
Abstract
Model-based analysis of regulation of gene expression (MARGE) is a framework for interpreting the relationship between the H3K27ac chromatin environment and differentially expressed gene sets. The framework has three main functions: MARGE-potential, MARGE-express, and MARGE-cistrome. MARGE-potential defines a regulatory potential (RP) for each gene as the sum of H3K27ac ChIP-seq signals weighted by a function of genomic distance from the transcription start site. The MARGE framework includes a compendium of RPs derived from 365 human and 267 mouse H3K27ac ChIP-seq data sets. Relative RPs, scaled using this compendium, are superior to superenhancers in predicting BET (bromodomain and extraterminal domain) -inhibitor repressed genes. MARGE-express, which uses logistic regression to retrieve relevant H3K27ac profiles from the compendium to accurately model a query set of differentially expressed genes, was tested on 671 diverse gene sets from MSigDB. MARGE-cistrome adopts a novel semisupervised learning approach to identify cis-regulatory elements regulating a gene set. MARGE-cistrome exploits information from H3K27ac signal at DNase I hypersensitive sites identified from published human and mouse DNase-seq data. We tested the framework on newly generated RNA-seq and H3K27ac ChIP-seq profiles upon siRNA silencing of multiple transcriptional and epigenetic regulators in a prostate cancer cell line, LNCaP-abl. MARGE-cistrome can predict the binding sites of silenced transcription factors without matched H3K27ac ChIP-seq data. Even when the matching H3K27ac ChIP-seq profiles are available, MARGE leverages public H3K27ac profiles to enhance these data. This study demonstrates the advantage of integrating a large compendium of historical epigenetic data for genomic studies of transcriptional regulation.
Cis-regulation of gene expression is an essential aspect of molecular biology that underlies developmental processes and disease etiology. Several genomic techniques, including ChIP-seq (Barski et al. 2007; Johnson et al. 2007; Mikkelsen et al. 2007), DNase-seq (Crawford et al. 2006; Hesselberth et al. 2009; Boyle et al. 2011; He et al. 2014), and ATAC-seq (Buenrostro et al. 2013), have been developed to experimentally identify cis-regulatory regions genome-wide. Attempts to use these data to understand gene expression have, however, been impeded by the following factors: Data for only a small subset of transcription factors (TFs) participating in any system can be generated in practice (Gerstein et al. 2012); not all TF binding sites necessarily play roles in gene regulation; mapping between enhancers and genes is still an open question; the regulatory environment that controls a gene may depend on a complex interaction of many factors at the promoter and enhancers that may act cooperatively or antagonistically (Montavon et al. 2011; Spitz and Furlong 2012); and technical biases in chromatin profiling data may obscure biologically relevant signal (Meyer and Liu 2014). Most importantly, chromatin profiling capabilities are available to a limited number of pioneering laboratories on select tissue samples, and only a minority of gene expression studies are interpreted in this perspective. Therefore investigations into the cis-regulation of gene expression have been carried out only by a limited number of groups in well-characterized systems.
Nevertheless, several developments in genomics suggest that information about gene regulation may be revealed using a combination of surrogate data and machine learning techniques. First, the transcription factor binding sites discovered in most ChIP-seq experiments tend to fall within a set of genomic regions that are DNase I-hypersensitive (Hesselberth et al. 2009; Neph et al. 2012b; Thurman et al. 2012; He et al. 2014). The union of DNase-seq (UDHS) peaks across a broad array of human cell types can therefore be used to define a superset of transcription factor binding loci in most cell types. Second, chromatin exists in several “states” (Barski et al. 2007; Heintzman et al. 2007; Ernst and Kellis 2010; Ernst et al. 2011; Hoffman et al. 2013) with a spectrum of functional properties that may be identified using ChIP-seq of histone modifications. In particular, transcription factor binding is associated with DNase I hypersensitivity and can be further characterized using the H3K27ac mark as “poised” or “active.” The active state has high levels of H3K27ac and is more strongly associated with gene expression than the poised one (Creyghton et al. 2010; Rada-Iglesias et al. 2011). Third, although specific long-range interactions between enhancers and promoters are important in the regulation of some genes, three-dimensional chromatin conformation maps show that the main quantitative trend in the frequency of chromatin interactions is decreased interaction as a function of genomic distance, as well as the existence of large topologically associating domains (TADs) that are conserved over cell lineages and even species (Lieberman-Aiden et al. 2009; Dixon et al. 2012). Finally, the accumulation of a large number of ChIP-seq profiles provides extensive information about the way in which chromatin environments vary across diverse cell types (Roadmap Epigenomics Consortium 2015).
