved through direct or indirect regulation by hormones or other sex-biased factors.
We speculated that one regulatory route to sex-biased miRNA expression might simply be coexpression with a sex-biased protein-coding gene, especially since it is known that many miRNAs, including both those generated via canonical and noncanonical pathways, are located within the introns of protein-coding genes and transcribed from the same promoter as their host (Rodriguez et al. 2004; Baskerville and Bartel 2005; Berezikov et al. 2007; Meunier et al. 2013). In our data, there was considerable potential for coexpression, with 9 of 33 (27%) MMGs with significantly sex-biased expression in mouse heart having at least one intronic member, and the corresponding numbers for mouse liver were 7 sex-biased MMGs of 19 (37%). However, when we compared the sex-biased expression of intronic miRNAs with that of their host genes, based on expression data from Marin et al. (2017) (see also Methods), we did not find any significant correlation between miRNA and host gene male-to-female (M:F) ratios in heart, and only a weak correlation in liver, which was no longer significant after correction for multiple tests (Fig. 2A). These results indicate that miRNA sex bias is not in general a secondary effect caused by sex-biased regulation of protein-coding genes in somatic tissues, although a stronger correlation was seen for the gonads (Fig. 2A). The male-biased expression of miR-1948-5p/3p and miR-455-5p/3p in mouse liver nevertheless appeared to constitute an exception, since the host genes Ttc39c and Col27a1 were male-biased to a similar degree (Fig. 2A). However, we found that both miRNA loci were associated with mouse liver DNase I hypersensitivity (DHS) sites identified by Ling et al. (2010) (Methods), which is an indication that they possess independent regulatory elements and can be transcribed separately from the host. Although the possibility remains that some miRNAs are transcribed from both miRNA-specific and host-associated promoters (Ozsolak et al. 2008), these observations suggest that the sex bias we observe for mouse miRNAs occurs in parallel to the sex bias of protein-coding genes, but for the most part, is not a direct consequence thereof. Similar decoupling of intronic miRNA expression from that of host genes due to independent transcription and/or post-transcriptional processing has previously been observed across mammalian tissues (Ozsolak et al. 2008; Wen et al. 2015).
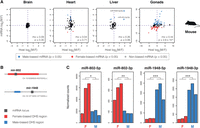
Transcriptional mechanisms underlying sex-biased miRNA expression. (A) Correlation between sex-biased expression of intronic miRNAs (y-axis) and their host genes (x-axis). Male-to-female (M:F) expression ratios were calculated with DESeq2. For each tissue, the Spearman correlation coefficient (rho) is given, together with its Benjamini-Hochberg corrected P-value (Benjamini and Hochberg 1995). (B) Overview of the two miRNA loci that were associated with sex-biased DHS regions (Ling et al. 2010) in mouse liver. In both cases, a sequence of 1 kb is depicted. The displayed miRNA precursor region corresponds to the 5p and 3p sequences with the intervening loop sequence. (C) Expression levels in mouse liver of the 5p and 3p mature miRNAs shown in B, for three female and three male replicates. Raw read counts were normalized with DESeq2. Statistical significance: (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
Although these analyses suggest that the majority of sex-biased miRNAs are transcribed from independent promoters, they do not address whether these promoters are themselves targets of sex-biased regulation. The DHS site that we were able to associate with the mir-1948 locus had previously been annotated as male-biased (Ling et al. 2010), which would be consistent with the male-biased expression of miR-1948-5p and -3p (Fig. 2A). This finding was nevertheless somewhat surprising, given that sex-biased regulation of protein-coding genes is thought to primarily take place at distally located regulatory elements rather than promoters, with 80% of all sex-biased DHS sites being located >100 kb away from sex-biased genes (Ling et al. 2010). To investigate the role of sex-biased promoters further, we intersected the coordinates of all 185 liver-expressed single-member MMGs (i.e., miRNAs for which most sequencing reads map to a single genomic locus) with genome-wide DHS annotations from Ling et al. (2010) (Methods). We could associate 95 miRNAs with 61 DHS sites within 5 kb upstream or overlapping the miRNA (P < 0.001 based on 1000 permutations of the DHS site coordinates), and were able to locate one additional miRNA-associated sex-biased DHS site, this time a female-biased site associated with the equally female-biased mir-802 locus (Fig. 2B). In spite of the limited sample size, the association between sex-biased DHS sites and sex-biased miRNA loci was significant (two of two sex-biased and six of 59 unbiased DHS sites were associated with sex-biased miRNAs, P = 0.015, Fisher's exact test), which suggests that, for a subset of miRNAs, sex-biased expression is likely associated with differential promoter accessibility in males and females.
Overall, our findings point to diverse transcriptional mechanisms behind sex-biased miRNA expression in the mouse, including potential piggybacking on sex-biased host genes in gonads and possibly liver, regulation at miRNA promoters, and presumably, regulation at distally located regulatory elements, such as enhancers. Conceivably, an additional level of regulation might be achieved through differential processing of miRNA transcripts in males and females (Bhat-Nakshatri et al. 2009; Nothnick et al. 2010). Given that 5p/3p pairs from the same miRNA locus frequently have highly similar M:F ratios (e.g., miR-1948-5p/3p, miR-455-5p/3p, and miR-802-5p/3p) (Fig. 2), we consider it more likely that such sex-biased regulation, if present, would affect the general efficiency of miRNA biogenesis rather than modulate the balance between 5p and 3p mature miRNAs through arm switching (Chiang et al. 2010).
