
Drought responsive gene expression regulatory divergence between upland and lowland ecotypes of a perennial C4 grass
- John T. Lovell1,
- Scott Schwartz1,
- David B. Lowry2,
- Eugene V. Shakirov1,3,
- Jason E. Bonnette1,
- Xiaoyu Weng1,
- Mei Wang4,
- Jenifer Johnson4,
- Avinash Sreedasyam5,
- Christopher Plott5,
- Jerry Jenkins5,
- Jeremy Schmutz4,5 and
- Thomas E. Juenger1
- 1Department of Integrative Biology, University of Texas at Austin, Austin, Texas 78712, USA;
- 2Department of Plant Sciences, Michigan State University, East Lansing, Michigan 48824, USA;
- 3Institute of Fundamental Medicine and Biology, Kazan Federal University, Kazan 42008, Republic of Tatarstan, Russia;
- 4Department of Energy Joint Genome Institute, Walnut Creek, California 94598, USA;
- 5HudsonAlpha Institute for Biotechnology, Huntsville, Alabama 35806, USA
- Corresponding author: johntlovell{at}gmail.com
Abstract
Climatic adaptation is an example of a genotype-by-environment interaction (G×E) of fitness. Selection upon gene expression regulatory variation can contribute to adaptive phenotypic diversity; however, surprisingly few studies have examined how genome-wide patterns of gene expression G×E are manifested in response to environmental stress and other selective agents that cause climatic adaptation. Here, we characterize drought-responsive expression divergence between upland (drought-adapted) and lowland (mesic) ecotypes of the perennial C4 grass, Panicum hallii, in natural field conditions. Overall, we find that cis-regulatory elements contributed to gene expression divergence across 47% of genes, 7.2% of which exhibit drought-responsive G×E. While less well-represented, we observe 1294 genes (7.8%) with trans effects. Trans-by-environment interactions are weaker and much less common than cis G×E, occurring in only 0.7% of trans-regulated genes. Finally, gene expression heterosis is highly enriched in expression phenotypes with significant G×E. As such, modes of inheritance that drive heterosis, such as dominance or overdominance, may be common among G×E genes. Interestingly, motifs specific to drought-responsive transcription factors are highly enriched in the promoters of genes exhibiting G×E and trans regulation, indicating that expression G×E and heterosis may result from the evolution of transcription factors or their binding sites. P. hallii serves as the genomic model for its close relative and emerging biofuel crop, switchgrass (Panicum virgatum). Accordingly, the results here not only aid in the discovery of the genetic mechanisms that underlie local adaptation but also provide a foundation to improve switchgrass yield under water-limited conditions.
Locally adapted populations outperform foreign populations in their native site but are less fit at other sites. There is a growing appreciation that this reciprocal home site advantage, which characterizes local adaptation, is driven by selection not only on coding sequence polymorphisms but also on gene expression regulatory variants (King and Wilson 1975; Hoekstra and Coyne 2007; Prud'homme et al. 2007; Stapley et al. 2010; Fraser 2011). Genotype-by-environment interactions (G×E), which underlie adaptive physiological variation and cause adaptation, have been partially resolved in a variety of species (e.g., Angert and Schemske 2005; Ågren and Schemske 2012; Anderson et al. 2012); however, the patterns of G×E at the gene expression level are less well understood (but see Hannah et al. 2006; Fraser 2011; Des Marais et al. 2013; Lasky et al. 2014).
Several studies have suggested that gene expression regulatory elements may be particularly likely to be involved in adaptive evolution (Wray et al. 2003; Wittkopp and Kalay 2011; Fraser 2013). This conclusion is supported by the observation that expression regulation can be tissue and environment specific while nonsynonymous mutations may alter protein function regardless of environmental cues (Wagner and Lynch 2008). Gene expression regulatory elements underlying adaptation may exist in close physical proximity to the target gene (cis) or in distant and often physically unlinked regions (trans). While cis-acting elements typically cause expression variation in a single gene, trans-acting modifiers may affect expression of several or, in the case of global transcription factors, thousands of genes. Mutations at loci with wide-ranging pleiotropic effects are typically deleterious (Keightley and Hill 1990; Kondrashov and Turelli 1992), leading to the hypothesis that trans-acting expression regulation may be subjected to purifying selection (Emerson et al. 2010), while cis-acting regulatory elements may respond to directional selection and contribute to adaptive differentiation (Schaefke et al. 2013). Indeed, many regulatory factors underlying physiological responses to drought and other stresses are conserved across broad phylogenetic distances (Matsukura et al. 2010; Des Marais et al. 2012; Mizoi et al. 2012).