In this study, we base our analysis on the H3K27ac modification, as several studies have found it to be among the most highly informative about gene regulation (Creyghton et al. 2010; Karlić et al. 2010; Rada-Iglesias et al. 2011), and there is a large and rapidly increasing number of published H3K27ac ChIP-seq profiles in diverse cell types. One way in which H3K27ac is understood to mediate RNA transcription rates is through its interaction with the bromodomain and extraterminal domain (BET) protein BRD4 (Dey et al. 2003). BRD4 facilitates transcriptional elongation by interacting with the positive transcription elongation factor b (p-TEFb), which phosphorylates the C-terminal domain of RNA polymerase II (Pol II), releasing it from negative elongation factors (Price 2000; Jang et al. 2005). Experiments in a variety of cell lines have shown that, although the BET-inhibitor JQ1 represses a large number of genes (Ott et al. 2012; Chapuy et al. 2013; Lovén et al. 2013), it does not inhibit all of them, and even activates some. Previous work has suggested that this incomplete inhibitory effect results from the preferential influence of BET-inhibitors on superenhancers, genomic intervals with exceptionally high levels of H3K27ac, BRD4, or MED1 enrichment (Chapuy et al. 2013; Lovén et al. 2013). This idea is used in the ROSE method that identifies superenhancers and assigns them to genes using a distance threshold (Lovén et al. 2013; Whyte et al. 2013). ROSE, however, frequently fails to identify BET-inhibitor suppressed genes, including some with high H3K27ac activities (Supplemental Fig. S1).
Without a clear understanding of gene regulatory mechanisms, different rules have been used to identify transcription factor target genes. One common approach is to map each TF ChIP-seq peak to the nearest TSS and to use a genomic distance threshold to decide whether or not that gene is a target of TF binding. Other methods consider the contribution of multiple binding sites weighted by the distance between the binding site and the TSS (Ouyang et al. 2009; Tang et al. 2011; Wang et al. 2013; Jiang et al. 2015). These approaches are motivated by the assumption that most genes are regulated through the integrated activity of multiple cis-regulatory elements (Hong et al. 2008; Frankel et al. 2010; Montavon et al. 2011; Perry et al. 2011; Ahn et al. 2014; Bender et al. 2015; Canver et al. 2015; Meyer et al. 2015). The large number of TF binding sites that are typically detected in mammalian cells (Gerstein et al. 2012) and the tendency of these sites to occur in clusters in the genome (Ji et al. 2006) suggest that integrated cis-element activity is likely to be a general regulatory principle. The above-mentioned methods do not take advantage of the large quantities of public ChIP-seq data derived from various cell types in consideration of their predictions. ChromImpute (Ernst and Kellis 2015), an imputation method that does make use of compendia of chromatin profiling data, does not make predictions about the regulation of differentially expressed genes.
To build a system that predicts the cis-regulation of differential gene expression, we explore the systematic use of H3K27ac ChIP-seq data in MARGE (model-based analysis of regulation of gene expression), a statistical modeling and machine learning framework for gene regulation studies. We use a compendium of human (Supplemental Table S1) and mouse (Supplemental Table S2) H3K27ac ChIP-seq profiles and DNase I-hypersensitive regions to make inferences about the cis-regulation of gene expression. MARGE defines regulatory potentials based on H3K27ac ChIP-seq data that serve as measures of the integrated cis-regulatory activities that impact gene expression. MARGE demonstrates how public H3K27ac ChIP-seq profiles can be used to infer differential gene expression and transcription factor binding in a variety of systems, not limited to those for which ChIP-seq data are available.