Somatic miRNA sex bias mirrors gonadal expression in mouse but not chicken
In addition to the sex-biased miRNA expression found in somatic tissues, we consistently found pronounced sex differences in the gonads (Fig. 1). The magnitude of these differences is not surprising, given that ovary and testis transcriptomes are shaped by sex-biased as well as tissue-biased factors (Discussion). Previous studies have shown that the X Chromosome of mouse and opossum is enriched for recently expanded testis-expressed miRNA families, whereas no similar enrichment exists for the chicken Z Chromosome (Guo et al. 2009; Meunier et al. 2013). The mechanistic basis of this phenomenon has been a matter of intense study (Song et al. 2009; Royo et al. 2015; Sosa et al. 2015). As previously noted, the surplus of X-linked, testis-biased miRNAs in both mouse and opossum was clearly visible also in our data (Fig. 1; Supplemental Table S4). Even so, the majority of ovary-biased and testis-biased miRNAs in our data set were autosomal, and a large proportion of them could be detected in at least one somatic tissue, although testis-biased autosomal miRNAs were significantly less likely to be somatically expressed (Fig. 3A). The relative lack of shared miRNA expression between testis and somatic tissues fits well with known expression patterns of mRNAs and long noncoding RNAs, which are more frequently testis-specific than ovary-specific in all three species investigated here (Necsulea et al. 2014). However, it should be noted that ovary samples contain a larger proportion of somatic cells, and the power to detect ovary-specific miRNAs therefore might be reduced.
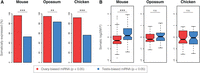
Somatic expression of ovary-biased and testis-biased miRNAs. (A) Proportion of autosomal MMGs with ovary-biased or testis-biased expression that are detected in at least one somatic tissue. (B) Somatic sex bias of ovary-biased and testis-biased miRNAs. If an MMG was detected in more than one somatic tissue, the most extreme M:F ratio was chosen. Statistical significance: (**) P < 0.01; (***) P < 0.001; (n.s.) not significant.
We were interested in whether miRNAs were consistently sex-biased between gonads and somatic tissues, as might be the case if they fulfilled sex-specific functions across a variety of conditions. To this end, we compared the degree of somatic sex bias for ovary-biased and testis-biased autosomal miRNAs (Fig. 3B), while choosing the tissue with the strongest sex bias if a miRNA was expressed in multiple somatic tissues (Methods). In mouse, we found that ovary-biased miRNAs tended to be female-biased in somatic tissues (median M:F ratio: −0.29), whereas testis-biased miRNAs tended to be male-biased (median M:F ratio: 0.13, P = 3.4 × 10−5, Mann-Whitney U test with Benjamini-Hochberg correction [Benjamini and Hochberg 1995]). A similar, although not significant trend was hinted at in the opossum data (P = 0.30), but was not observed in chicken (P = 0.93). These findings thus point to a more general coordination of sex-biased expression in mouse, which is largely absent in the other species, especially chicken, although we did notice that a novel, autosomal miRNA (gga-miR-2184a-5p) was the second most female-biased miRNA in chicken liver and was also significantly female-biased in heart and gonads (Supplemental Table S4). Although we were unable to address the root of the apparent discrepancy between mouse and chicken with the present data, it is tempting to speculate that hormonal regulation might facilitate joint control of miRNA expression across tissues. Indeed, many mammalian miRNAs are known to be regulated by gonadal hormones (Yang and Wang 2011; Klinge 2012). The majority of these studies were carried out in human cell lines, but there is considerable overlap with the miRNAs for which we observe significant sex-biased expression in our mouse data, including miR-17-3p and miR-19a-3p from the mir-17-92 cluster (Castellano et al. 2009), and miR-451a-5p (Bergamaschi and Katzenellenbogen 2012). In contrast to the seemingly widespread contributions of gonadal hormones to miRNA regulation in placental mammals, they play a reduced role in the establishment of male and female somatic tissues in chicken (Zhao et al. 2010), which would be consistent with the lack of coordinated sex bias in this species. Global studies of the hormonal impact on miRNA expression will however be required to determine whether the coordination of miRNA sex bias is driven by gonadal hormones and whether the effect is direct or indirect.