Differentiation among plant populations across abiotic stress gradients provides some of the most compelling and best understood examples of adaptation in nature (e.g., Clausen et al. 1948; Stebbins 1950; McMillan 1959; Rieseberg and Willis 2007). In particular, natural selection imposed by differences in soil water availability is one of the most common and strongest drivers of local adaptation among plant populations (Stebbins 1952; Juenger 2013). To survive and reproduce in regions with a threat of periodic droughts, plants must alter their physiology quickly and effectively (Bohnert et al. 1995; Tisné et al. 2010). Drought responses at both the gene expression and whole-plant levels are implicated as major forces underlying local adaptation in a variety of plant species (Ramanjulu and Bartels 2002; Chaves et al. 2003). Given these strong selective forces, it is not surprising that many plant species have evolved “upland” and “lowland” ecotypes adapted to xeric and mesic environments, respectively (Porter 1966; Yu and Nguyen 1994; Kumar et al. 2008).
Panicum hallii variety hallii and var. filipes represent an ideal system to study the genetics of adaptation to drought. P. hallii is a genetically tractable diploid model system for C4 perennial grasses with complex genomes (e.g., P. virgatum/Switchgrass). Accordingly, a variety of genomic tools have been recently developed in P. hallii (Lowry et al. 2012, 2013). Importantly, the two varieties display striking ecological divergence, where the lowland var. filipes is primarily found in mesic regions of the Gulf Coast and Rio Grande Valley of Texas and northern Mexico, while the upland var. hallii’s range extends into dry regions of the Chihuahuan and Sonoran deserts (Waller 1976; Lowry et al. 2013).
Here, we define the extent and direction of drought-responsive gene expression at both whole-transcriptome and allele-specific expression (ASE) levels. Specifically, we tested three hypotheses: (1) divergence between upland and lowland ecotypes is characterized by drought-responsive differential gene expression; (2) such gene expression G×E is regulated primarily by cis factors; and (3) trans-regulated genes will be associated with regulatory elements that are known to respond to abiotic stress. To assess these hypotheses, we conducted a combined analysis of parental genotypes of each variety and their hybrid progeny across drought and drought recovery conditions. In doing so, we characterize the effects of cis-, trans-, and drought-responsive gene expression regulation that may underlie physiological divergence between the upland and lowland ecotypes of P. hallii.
Results and Discussion
Climatic context of the experimental drought
The two parental genotypes used in this experiment, HAL2 (var. hallii) and FIL2 (var. filipes) (Fig. 1A), represent the reference genomes for each variety. The HAL2 accession was collected from a population in xeric oak-shrub savanna at the Ladybird Johnson Wildflower Center (Austin, TX, USA 97.87°W, 30.18°N), while the FIL2 accession was collected from the Corpus Christi Botanical Gardens (Corpus Christi, TX, USA, 97.40°W, 27.65°N). These sites are representative of xeric hill country savanna and mesic coastal plains habitats.
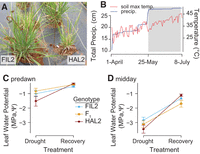
Climatic conditions and leaf-level responses across a natural drought and recovery. (A) An image of FIL2 (left) and HAL2 (right) that was taken prior to the onset of drought demonstrates the reduced growth rate of HAL2 compared to its lowland relative. From May 25 through July 5, 2013, <1 cm of rain fell at the site of the experiment. (B) Cumulative rainfall is presented along with daily maximum soil temperature. Leaf water potential (MPa, Ψ) was measured on July 5 (drought) and again on July 8 after irrigation and natural rainfall (recovery); means ± SE are presented for Ψ at predawn (C) and midday (D).
We grew the two ecotypes and their F1 hybrid progeny (“F1”) in a replicated common garden. The genotypes were vegetatively cloned (HAL2: n = 28, FIL2: n = 35, and F1: n = 29), planted in the field in October 2012, and subjected to a 41-d natural drought at the Ladybird Johnson Wildflower Center in 2013 (Fig. 1B). The 2013 drought represented the driest period from May 25 to July 5 since 1946 in Austin, TX (Supplemental Fig. S1A,B). We utilized this natural drought to test for responses to soil moisture variation by comparing droughted plants to those subjected to an experimental rewatering treatment. On July 5, 2013, after 41 d of drought, we collected leaf tissue and measured leaf water potential. On July 7, 2013, each plant was irrigated with 4 L of water. On July 8, 2013, leaf water potential was again assayed, and leaf tissue was collected on an independent subset of plants. Samples collected from the first harvest constituted the “drought” treatment, while the subsequent harvest, which followed watering, represented the “recovery” experimental treatment.