Results
Method overview
The MARGE framework includes three main functions: MARGE-potential, MARGE-express, and MARGE-cistrome. The first function, MARGE-potential, computes the regulatory potential (RP), a measurement of the cis-regulatory environment surrounding the transcription start site of a gene. Comparison of regulatory potentials from user-provided H3K27ac ChIP-seq samples with this compendium can identify genes that have unusually high regulatory potentials in these samples. The second MARGE function, MARGE-express, uses regression to link gene expression perturbations with regulatory potentials derived from a small subset of H3K27ac ChIP-seq data from the full compendium. In this way, MARGE determines changes in regulatory potentials that are predictive of gene expression changes. The H3K27ac patterns identified by MARGE-express are used in a third function, MARGE-cistrome, to predict cistromes, the genome-wide binding sites of trans-acting factors that regulate given gene sets. MARGE-cistrome identifies patterns of perturbations in the cis-elements that are consistent with perturbations in the H3K27ac regulatory potentials identified by MARGE-express. Investigators who wish to understand how particular genes in their gene set are regulated can use MARGE-cistrome to identify candidate cis-regulatory elements, even when chromatin profiling data are not available in their system.
H3K27ac defined regulatory potentials identify genes suppressed by BET-inhibitors
We first demonstrate with MARGE-potential how H3K27ac ChIP-seq profiles, summarized as a regulatory potential for each gene, can be a useful predictor of gene expression changes. In this analysis, we focus on systems in which gene expression changes are induced through treatment with BET-inhibitors. It has been proposed that the genes perturbed by these drugs are primarily those associated with superenhancers (Chapuy et al. 2013; Hnisz et al. 2013; Lovén et al. 2013). We assess this regulatory potential metric relative to the ROSE superenhancer-based method (Chapuy et al. 2013; Hnisz et al. 2013; Lovén et al. 2013).
ChIP-seq of H3K27ac reveals a complex profile comprised of a mixture of broad domains and narrow peaks that are likely the result of multiple distinct biological processes (Fig. 1A). Regardless of the fine-scale pattern of H3K27ac signal, we assume that H3K27ac ChIP-seq tag counts reflect an activating chromatin environment. Instead of calling peak regions or domains of enrichment, we define a regulatory potential, pi, for each gene i that integrates H3K27ac signal within 100 kb, both upstream and downstream, from the transcription start site (TSS) (Fig. 1A). pi is the weighted sum of H3K27ac ChIP-seq reads sk at genomic position k, where the weight decreases with distance from the TSS of gene i: . In this definition, , and ti is the genomic position of the TSS of gene i. Throughout this study, the parameter µ, which determines the decay rate as a function of distance from the TSS, is set so that a H3K27ac read 10 kb from the TSS contributes one-half of that at the TSS (Fig. 1A). This parameter was determined empirically, based on the observed performance of predicting BET-inhibitor repressed genes as described below (Supplemental Fig. S2) and is used consistently throughout this paper, although MARGE has robust performance over a wide range of parameter settings.