To search for functional commonalities of sex-biased miRNA regulation across mouse tissues, we analyzed Gene Ontology annotations of miRNA target genes using target predictions from TargetScan (Agarwal et al. 2015) and the GOrilla tool (Eden et al. 2009) (Methods). Only protein-coding genes with a minimum expression of 1 FPKM (fragments per kilobase of transcript per million mapped reads) in the tissue of interest were considered based on data from Marin et al. (2017). To avoid the frequent false positives associated with functional analyses of miRNA targets (Bleazard et al. 2015), we compared targets of sex-biased miRNAs only to targets of unbiased miRNAs instead of to all genes expressed in the tissue of interest (Methods). We further chose to focus on mouse heart and liver for this analysis, since the extent of sex-biased miRNA expression was too limited in brain and too extensive in gonads to permit a reasonable division of protein-coding genes into targets of male-biased, female-biased, and unbiased miRNAs, respectively. Although we did not observe any significant functional enrichment for targets of female-biased miRNAs, we did identify a number of Gene Ontology terms associated with male-biased miRNAs in both heart and liver (Supplemental Table S5). These terms were highly similar for the two tissues, with the top liver category “regulation of cell communication” being the eleventh most enriched term in heart, and the top heart category “positive regulation of cellular process” being the fourth most enriched term in liver. The potential functional parallels between targets of sex-biased miRNAs in heart and liver are noteworthy in light of the apparent coordination of sex-biased miRNA expression in the mouse and underline the value of conducting screens for sex-biased genes across multiple tissues.
Gene retention on the chicken W Chromosome allows nonbiased expression of miR-122-5p
Unlike mouse and opossum, chicken does not have a global mechanism of dosage compensation, meaning that Z-linked protein-coding genes generally tend to be more highly expressed in males compared to females (Ellegren et al. 2007; Itoh et al. 2007; Mank and Ellegren 2009; Julien et al. 2012). The most prominent example of a Z-linked, male-biased miRNA was miR-2954-3p (Table 1; see next section), but the association between Z linkage and male-biased expression did not hold for all miRNAs on the chicken Z Chromosome. A particularly striking exception was miR-122-5p, which was the most abundant miRNA in the chicken liver and almost completely unbiased between males and females (Fig. 1; Supplemental Table S4). The expression pattern of miR-122-5p in chicken is thus similar to miR-122-5p expression in mouse and opossum (Supplemental Table S4), although the mir-122 locus is autosomal in these species. Given that miR-122-5p is expressed at extreme levels (miR-122-5p contributed >60% of all miRNA-mapped reads per chicken liver library), it is difficult to imagine how sufficient dosage compensation could be achieved through regulatory means.
However when we remapped miRBase miRNAs to the most recent chicken genome release (galGal5) as part of our annotation procedure, we were able to locate a copy of the mir-122 locus on the updated W Chromosome, the presence of which likely grants equal miR-122-5p dosage in both sexes (a third mir-122 copy is present on contig chrZ_NT_463593v1_random, although this might be an assembly error). This miRNA therefore joins the highly select group of dosage-sensitive genes that were retained on the W Chromosome (Bellott et al. 2017). We did however detect significant female bias for miR-122-5p in the chicken heart, which could be due to sex-specific control or may indicate slight regulatory diversification of the Z-linked and W-linked copies in this tissue. Similar sex bias of miR-122-5p was previously observed in the zebra finch heart based on a comparison of one male and one female individual (Luo et al. 2012). In addition, remapping confirmed W linkage of the mir-7b locus (Ayers et al. 2013), which belongs to a family with both Z-linked and autosomal members. Given that the chicken W Chromosome is thought to host only 28 protein-coding genes (Bellott et al. 2017), miRNAs thus make up a sizable fraction of the total W-linked gene repertoire.
Beyond mir-122 and mir-7b, we did not detect any additional W-linked or Y-linked miRNAs in any species using remapping of known miRNAs (Methods) or de novo annotation with miRDeep2, although the latter did predict two novel miRNAs (gga-mir-12266-2 and gga-mir-12266-3) on W-labeled contigs. Because these methods would not pick up miRNAs that lack an equivalent on the X or Z Chromosome and were generated through an atypical mechanism (Yang et al. 2010), we additionally probed the data set for sex-specific reads derived from the Y or W Chromosomes (Methods). However, although we successfully identified the mir-7b locus in this manner, we did not recover reads from the putatively W-linked loci mentioned above, suggesting they do not represent bona fide W-linked miRNA genes (Supplemental Fig. S2). Although we did identify a number of male-specific reads in mouse and opossum, many male-specific reads were also present in chicken, indicating that most such reads might not be Y-derived, but instead a consequence of the transcriptional complexity of the testis (Soumillon et al. 2013). Indeed, when we mapped the male-specific reads from mouse to the Y Chromosome, not a single match was found (Supplemental Fig. S2). We therefore consider it unlikely that additional miRNAs, unique to the Y or W Chromosomes, are robustly expressed in the tissues studied here.