Leaf water potential (Ψ) was 2.2× lower in the drought treatment (F(df=1) = 78.7, P < 0.001) (Fig. 1C,D) regardless of genotype, indicating that our recovery treatment sufficiently relaxed the leaf-level physiological stresses of drought. While there was little evidence of an additive effect of genotype on Ψ (F(df=2) = 0.8, P > 0.1), there was a significant genotype-by-environment interaction (G×E) (F(df=2) = 3.43, P < 0.05) (Fig. 1C,D). This G×E was driven by strong drought responses of the HAL2 genotype in both predawn and midday leaf water potential measurements. Increased phenotypic plasticity to drought may contribute to the whole-plant drought adaptation characteristic of var. hallii (Lowry et al. 2015).
Drought-responsive gene expression divergence between upland and lowland ecotypes
Leaf tissue was harvested and RNA was extracted from the most recently expanded leaf from 17 FIL2, 12 HAL2, and 14 F1 clones in the drought treatment and 17 FIL2, 13 HAL2, and 15 F1 clones in the recovery treatment. Whole RNA sequencing yielded 22,256 genes with mean counts >5 (Supplemental Fig. S2). These expressed genes constituted 59.3% of the total gene models, indicating that a majority of genes were expressed in the most recently expanded leaves under natural field conditions. We fit a generalized linear model via DESeq2 (Love et al. 2014) to the transcript count data and calculated Wald test contrast P-values for genotype, treatment, and G×E model terms. For each contrast, we applied multiple testing corrections via Q-values calculated from the P-value distributions (Storey 2002). Genes with Q-values ≤0.05 were determined to have a significant effect. We also estimated the proportion of true null hypotheses (π0) (Storey et al. 2004) as an alternative approach to count the number of genes that were affected by an experimental factor.
Across all genes, 15,332 (π0 = 0.264) and 16,766 (π0 = 0.224) genes were affected by treatment and genotype, respectively (Fig. 2A). While the number of genes affected by each factor was relatively similar, the effect size (calculated as the mean absolute value of the log2 fold change [LFC]) due to the genotype term (1.59) was >2.5× that of treatment (0.63). Consistent with this observation, the most common expression profile was found among genes regulated in the same direction across both treatments between HAL2 and FIL2 (Fig. 2A). However, 5471 genes were differentially expressed exclusively in the drought or drought-recovery treatment. Interestingly, these drought-responsive genes tended to be up-regulated (HAL2 > FIL2) in drought (52%, binomial P = 0.01) but down-regulated in the recovery treatment (58%, binomial P < 0.0001) (Fig. 2A).
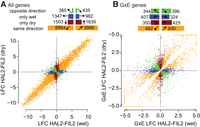
Distribution of differential expression of total counts between HAL2 and FIL2 parents in drought and recovery treatments. Log2 fold changes (LFC) between HAL2 and FIL2 were calculated for all genes independently in each experimental treatment. (A) All genes with significant genotype effects are presented. (B) Only those genes with significant G×E effects are presented; those G×E genes with log2 fold changes greater than 5 or less than −5 are shrunk to 5 (−5) for plotting purposes. In each panel, genes were binned by the direction and significance of the LFC. The numbers of genes belonging to each bin are displayed as horizontal bar plots at the top of each panel.
We also observed significant (q ≤ 0.05) patterns of genotype-by-environment interactions (G×E) among the expression of 3907 genes (Fig. 2B). Nearly half of the G×E genes (1795, 46%) were driven by differential expression in both treatments (orange points, Fig. 2B). However, the experimental treatment drove differential expression across many of the remaining genes: 33.6% of the genes declared to have significant G×E effects were differentially expressed in one treatment but not the other. Finally, 20.5% of G×E genes had opposite response directions (green points, Fig. 2B). These rank- and sign-changing G×E genes demonstrate phenotypic trade-offs between environments and represent an interesting and potentially evolutionarily important subset of loci that may underlie adaptive differences in response to drought between var. hallii and var. filipes (Des Marais et al. 2013; Juenger 2013; Lasky et al. 2014).