Regulatory potential is predictive of BET-inhibited differential gene expression. (A) The H3K27ac regulatory potential of a gene (in this instance, CD48) is the sum of H3K27ac ChIP-seq reads weighted by a function (pink) that decreases with distance from the transcription start site. All H3K27ac signal is included, irrespective of whether the signal falls within annotated exons, introns, or promoters. (B) Receiver-operator characteristic (ROC) curves show the H3K27ac regulatory potential performs better than the ROSE superenhancer based approach in the identification of genes down-regulated by the BET-inhibitor JQ1 in the diffuse large B-cell lymphoma (DLBCL) derived cell line LY1. Areas under the ROC curves are shown in parentheses. The relative regulatory potential, defined as the ratio of the regulatory potential to the median regulatory potential across all compendium samples, performs consistently better than the other approaches. H3K27ac ChIP-seq read counts in a 2-kb promoter region centered on the transcription start site performs better than superenhancers but not as well as the regulatory potential based methods. (C) The area under the ROC curve performance summaries of the regulatory potential, relative regulatory potential, promoter-based approach, and ROSE superenhancers in five DLBCL cell lines, one liver cancer cell line (HepG2), and one malignant peripheral nerve sheath tumor cell line (90-8TL), are consistent with the result observed in LY1. (D) The distribution of median regulatory potentials across all H3K27ac ChIP-seq samples varies between JQ1 up-, down-, and nonregulated genes. The median regulatory potential of JQ1 up-regulated genes is higher than the rest (Wilcoxon rank-sum test P-value < 10−15), indicating that these genes are likely to be constitutively expressed across a variety of cell types. (E) The median regulatory potential is associated with the CpG/CG ratio of gene promoters. The high CpG genes tend to have the higher median regulatory potentials (Wilcoxon rank-sum test P-value < 10−15).
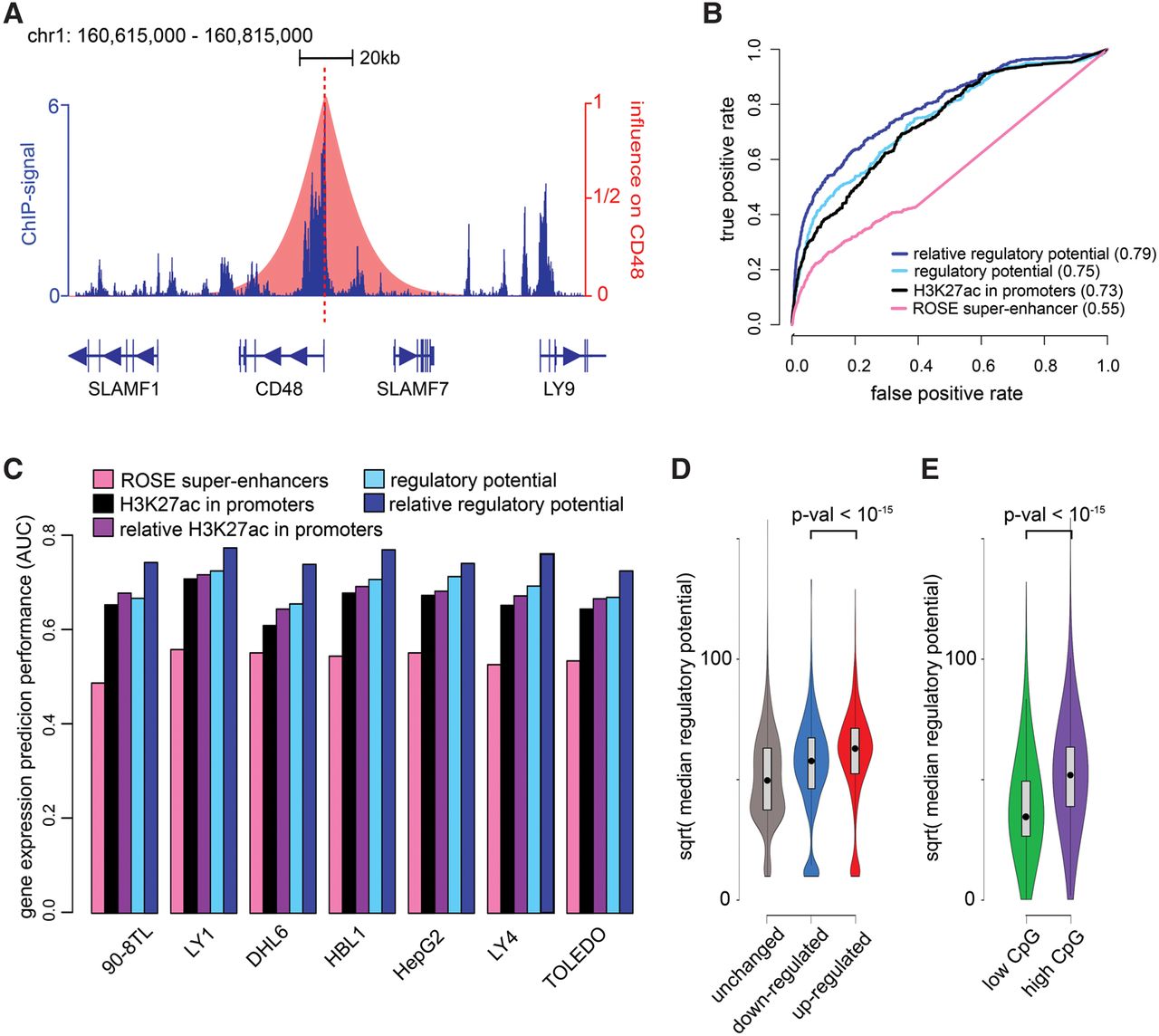
To test if our definition of regulatory potential could predict BET-inhibitor repressed genes, we examined five diffuse large B-cell lymphoma (DLBCL) cell lines (Chapuy et al. 2013), one liver cancer cell line (HepG2) (Picaud et al. 2013) and one malignant peripheral nerve sheath tumor cell line (90-8TL) (De Raedt et al. 2014) in which BET-inhibitor effects were measured using expression microarrays and H3K27ac ChIP-seq (Chapuy et al. 2013; Picaud et al. 2013; De Raedt et al. 2014). Using H3K27ac ChIP-seq data in the pre-treatment condition, we predicted BET-inhibitor repressed genes in three different ways: (1) calling H3K27ac peaks using MACS2 (Zhang et al. 2008) and identifying superenhancers and target genes using ROSE (Lovén