Male-biased expression of miR-2954-3p serves as a gene-specific dosage compensation mechanism in birds
Similar to previous reports (Zhao et al. 2010; Luo et al. 2012; Lin et al. 2014), we observed male-biased expression of the Z-linked miR-2954-3p (Fig. 4A) in all chicken tissues. Although the mir-2954 locus is located within an intron of the XPA gene, XPA is not male-biased to the same extreme degree (Methods; Marin et al. 2017). Notably, the male bias of miR-2954-3p is considerably more pronounced (up to almost eightfold in chicken liver) than the maximally twofold difference that is expected between males and females due to gene dosage alone (Lin et al. 2014). The high expression in chicken males is therefore likely a result of increased production and/or stability of miR-2954-3p, in combination with the dosage effect. The biological relevance of such extreme male bias is nevertheless not currently understood. Although previous work in zebra finch has elucidated the involvement of miR-2954-3p in song response (Gunaratne et al. 2011; Lin et al. 2014), these observations cannot explain why miR-2954-3p is male-biased in neural as well as non-neural tissues both in zebra finch and a non-songbird such as chicken (Fig. 4A). To understand the functional impact of miR-2954-3p, we therefore used TargetScan (Agarwal et al. 2015) to predict target genes in the chicken genome (Methods). Consistent with an earlier study (Luo et al. 2012), targets of miR-2954-3p were markedly enriched on the Z Chromosome (Fig. 4B), particularly when we considered only 8-mer sites, which are the strongest type of target site (Agarwal et al. 2015). For this site type, 47 of the 129 (36.4%) predicted miR-2954-3p targets were on the Z Chromosome, whereas the corresponding number for targets of all autosomal miRNAs was 327 of 7185 (4.6%), corresponding to an eightfold enrichment (P = 2.9 × 10−58, χ2-test).
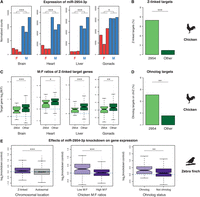
Role of miR-2954-3p in dosage compensation. (A) Expression of miR-2954-3p in chicken tissues. Raw read counts were normalized with DESeq2. (B) Proportion of Z-linked genes among the genes that were predicted to be targets of miR-2954-3p and autosomal miRNAs. Target prediction was performed with TargetScan and only 8-mer sites were considered. (C) Log2-transformed M:F expression ratios for Z-linked target genes of miR-2954-3p or autosomal miRNAs. (D) Proportion of annotated ohnologs between Z-linked targets of miR-2954-3p and autosomal miRNAs. (E) Log2-transformed expression ratios for protein-coding genes following miR-2954-3p knockdown compared to control. Genes that are down-regulated by miR-2954-3p are expected to have positive ratios. The first panel shows Z-linked genes in purple and autosomal genes in gray. The following two panels show Z-linked genes predicted to be more (light purple) or less (dark purple) dosage-sensitive based on chicken expression data and ohnolog annotations. Only genes that were 1-to-1 orthologs and located on the Z Chromosome in both chicken and zebra finch were included in the analyses. Statistical significance: (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
Although there is no chromosome-wide dosage compensation in chicken, Z-linked genes differ in their M:F expression ratios, with a tendency for dosage-sensitive genes to be more equally expressed between the sexes (Zimmer et al. 2016), but the regulatory means through which this gene-specific dosage compensation is achieved are not fully understood (Discussion; Melamed and Arnold 2007; Mank and Ellegren 2009; Itoh et al. 2010; Livernois et al. 2013; Wang et al. 2017). We reasoned that miR-2954-3p, with its strong male bias, broad expression, and preference for Z-linked genes might provide an alternative path to gene-specific dosage compensation. In order to evaluate whether miR-2954-3p regulation is indeed associated with more similar expression patterns in males and females, we calculated M:F ratios for all Z-linked protein-coding genes based on expression data from Marin et al. (2017) and compared target genes of miR-2954-3p to target genes of autosomal miRNAs (Methods). Consistent with the dosage compensation hypothesis, we observed significantly smaller M:F ratios for miR-2954-3p targets (Fig. 4C), indicating that the miRNA compensates for gene dosage differences, leading to more similar expression levels between males and females. As expected, we did not observe a significant difference between the two types of targets when we repeated the analysis for autosomal genes (Supplemental Fig. S3A).
If down-regulation by miR-2954-3p serves as a dosage compensation mechanism, we would furthermore expect an enrichment of dosage-sensitive genes among its targets. A recent study demonstrated that one class of dosage-sensitive genes known as ohnologs (gene duplicates retained after whole genome duplication) are preferentially dosage-compensated compared to other genes on the chicken Z Chromosome (Zimmer et al. 2016). Drawing on this observation, we counted the number of reported ohnologs (Singh et al. 2015) that were identified in our target prediction analysis. We found that 25.5% (12 of 47) of Z-linked miR-2954-3p targets were indeed ohnologs, whereas only 7.8% (15 of 192) of Z-linked genes targeted by autosomal miRNAs belonged to this category (P = 0.0015, χ2-test) (Fig. 4D). No significant difference was found among autosomal target genes (Supplemental Fig. 3B). As expected under the dosage compensation hypothesis, our data thus show that miR-2954-3p preferentially targets Z-linked dosage-sensitive genes.