To determine the identity and characteristics of the significantly differentially expressed genes, we calculated the overrepresentation of gene ontology (GO) terms and sequence motifs in the promoter (Supplemental Tables S1, S2). Across all G×E genes that were differentially expressed in drought (red points, Fig. 1A), there was a significant enrichment of several hundred GO terms (Supplemental Table S1). Some of the most interesting were those that were responsive to heat, drought, and reactive-oxygen species (Supplemental Table S1). Additionally, GO terms enriched among the opposite direction G×E genes (green points, Fig. 2B) included responses to heat, flowering, and guard-cell development. Physiological divergence of these traits has been observed among differentially drought-adapted genotypes in other systems (Lebaudy et al. 2008; Wilczek et al. 2009). Interestingly, the most highly enriched motif in HAL2 up-regulated genes is specific to an abscisic acid (ABA) responsive transcription factor (ABRE) (ABADESI2, P < 1 × 10−21). ABREs are known to increase expression in drought and elevated ABA concentrations (Lam and Chua 1991; Busk and Pagès 1998; Narusaka et al. 2003), driving adaptive drought responses (Des Marais and Juenger 2010).
Impact of drought on gene expression heterosis
The cis- and trans-acting regulatory elements that cause environmental responses are thought to contribute to patterns of heterosis at both the gene expression and whole-plant levels (Hochholdinger and Hoecker 2007; Chen 2013). We explored patterns of gene expression heterosis between var. hallii and var. filipes by contrasting total transcript abundance of each gene among HAL2, FIL2, and their F1 hybrid. Each gene was classified into one of seven expression categories based on Q-values (α = 0.05) for three separate contrasts (Table 1; Stupar and Springer 2006; Paschold et al. 2012): (1) no differential expression; (2) additive effects; (3) high-parent heterosis (hp); (4) low-parent heterosis (lp); (5) above high-parent heterosis (>hp); (6) below low-parent heterosis (<lp); and (7) ambiguous expression patterns (Table 1). It is important to note that these categorizations are conducted on the log2 scale, so genes with “additive” effects may not appear to be linear on an untransformed scale.
Heterosis categorization of all expressed genes
Across environmental treatments, a slight majority of differentially expressed genes showed additive gene expression patterns (Table 1). Additive expression patterns were found almost exclusively among the genes exhibiting significant differential expression in both environments. In fact, only 125 (1.9%) of the additive genes were differentially expressed in a single environment, and four (0.08%) additive genes were differentially expressed in opposite directions across environments (Supplemental Fig. S3).
The extreme scarcity of additive genes that were differentially expressed in only a single environment was balanced by a significant overrepresentation of genes displaying both G×E and heterosis. Hybrids exhibited the expression values of the high parent or low parent across 3365 (19.8%) and 4436 (26.1%) genes, respectively (Table 1; Supplemental Fig. S3). This bias toward below-mid-parent heterosis was highly significant (binomial test P < 0.0001). Among these 7801 genes, 3431 (44.0%) displayed significant differential expression in one environment but not the other. As such, genes with high- or low-parent heterosis represented 93% of all genes with G×E (Supplemental Fig. S3). Physiological G×E effects often underlie the fitness G×E that constitute local adaptation (Clausen et al. 1948; Ågren and Schemske 2012; Juenger 2013) and may be a result of G×E at specific loci (Hall et al. 2010; Des Marais et al. 2012; Ågren et al. 2013). Therefore, it is plausible that such heterotic G×E genes contribute to the adaptive divergence observed between these upland and lowland ecotypes.
Heterotic genes that significantly exceeded the range of the parents comprised an extreme minority. Only 104 (0.4%) and 253 (1.1%) genes were found to have above high-parent and below low-parent heterosis, respectively. Many of the heterotic genes were initially classified as conserved (Fig. 2, gray points). However, genetic variation was present for these genes between HAL2 and FIL2 but only was revealed within the F1 hybrid, implicating multiple antagonistic loci, epistatic interactions, or other forms of trans-acting gene expression regulation (Rieseberg et al. 1999; Li et al. 2008; Chen 2010).