et al. 2013); (2) using H3K27ac read counts in gene promoters (1 kb upstream of and downstream from the TSS); and (3) using the regulatory potential defined above. We used genes down-regulated under BET-inhibitor treatment over control conditions (FDR ≤ 0.01, fold-change ≤0.5) to define the true set of BET-inhibitor suppressed genes. All other genes were labeled as nonsuppressed. The receiver operator characteristic (ROC) curves indicate that, while ROSE is better than a random prediction, the regulatory potential is far more predictive of down-regulated genes (Fig. 1B,C; Supplemental Fig. S3) than both ROSE and the promoter-based prediction.
We investigated whether filtering out reads that are not in MACS2 detected H3K27ac ChIP-seq peaks could reduce noise and improve the performance over the all-inclusive regulatory potential. We found, however, that the peak-calling step has no significant impact on performance (Supplemental Fig. S4). Including information about topologically associating domains also does not have a significant impact on performance (Supplemental Fig. S3), so it was excluded from the current model. Although more sophisticated modeling of regulatory potentials using cell-type–specific Hi-C data might improve performance, we do not investigate this here, as high resolution Hi-C data are available only in a small number of cell lines. The regulatory potential that we have defined without recourse to chromatin interaction data is a useful summary of the H3K27ac defined cis-regulatory environment surrounding a gene and predicts genes that are responsive to BET-inhibitor treatment.
Baseline H3K27ac regulatory potential improves prediction of genes repressed through BET-inhibition
Although most genes are down-regulated in response to BET-inhibitor treatment, expression analyses show that a small number of genes are apparently up-regulated. Computing the median regulatory potential of 365 human H3K27ac data sets across diverse cell types, we discovered that BET-inhibitor up-regulated genes tend to have significantly higher regulatory potentials than down-regulated genes, which in turn have significantly higher regulatory potentials than nonregulated genes (Fig. 1D). Furthermore, the median regulatory potential of a gene across many cell types can distinguish between different types of response. Genes with high regulatory potentials over a large number of cell types are more likely to have universally essential functions and are less likely to be inhibited than those with cell-type–specific regulatory potentials (Supplemental Fig. S5). In fact, genes with high median regulatory potentials have a greater chance of being up-regulated by BET-inhibitor treatment and tend to have CpG rich promoters, suggesting they may be controlled through an alternative regulatory mode (Fig. 1E).