The results described above make a compelling case for the involvement of miR-2954-3p in gene-specific dosage compensation, but it should be noted that the targeting properties of miR-2954-3p were so far determined based solely on computational predictions. To extend our results to targets with experimental support, we turned to a data set generated by Lin et al. (2014), who measured mRNA expression following knockdown of miR-2954-3p in a male zebra finch cell line. We first wished to verify the Z preference of miR-2954-3p and therefore analyzed the degree of up-regulation of Z-linked and autosomal mRNAs following miR-2954-3p knockdown. Consistent with our target predictions in chicken, we found that Z-linked genes in zebra finch were significantly more up-regulated than autosomal genes (Fig. 4E). The enrichment of Z-linked miR-2954-3p targets was further underlined when we considered only mRNAs with significant up- or down-regulation following miR-2954-3p knockdown (FDR 5%): Among the autosomal genes, only 45.2% (1296 of 2868) were up-regulated after knockdown, suggesting that many autosomal genes might not be direct targets of miR-2954-3p, whereas 62.0% (93 of 150) of Z-linked genes were up-regulated, as expected if some Z-linked genes are directly suppressed by miR-2954-3p (P = 8.0 × 10−5, χ2-test). Our findings thus demonstrate that miR-2954-3p disproportionally targets Z-linked genes in both chicken and zebra finch, which separated over 70 million years ago (Prum et al. 2015).
As miR-2954-3p-induced dosage compensation appears to be a conserved feature of bird genomes, we speculated that predictions of dosage sensitivity derived from chicken data should be reflected in the response to miR-2954-3p knockdown in zebra finch, in spite of the evolutionary distance between the two species. We therefore divided genes into those with above-median and below-median M:F ratios, based on expression in the chicken brain, and evaluated their response to miR-2954-3p knockdown in the zebra finch cell line, which was derived from a non-neural head tumor (Lin et al. 2014). We found that genes with low M:F ratios in chicken were more strongly up-regulated in zebra finch after knockdown (P = 2.2 × 10−4) (Fig. 4E), consistent with a conserved role of miR-2954-3p in gene-specific dosage compensation. We confirmed that the same effect was evident when we repeated the analysis based on zebra finch M:F ratios (P = 0.0027) (Supplemental Fig. S4), again based on gene expression in the brain (Itoh et al. 2010). Further to the conserved association with chicken M:F ratios, we found that ohnolog annotations from the chicken genome also correlated with the effects of miR-2954-3p in zebra finch, with ohnologs being more strongly up-regulated after miR-2954-3p knockdown (P = 0.0039) (Fig. 4E). Together, our findings thus demonstrate that the strongly male-biased, ubiquitously expressed miR-2954-3p preferentially targets and down-regulates dosage-sensitive genes on the bird Z Chromosome. To our knowledge, this is the first report of miRNA regulation as a means of dosage compensation.
Discussion
Sex-biased gene expression has important implications for phenotypic variation between males and females (Grath and Parsch 2016; Mank 2017). Here, we have performed the first large-scale study of sex-biased miRNA expression, by analyzing somatic and gonadal tissues from mouse, opossum, and chicken. We chose to focus on adult samples, which we consider especially suited for the study of sex-biased miRNAs in somatic tissues that are largely similar in males and females. Although we also identified ample examples of ovary-biased and testis-biased miRNAs, their designation as sex-biased is less straightforward. In evolutionary terms, these miRNAs are probably best viewed as sex-biased, as their expression pattern likely causes them to have a higher impact on fitness in one of the sexes. However, from a regulatory perspective, ovary-biased and testis-biased miRNAs may be better described as tissue-biased, since male and female gonads start diverging from a common tissue precursor in early development and have highly distinct transcriptional programs in adulthood (Mank et al. 2010; Necsulea et al. 2014). To study the regulatory origins of gonadal miRNA expression, a more detailed investigation of sex-biased miRNA expression throughout development would be a useful first step (Bannister et al. 2009).
Our work revealed divergent regulatory regimes between species, with an overarching coordination of sex bias across tissues in mouse, which was not significant in opossum and fully absent in chicken. Potentially these differences reflect the reduced reliance on gonadal hormones to establish sex identity in chicken (Zhao et al. 2010). Given the regulatory divergence between the species studied here, it is perhaps not surprising that we observe very limited evolutionary conservation of sex-biased miRNA expression across species. In fact, we were not able to identify a single case in which an orthologous (Kozomara and Griffiths-Jones 2014; Fromm et al. 2015) mature miRNA showed significant bias in the same direction and in the same somatic tissue in two species (Supplemental Fig. S5). Among the overwhelming numbers of ovary- and testis-biased miRNAs, we did find some shared instances, such as miR-34a-5p, which was testis-biased in all three species (Supplemental Table S4). However, we also found multiple cases of miRNAs that had radically changed their expression patterns, including miR-34b-5p and miR-34c-5p, which were testis-biased in opossum and mouse, in which knockout of the mir34b/c cluster has been shown to disrupt spermatogenesis (Wu et al. 2014), but which were strongly ovary-biased in chicken. Thus it appears that the evolutionary turnover of sex-biased miRNA expression is relatively fast, similar to what has been observed for the expression of protein-coding genes (Harrison et al. 2015).