Inference of cis- and cis-treatment gene expression G×E via a linear model
We quantified allele-specific expression for 16,465 genes by the presence of HAL2- or FIL2-specific single nucleotide polymorphisms. We then partitioned the variance of ASE into that attributable to allelic imbalance in the F1 generation (cis) and differential allelic imbalance across parental (F0) and F1 generations (trans) using a linear modeling approach (Figs. 3, 4A,B). This methodology is analogous to the binomial and χ2 tests (Supplemental Figs. S4, S5) that have been traditionally used to assess cis- and trans-acting gene expression regulation (Wittkopp et al. 2004; McManus et al. 2010; Bader et al. 2015); however, linear modeling affords several benefits, including the capability to incorporate biological replicates and to control for experimental design variables (i.e., blocking, treatment, and other covariates). We tested the effect of cis, trans, experimental treatments, and all additive and interactive combinations therein using a linear model specification within the DESeq2 negative binomial framework (Bader et al. 2015) (see the Supplemental Material for comparisons between the DESeq2 model specification, mixed models, and traditional cis-trans test methods).
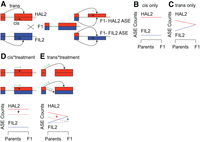
Description of the F0/F1 ASE test for cis- and trans-acting gene expression regulation. (A) Each inbred parent possesses only the cis and trans factors associated with that genotype, while the F1 hybrid possesses both. (B) In the simplest case where ASE patterns are affected by a single cis element, allelic imbalance ratios are identical across generations. (C) In the case where a single trans factor causes ASE variation, the F1 ASE counts should be identical for both alleles since both trans factors are acting on each allele. To illustrate the effects of cis- and trans-by-treatment interactions, take the case where an experimental treatment induces the expression of a HAL2-specific cis- or trans-acting repressor (green symbol, dashed lines). The resultant expression patterns would shift so that the difference between alleles (cis-by-treatment, D) or the slope of the genotype*generation interaction depends on the treatment (trans-by-treatment, E). The scheme presented here is a simplification, and all combinations of cis, trans additive effects and interactions with the environment are possible.
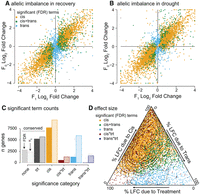
Distribution of cis, trans, and environmental interaction effects. The log2 fold change of the FIL2 vs. the HAL2 allele in the F0 and F1 generations is plotted for the recovery (A) and drought treatments (B). Coloration is derived from the significance of terms (allele—cis, allele*generation—trans). (C) All ASE genes were categorized by the model terms that caused significant differential expression. The number of genes in each bin was determined by a strict FDR threshold (left, darker bars) and the estimated number of true null hypotheses (π0, right, transparent bars) methods. Where both additive and interactive terms were significant for cis or trans (e.g., best model = y ∼ cis + cis*treatment), the model was binned into the interactive effect category. (D) For those genes with significant genetic effects (cis and/or trans), the proportions of log2 fold change due to cis, trans, and treatment are plotted in the ternary diagram. Total variance is the sum of the three components and variance attributable to interaction terms is ignored (but see Fig. 5).
We found 17,524 (78.7%) genes that were differentially expressed when contrasting the parents; however, only 8086 (49.8%) of the 16,465 genes that had detectable ASE had significant cis- or trans-regulated allele-specific expression (Fig. 4A–C). This discrepancy was primarily a result of larger allelic imbalance effect sizes in the F0 compared to the F1 generation (Fig. 4A,B; Supplemental Fig. S6; Supplemental Material). Nonetheless, our experiment had surprisingly strong power to detect cis effects. We found that >47% (7699) of gene expression patterns were affected by cis factors, a finding that exceeded other published studies using linear modeling to test for cis effects, which ranged from 15% to 30% (Cubillos et al. 2014; Bader et al. 2015). The increased power is likely attributable to significant genomic divergence between the parents and individual, rather than pooled, sequencing of each biological replicate (Liu et al. 2014).
Combined, the additive effects of cis factors and treatment explained the bulk of differential ASE (Fig. 4D); however, many genes also displayed complex G×E patterns (Supplemental Fig. S6). Across the 8379 genes with genotype-driven ASE, we estimated that expression was affected by a cis-by-treatment term across 2030 genes (π0 = 0.76); however, more stringent methods revealed many fewer genes (562, Q-value ≤0.05) (Fig. 4C,D). As in the G×E allelic imbalance tests, the most common patterns of cis-by-treatment effects were found in cases where both expression responses had the same sign and the alleles retained their relative ranks across treatments but the difference between means shifted by treatment (e.g., HAL2 >> FIL2 in wet, HAL2 > FIL2 in dry) (Supplemental Fig. S6).