We investigated the prediction performance of the relative regulatory potential of gene i in sample j, , defined as the ratio of the regulatory potential in sample j over the median regulatory potential of that gene across all H3K27ac compendium samples. Using this relative regulatory potential, we were able to significantly improve our prediction of BET-inhibitor down-regulated genes in all seven cell types tested (Fig. 1C). A relative promoter signal, defined as the ratio of the promoter signal over the median promoter signal across H3K27ac samples, produced slight gains in performance over the absolute promoter signal (Fig. 1C) but could not reach the performance of the relative regulatory potential. Precision-recall analysis shows that the relative regulatory potential has far higher precision than superenhancers, even at low recall rates (Supplemental Fig. S6). Further analyses show that relative regulatory potential outperforms the absolute regulatory potentials, mainly in the prediction of the most significantly down-regulated genes (Supplemental Fig. S7) with the greatest fold-changes (Supplemental Fig. S8). These results suggest that the regulatory potential across diverse cell types is a rich source of information that can be used broadly across cis-regulatory studies. As a genomics resource, we provide tables of relative regulatory potentials for RefSeq genes in 365 H3K27ac human and 267 mouse data sets (Supplemental Tables S3, S4; http://cistrome.org/MARGE/).
Relative regulatory potential identifies cell-type–specific genes
It has been suggested that superenhancers regulate key tissue-specific genes (Hnisz et al. 2013). We assessed whether the relative regulatory potential, p*, could be used for similar purposes. We computed p* in cells derived from 14 diverse tissues and carried out Gene Ontology (GO) enrichment analysis based on the 500 genes in each cell type with the highest relative regulatory potentials. Many GO categories pertinent to the specific biological functions of these cell types are enriched among these genes with the highest p* values (Supplemental Fig. S9), such as skeletal system development genes in skeletal muscle, blood vessel development in aorta and umbilical vein, and immune response in B-cells. In a comparison with the same number of ROSE superenhancer targets from the same data, we found that MARGE-defined high relative regulatory potential genes showed much better tissue-specific GO enrichment (in GO categories defined by Hnisz et al. 2013) than ROSE superenhancer-associated genes (Fig. 2A). The GO categories that were more highly enriched in superenhancer-associated genes included categories that are not cell-type specific, such as regulation of cell proliferation and regulation of transcription. Therefore, the relative regulatory potential appears to be a better way of identifying tissue-specific genes than the ROSE superenhancer based approach.
Regulatory potentials in the identification of key tissue-specific genes. (A) Gene ontology analysis of the genes with the highest relative regulatory potential (lower right triangles) in a variety of cell types shows functional enrichment to correspond with the known function of the different cell types. The pattern of enrichment of ROSE superenhancer-associated genes (upper left triangles) shows these genes to be less enriched in several tissue-specific gene categories and more enriched in some more generic categories, for example, “regulation of transcription.” (B) The regulatory potentials in diverse cell types cluster in a way that is mostly consistent with cell types. Known regulators of several cell types can be clearly identified through regulatory potential analysis.