One clear example of an evolutionarily conserved sex-biased miRNA, the male-biased avian miRNA miR-2954-3p, was nevertheless evident in the data presented here (Table 1) and in previous studies (Zhao et al. 2010; Luo et al. 2012). Our analyses of miR-2954-3p targets in chicken and zebra finch revealed that the miRNA plays a role in dosage compensation of the bird Z Chromosome, by preferentially down-regulating Z-linked dosage-sensitive genes in males (Fig. 4). The need for dosage compensation arises during sex chromosome evolution when increasingly larger regions of the originally identical sex chromosomes cease to recombine, leading to the gradual decay of one member of the pair, e.g., the female W Chromosome in birds (Graves 2016). Although some genes, such as the mir-122 locus, are retained on the W Chromosome and therefore are present in equal numbers in males and females, the number of W-linked genes in chicken is vanishingly small (Bellott et al. 2017) and most Z-linked genes are therefore present in a single copy in ZW females and double copies in ZZ males. The reduced gene dosage in ZW females, from two copies prior to W decay to the present single copy, is potentially problematic, since Z-linked genes might interact with autosomal genes that remain in their ancestral double-copy state in both sexes. As a coping strategy, many lineages have evolved various mechanisms of dosage compensation, which typically involve equalization of gene expression in males and females in conjunction with the evolution of adjusted expression ratios between sex-linked genes and their autosomal partners (Julien et al. 2012; Graves 2016). In mammals, differences between males and females are reduced through female global X inactivation (Deng et al. 2014), but birds lack any such chromosome-wide dosage compensation mechanism, and the resulting male-biased expression of Z-linked genes (Ellegren et al. 2007; Itoh et al. 2007; Mank and Ellegren 2009; Julien et al. 2012) can therefore be assumed to prevent Z-linked and autosomal gene expression levels from being optimally adjusted in both sexes. That said, previous work has demonstrated that M:F expression ratios vary across Z-linked genes, such that the intrinsic male bias is counteracted for a subset of dosage-sensitive genes (Zimmer et al. 2016), but it has not been clear how such gene-specific dosage compensation is achieved. A handful of Z-linked genes may be affected by local Z inactivation (Livernois et al. 2013), although more extensive Z inactivation is not supported by patterns of allele-specific expression (Wang et al. 2017). Additionally, it has been suggested that a small region of the chicken Z Chromosome might be enriched for dosage-compensated genes (Melamed and Arnold 2007), but the pattern was not recapitulated in another study (Mank and Ellegren 2009) and does not appear to be evolutionarily conserved (Itoh et al. 2010).
Our finding that miR-2954-3p acts to equalize male and female expression levels of Z-linked, dosage-sensitive genes in two distantly related bird species presents an alternative solution to this long-standing problem and suggests that miR-2954-3p could be an ancestral avian dosage compensation mechanism, which might nevertheless be complemented by additional means of regulation, such as local Z inactivation in males or Z up-regulation in females, in individual lineages. Notably, miRNA targeting properties might also provide a certain level of evolutionary flexibility, given that the short target site sequences required for miR-2954-3p recognition could evolve de novo or deteriorate quite easily, thus providing an opportunity to fine-tune the extent of dosage compensation on a gene-by-gene, or even transcript-by-transcript, basis. In addition, the versatility of miRNA regulation might allow this strongly male-biased miRNA to be co-opted for sex-specific functions in individual lineages, as suggested by its involvement in the zebra finch song response (Gunaratne et al. 2011; Lin et al. 2014). Careful investigation of miR-2954-3p targets across a range of bird species therefore promises to yield valuable insights not only into the evolutionary dynamics of partial dosage compensation, but also into the genetic architecture underlying sex-specific characteristics.
Methods
Sequencing and processing of small RNA libraries
Total RNA for adult male and female tissue samples from mouse (Mus musculus), gray short-tailed opossum (Monodelphis domestica), and chicken (Gallus gallus) was extracted from 10 to 20 mg of tissue using the RNeasy Micro Kit (Qiagen) with 350 µL lysis buffer RLT containing 7 µL 2 M Dithiothreitol and wash buffer RWT, and the miRNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. Total RNA was quantified by a NanoDrop spectrophotometer (Thermo Scientific). The quality of total RNA was measured with a Fragment Analyzer (Advanced Analytical Technologies). Small RNA 15–45 nt fraction was purified from 0.5–2 µg total RNA by denaturing 15% TBE-Urea polyacrylamide gel electrophoresis, treated with 0.3 M sodium chloride for 4 h at room temperature, and ethanol precipitated. Libraries were generated using the NEXTflex Small RNA-Seq Kit (Bioo Scientific) according to the manufacturer's protocol. As a comparison, libraries were also prepared from the small RNA fraction purified from 2 µg total RNA of one mouse brain and one mouse liver sample using the lllumina TruSeq Small RNA Library Prep Kit (Illumina) according to the manufacturer's instructions, as well as using the HD protocol developed by Sorefan et al. (2012). The libraries were sequenced at the Lausanne Genomic Technologies Facility on an Illumina HiSeq 2500 System (Illumina). Adapters were removed from the raw sequencing reads, and only trimmed reads in the size range 15–28 nt with a minimum quality score of 20 at all positions were retained for further analysis.