Trans and trans-by-treatment expression regulatory divergence
In the linear model, inference of the trans effect is captured by an allele-by-generation interaction (Fig. 3; Bader et al. 2015). We estimated that the expression of 5969 (π0 = 0.63) of the 16,465 genes with quantifiable ASE were affected by trans factors; however, in many cases the trans effect size was relatively small (Fig. 4D). Therefore, it is not surprising that more stringent Q-value thresholding found only 1285 trans-regulated genes (19.8%) (Fig. 4C), most of which were also regulated by cis factors (Fig. 4D). Of these genes, 154 (Supplemental Fig. S7) showed significant homogenization in the F1 generation, a pattern potentially driven by the complementation of nonfunctional trans factors in the F1 (Supplemental Fig. S7; Paschold et al. 2012).
Among the trans-regulated genes, we observed patterns indicative of compensatory evolution in the expression of 312 genes (Supplemental Fig. S7). Expression of these genes was conserved in the parents but divergent in the F1. Such compensatory evolution is likely a result of co-evolution among cis and trans factors (Landry et al. 2005). In this case, selection may have favored consistent expression among genotypes, but the trans-acting factors producing such consistency were not identical-by-state in each genotype.
Transcription factors and other regulatory elements are expected to drive the expression of trans-regulated genes. As such, we expected that many transcription-factor target motifs would be highly enriched among compensatory and other trans-regulated genes. Since differential drought adaptation is one of the primary factors characterizing HAL2-FIL2 divergence, we hypothesized that genes that have diverged due to trans regulation would tend to contain drought-responsive element binding factor target motifs. The data strongly support this hypothesis: Three of the four most enriched motifs among all compensatory trans-regulated gene promoters corresponded to the binding sites of ABA-responsive transcription factors (Supplemental Table S2), including the above mentioned ABADESI2 and two other ABRE members.
We found that many genes (1287, π0 = 0.812) were marginally affected by trans-by-treatment interactions (Fig. 4C). However, FDR thresholding reduced this number to only nine significant genes (Fig. 4C,D). This discrepancy was driven by the relatively small effects of trans-by-treatment interactions (Fig. 4D; Supplemental Fig. S8) and decreased power to detect interaction terms relative to main effects in linear models. The nine genes with expression affected trans-by-treatment interactions (Table 2; Fig. 5; Supplemental Fig. S8) had the strongest effects and spanned a diverse array of possible G×E expression patterns (Fig. 5; Supplemental Fig. S9). Due to the small number of genes affected by trans-treatment interaction, neither GO nor motif enrichment analyses can be reasonably applied. We therefore examined the Arabidopsis annotations of these genes directly. Interestingly, five of these nine were orthologs of Arabidopsis or rice genes with annotations related to drought or other stress responses: (1) UTG85A2 (Fig. 5A)—such glycosyltransferase family genes have been shown to disrupt the abscisic acid responsive pathway (Priest et al. 2006), affecting drought responses (Tognetti et al. 2010); (2) TINY2, a DREB family transcription factor (Fig. 5B)—DREBs are the most extensively studied drought-responsive group of genes in Arabidopsis (Sakuma et al. 2002; Agarwal et al. 2006; Xianjun et al. 2011); (3) FT, a central gene in the flowering pathway (Fig. 5C)—several recent studies have implicated a physiological and genetic tie between drought-responsive and flowering pathways (McKay et al. 2003; Lovell et al. 2013, 2015; Riboni et al. 2013; Kimura et al. 2015); (4) RLK1, the rice ortholog (OsLecRK4) which improves resistance to insect herbivory (Liu et al. 2015), and (5) a rice ortholog (OSMate2) of the MATE efflux family gene AT1G71140, which, when overexpressed in A. thaliana, alters a variety of abiotic and biotic stress responses (Tiwari et al. 2014).
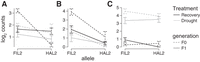
Expression patterns of three genes with significant trans-treatment interactions. Mean log2 transformed and library size corrected counts are plotted for Pahalv11b006276 (A), Pahalv11b030791 (B), and Pahalv11b035347 (C). The Arabidopsis TAIR gene identifiers and pseudonyms for each gene can be found in Table 2.