Annotation of novel miRNAs
All analyses were carried out on the following genome releases, which were downloaded from the UCSC Genome Browser (Tyner et al. 2017): mm10 (mouse), monDom5 (opossum), and galGal5 (chicken). For each library, we annotated novel miRNAs using miRDeep2 and standard settings, including the randfold step (Friedländer et al. 2012). Known miRNAs were downloaded from miRBase release 21 (Kozomara and Griffiths-Jones 2014). We found that the miRDeep2 algorithm performed better (detected more miRNAs at a given signal-to-noise cutoff) for single libraries compared to when we pooled all reads from a species together. We therefore assessed each library individually and set a cutoff score of 5, which corresponded to a signal-to-noise ratio of 9.6–23.3, with the exception of the three chicken testis libraries in which the ratio was 4.6–5.2. Using a higher cutoff score for the chicken testis libraries did not improve the signal-to-noise ratio. Instead, we opted to improve the quality of our annotations further by retaining only those miRNA precursors with a miRDeep2 score above the cutoff in at least three tissue replicates (male and female). Ovary and testis were considered the same tissue for the purpose of this filtering step. Overlapping precursors on the same strand were merged provided that the mature and star sequences predicted by miRDeep2 also overlapped. In addition to the score, miRDeep2 also provides graphical representations of predicted miRNA precursors. A subset of these, corresponding to all novel miRNA precursors that are further discussed in the Results section, are provided in Supplemental File S1. The full set of PDF files is available at http://www.zmbh.uni-heidelberg.de/Kaessmann/Data_Resources.html, and may be generated by rerunning miRDeep2 for each sample, using standard settings as described above.
Before combining the annotations for known and novel miRNA precursors, we trimmed the coordinates for known precursors to only include the 5p and 3p mature sequences as well as the loop sequence. In addition, we mapped all known trimmed precursor sequences to the respective genomes with Bowtie release 1.1.2 (Langmead et al. 2009), with no allowed mismatches, in order to remove the handful of known precursors that lacked a perfect match in the genome and include known precursor sequences that either lacked coordinates in miRBase or in which the coordinates differed between miRBase and our mapping. Finally, we removed all precursors that had been flagged by miRDeep2 as potentially belonging to a different RNA class, as well as all precursors that overlapped with annotated RNAs, including transfer RNAs (tRNAs), ribosomal RNAs (rRNAs), and small nuclear RNAs (snRNAs), from the UCSC Genome Browser RepeatMasker tracks for each species (Tyner et al. 2017). Genome coordinates were intersected with BEDTools (Quinlan 2014). The identified set of putative novel miRNA precursors, together with known precursors from miRBase, formed the basis for all subsequent analyses.
Mature miRNA coordinates for both novel and known miRNA precursors were defined by first mapping reads from all libraries jointly to the relevant genome with Bowtie without allowing any mismatches and then choosing the most frequent, perfectly mapping read corresponding to the 5p and 3p ends of the each precursor. If one of the 5p and 3p ends contributed <0.1% of all mature reads for the precursor or had fewer than 100 perfect reads in total, no mature miRNA coordinates were chosen.
Detection of sex-biased miRNA expression
We mapped all reads to the relevant genomes with Bowtie release 1.1.2 (Langmead et al. 2009), using a series of decreasingly stringent settings to allow for post-transcriptional modifications (Kawahara et al. 2007; Burroughs et al. 2010; Chiang et al. 2010; Warnefors et al. 2014). In the first round, we allowed for zero mismatches and did not trim any bases from the 3′ end. Next, we mapped all the unmapped reads from the previous round, after trimming first 1 and then 2 bases from the 3′ end. Finally, we repeated the procedure while allowing for one mismatch. Reads that could not be mapped with any setting and reads with more than 10 hits (using the first setting where any hits were found) were discarded.
Because many miRNAs occur in multicopy families, the assignment of miRNA reads to individual genomic loci is not trivial, especially in the presence of post-transcriptional modifications and sequencing errors. A previous study estimated that up to two-thirds of all reads in a miRNA sequencing experiment mapped ambiguously if one mismatch was allowed (de Hoon et al. 2010). Because most tools for miRNA quantification do not offer a robust procedure for dealing with ambiguously mapped reads, we adapted the strategy of multimap groups (MMGs), which was previously developed for protein-coding genes (Robert and Watson 2015). Mature miRNAs were grouped into MMGs based on all mapped reads from a given species and a cutoff of 5%, i.e., if a given mature miRNA shared at least 5% of its reads with another miRNA, the two were assigned to the same MMG. We then summed the read counts per library for each MMG. Any remaining multimapping reads below the cutoff were distributed equally between the mapped miRNA loci.
We estimated differential expression between males and females using DESeq2 (Love et al. 2014) and default settings, with the exception that we turned off independent filtering of lowly transcribed miRNAs. Instead, we performed our own expression filtering such that only MMGs that were represented by at least 10 transcripts per million miRNA-mapped reads (TPM) in at least three of six tissue replicates were included in the analysis. P-values were adjusted jointly for all somatic tissues from all species using the Benjamini-Hochberg method (Benjamini and Hochberg 1995). We opted to perform the P-value correction for the gonad samples separately, as we found that the large number of small P-values associated with these samples caused us to call more miRNAs significant for the somatic samples.
Calculation of M:F expression ratios for protein-coding genes
Mouse and chicken expression data for protein-coding genes were provided by Marin et al. (2017), who performed RNA sequencing for two male and two female individuals and each of the tissues for which we had miRNA data. We calculated M:F ratios for all expressed genes (>1 FPKM in all replicates from a given tissue) with the DESeq2 package (Love et al. 2014) based on raw read counts.