Description of the nine genes with significant trans-by-treatment interactions
We found support for similar physiological roles of these genes in Arabidopsis and P. hallii. For example, in the drought treatment, alleles of the P. hallii TINY2 ortholog were strongly diverged in the F0 generation but had identical expression in the F1. However, in the recovery treatment, the transcripts were barely quantifiable, regardless of the genotype or generation. This pattern, which yielded a strong trans-by-treatment effect in our experiment, is consistent with the documented expression patterns of TINY2 in A. thaliana (Wei et al. 2005). Finally, ABRE cis elements are enriched within promoters of DREB2A downstream genes (Sakuma et al. 2006). Since TINY2 is a member of the DREB family of transcription factors and ABRE motifs are significantly overrepresented in the promoters of trans-regulated genes observed here, it is plausible that the P. hallii ortholog of TINY2 may be one of the transcription factors causing such trans regulatory divergence. These observations may provide a fertile avenue for future research about the genetic networks that have diverged to cause differential drought adaptation of upland and lowland ecotypes of P. hallii.
Conclusions
Across all genes, treatment and cis effects accounted for the bulk of variation attributable to experimental factors (Figs. 2A, 4D). This extreme prevalence of cis-acting variation over trans has been observed in several recent analyses using similar approaches (Wittkopp et al. 2004; McManus et al. 2010; Cubillos et al. 2014; Bader et al. 2015). Nonetheless, we estimated that an additional 1468 cis-by-treatment, 2246 trans, and 1436 trans-by-treatment effects existed but were not of sufficient strength to be detected by FDR thresholding methods (Fig. 4C). These results indicated that many genes were influenced primarily by cis factors and secondarily by trans factors and by both cis and trans interactions with the environment. Importantly, several drought-responsive transcription factor-binding sites were highly enriched among genes that responded to drought or were trans regulated, indicating that drought adaptation in P. hallii may be due to similar genetic networks found in model species. Combined, these observations provide a strong foundation for further inquiry into the evolution of drought tolerance in this important genomic model for the emerging biofuel crop, switchgrass (P. virgatum).
Methods
Germplasm and experimental design
The parents, HAL2 (var. hallii; Austin, TX; 30.19° N, 97.87° W) and FIL2 (var. filipes; Corpus Christi, TX; 27.65° N, 97.40° W), were originally germinated from seeds at the University of Texas at Austin. The F1 hybrid was made through a controlled cross, as described in Lowry et al. (2015). The parents and the F1 hybrid were cloned through vegetative propagation to produce replicate plants for the experiment.
We conducted sampling of the plants in the drought treatment on July 5, 2013. Half of the parental and F1 plants were assigned randomly to the drought treatment prior to leaf collections. For each plant, we measured predawn leaf water potential (LWP) between 4:30 a.m. and 6:40 a.m. We then collected leaf tissue for RNA simultaneously with leaf tissue collection for mid-day LWP using a Scholander-type pressure bomb (PMS Instruments, model 1000) between 11:00 a.m. and 1:30 p.m. RNA was extracted from the most recently expanded leaf on a tiller that was representative of the majority of tillers at the time of sampling. The time of leaf collection was recorded to the nearest minute. Leaves for RNA extractions were flash-frozen with liquid nitrogen and packed immediately in dry ice. To simulate a large rainfall event, all plants were rewatered on July 7, 2013. Each plant received 4 L of water, applied by hand to the base of the plant. On July 8, we conducted sampling from the parental and F1 plants that had not been sampled on July 5. These rewatered plants were sampled for predawn and midday LWP and leaf tissue following the same sampling protocol as on July 5.
RNA extraction
P. hallii leaf samples (50–200 mg) were homogenized in Eppendorf tubes with steel beads on a Geno/Grinder 2000 (Spex SamplePrep). Total RNA was extracted with RNeasy Plant Mini kits (Qiagen) and treated with DNase I to remove contaminating genomic DNA. RNA concentration was quantified with Qubit (Invitrogen). Three micrograms of each RNA sample passing quality control (RIN of 5 or greater) were sequenced.
Sequencing effort
Plate-based RNA sample prep was performed on the PerkinElmer SciClone NGS robotic liquid handling system using Illumina's TruSeq Stranded mRNA HT Sample Prep kit following the protocol outlined by Illumina in their user guide and with the following conditions: total RNA starting material was 1 µg per sample, and 10 cycles of PCR was used for library amplification. The prepared libraries were then quantified using KAPA Biosystem's next-generation sequencing library qPCR kit and run on a Roche LightCycler 480 real-time PCR instrument. The quantified libraries were multiplexed into pools of four libraries each, and the pool was prepared for sequencing on the Illumina HiSeq sequencing platform utilizing a TruSeq Paired-End Cluster kit v3 and Illumina's cBot instrument to generate a clustered flowcell for sequencing. Sequencing of the flowcell was performed on the Illumina HiSeq 2000 sequencer using a TruSeq SBS sequencing kit (200 cycles, v3, following a 2 × 150 indexed run recipe).
Raw reads were trimmed, then mapped to the draft v1.1 P. hallii var. filipes genome (which is from the FIL2 accession), and counts were called for each of the 37,638 annotated gene models. There was no evidence of mapping bias toward the FIL2 allele (binomial test P > 0.1) (Supplemental Fig. S2).
Generation of counts and ASE data
Histograms of 50mer frequency were generated for the HAL2 and FIL2 parents, and 50mers were extracted within a frequency range bounded by full width at the half maximum of the genome peak of the individual parents. HAL2/FIL2-shared 50mers were excluded, and the 50mers for the HAL2/FIL2-specific parents were aligned to the FIL2 (v1.1) reference genome using the BWA (Li and Durbin 2009) short read aligner. Uniquely aligning HAL2 and FIL2 50mers (FIL2 aligning perfectly and HAL2 aligning with one mismatch) that aligned to the same location in the FIL2 reference were used to define a set of markers that discriminated between the parental lines. The 50mer marker pairs were then used to classify reads as either HAL2, FIL2, or undetermined. The final set of classified reads were aligned to the P. hallii FIL2 reference genome (v1.1) using short read aligning program GSNAP (Wu and Nacu 2010). HTSeq v0.6.1 (Anders et al. 2015), a Python package, was used to count the reads mapped to annotated genes in the reference genome. Outliers among the biological replicates were identified using Pearson's correlation coefficient, R2 ≥ 0.95 and multidimensional scaling and were not considered for further analysis.
Modeling differential expression
Differential expression was inferred by analyzing raw counts using the DESeq2 package (Love et al. 2014) in the R Environment for Statistical Computing (R Core Team 2012), which infers differential expression after accounting for library size variation and mean-variance structure. Details about the model specification can be found in the Supplemental Material.
Gene ontology and motif enrichment analyses
To qualify the identity and characteristics of the significantly differentially expressed genes, we utilized de novo InterPro (Mitchell et al. 2015), Arabidopsis thaliana, and Oryza sativa ortholog GO terms. We then tested for enrichment of terms in each of the direction and significance groupings (bar plot categories in Fig. 2A) using the classic Fisher's exact test available in the topGO package (http://bioconductor.org/packages/release/bioc/html/topGO.html). To assess patterns of transcription factor binding motifs, promoter sequences (2 kb upstream) for each gene were downloaded from the Phytozome online portal (http://phytozome.jgi.doe.gov/pz/portal.html). We then calculated the statistical enrichment of a set of 485 motifs found in plant species from the newPLACE (http://www.dna.affrc.go.jp/PLACE/) database using the R package PWMEnrich (Frith et al. 2004).
Data access
The sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under BioProject PRJNA306692. Sample information and accession numbers can be found in Supplemental Table S3.
Acknowledgments
J. Heiling and B. Whitaker assisted in propagating plants and planting the experiment. Many members of the Juenger laboratory assisted in harvesting leaf tissue and measuring leaf water potentials. We thank M. Simmons, M. Bertelsen, and the Ladybird Johnson Wildflower Center for facilitating our field experiment. Computational analyses were completed on the Stampede system with allocations from the Texas Advance Computing Center. Earlier versions of this manuscript were greatly improved following comments from J.R. Lasky, D. Bolnick, and three anonymous reviewers. J.T.L. was supported by a National Science Foundation IOS fellowship (IOS-1402393). D.B.L. was supported by a Department of Agriculture National Institute of Food and Agriculture (USDA NIFA) fellowship (2011-67012-309969). E.V.S. was supported in part by the Russian Government Program of Competitive Growth of Kazan Federal University. Funding for this project came from grants to T.E.J. from the National Science Foundation (IOS-0922457) and the Department of Energy (DOE) (DE-SC0008451). The work conducted by the DOE Joint Genome Institute was supported by the Office of Science of the DOE under contract DE-AC02-05CH11231.
Author contributions: All authors contributed significantly to this work. J.T.L., S.S., D.B.L., X.W., J.S., and T.E.J. wrote the paper and conducted the statistical analysis. D.B.L. and T.E.J. designed the experiment. D.B.L. and J.E.B. conducted the fieldwork. E.V.S., J. Johnson, and M.W. conducted RNA sequencing. J. Jenkins, C.P., and A.S. generated annotations and processed raw data.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.198135.115.
- Received August 13, 2015.
- Accepted January 26, 2016.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