Zebra finch data were available from the NCBI Gene Expression Omnibus (GEO) under accession number GSE20035. Genomic coordinates for GenBank ESTs that were represented on the custom-made microarray were downloaded from Ensembl and intersected with exons of annotated zebra finch genes. ESTs that matched multiple genes were excluded. For each gene, M:F ratios were calculated as the ratio between the averages of six male and six female replicates.
Association of miRNAs with upstream regulatory elements
Intronic miRNAs were identified based on positional overlap with protein-coding genes in Ensembl version 88 (Aken et al. 2017). Coordinates of 72,862 DHS sites in mouse liver, including 3378 sites that were identified as sex-biased with “standard stringency” settings, were provided by Ling et al. (2010) and converted to the mm10 genome release with the liftOver from the UCSC Genome Browser (Tyner et al. 2017). DHS sites were considered likely miRNA promoters if they occurred within 5 kb upstream of, or overlapped, a liver-expressed intronic or intergenic miRNA (>10 TPM in at least three replicates). This is a conservative cutoff, given that some miRNA transcription start sites are located 20 kb upstream of the miRNA precursor sequence (Ozsolak et al. 2008). To ensure that we associated DHS sites with transcribed miRNA loci, only miRNAs that belonged to single-member MMGs were considered for this analysis. For the permutation test, 1000 sets of randomized DHS coordinates were created with the shuffleBed tool from BEDTools (Quinlan 2014).
Gene Ontology analysis of targets of sex-biased miRNAs in mouse
Predicted targets with context++ scores below –0.1 were downloaded from TargetScanMouse release 7.1 (Agarwal et al. 2015). For each tissue, we first filtered out target genes without robust expression (<1 FPKM), based on data from Marin et al. (2017), and then divided the remaining target genes into targets of male-biased (targeted by at least one male-biased miRNA, but no female-biased miRNAs), female-biased, and unbiased miRNAs. Genes that were predicted targets of both male-biased and female-biased miRNAs were discarded. Enrichments of Gene Ontology categories among targets of male-biased or female-biased miRNAs, compared to all expressed genes targeted by sex-biased and/or unbiased miRNAs, were investigated with the GOrilla tool (Eden et al. 2009).
Detection of putative Y-linked and W-linked miRNAs
We searched for putative Y-linked and W-linked miRNAs using a transcriptome subtraction approach (Cortez et al. 2014), in which we removed all reads that were found in both sexes and focused on reads that were specific to the heterogametic sex (XY males or ZW females), while using reads specific to the other sex as a control. For a sequence to be included in the analysis, it had to be represented by at least 100 reads in total and be detected in all three sex-specific replicates from at least one tissue. Next, we mapped the filtered reads using our sequential Bowtie pipeline to the respective genome (while excluding the Y and W Chromosomes, if present) to remove all reads that mapped loci that are present in both males and females. For mouse and chicken, for which Y/W Chromosome assemblies are available, we finally checked whether the recovered reads were Y/W-linked by mapping them to the full genome, again relying on the sequential Bowtie pipeline.
Analysis of miR-2954-3p targets in chicken and zebra finch
We downloaded chicken 3′ UTR sequences for Ensembl genes from the UCSC Genome Browser (Tyner et al. 2017) (galGal5) and predicted target genes for all chicken miRNAs with TargetScan version 7.0 (Agarwal et al. 2015). M:F expression ratios for target genes were calculated as described above. Ohnolog status (strict data set) for Z-linked genes was provided by Singh et al. (2015). Expression data from the miR-2954-3p knockdown in zebra finch was provided by Lin et al. (2014). For clarity, we reversed the sign of their reported expression fold changes to be consistent with the main text. Data were compared between chicken and zebra finch based on 1-to-1 orthologs from Ensembl release 88 (Aken et al. 2017) that were Z-linked in both species. All statistical analyses were performed in R version 3.2.2 (R Core Team 2015).
Data access
Raw and processed data sets from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE102062. Novel miRNAs have been submitted to miRBase (Kozomara and Griffiths-Jones 2014).
Acknowledgments
We thank the Lausanne Genomics Technology Facility for high-throughput sequencing support, Margarida Cardoso Moreira and Ray Marin for assistance with the expression data for protein-coding genes, and Zhongyi Wang and all members of the Kaessmann group for helpful discussion. This research was supported by grants from the European Research Council (Grant 615253, OntoTransEvol) and Swiss National Science Foundation (Grant 146474) to H.K.
Author contributions: M.W. designed and performed all computational analyses. K.M. and J.H. prepared sequencing libraries. T.S. assisted with the computational analyses. J.L.V. provided high-quality, gray short-tailed opossum samples and key biological expertise regarding this species. I.L., A.F., and P.J. provided high-quality, red jungle fowl samples and key biological expertise regarding this species. H.K. supervised the study. M.W. wrote the paper with input from all authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.225391.117.
-
Freely available online through the Genome Research Open Access option.
- Received May 22, 2017.
- Accepted September 21, 2017.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