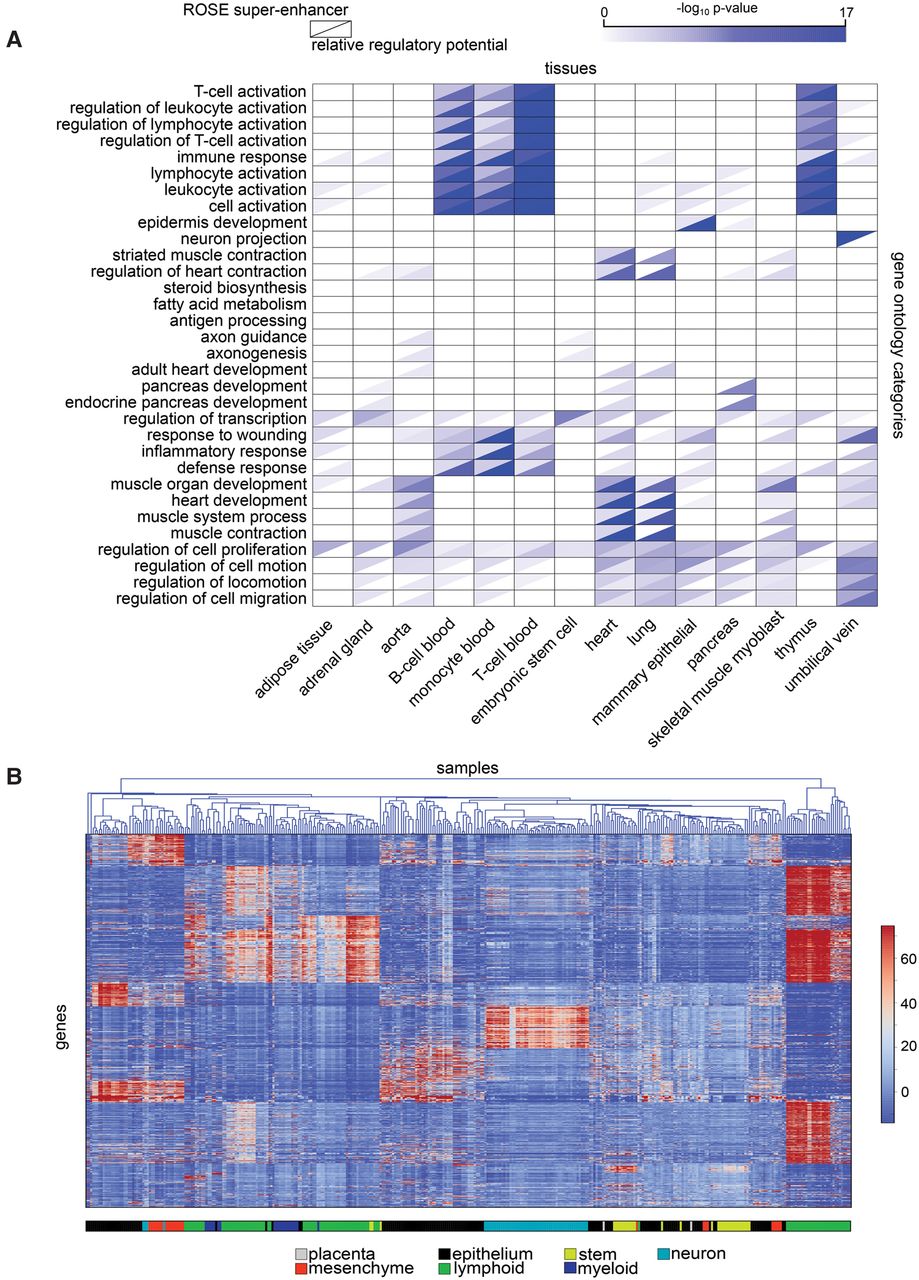
We carried out a clustering analysis of regulatory potentials across 365 human H3K27ac samples. We computed the H3K27ac ChIP-seq-defined regulatory potential for each gene in every sample, filtered out uninformative genes with low regulatory potentials across all samples, selected the 2000 genes with the largest coefficients of variation across samples, and carried out hierarchical clustering on samples and k-means clustering on genes. From this clustering (Fig. 2B), we observed the tendency for tissues of the same type to cluster together. We hypothesized that key regulators of a cell type could be identified accurately by determining the factors with the highest relative regulatory potentials across multiple samples of that type. We tested this by determining the transcription factors, chromatin regulators, and kinases with the highest median of relative regulatory potentials across neuronal, lymphoblastoid, and embryonic stem cell types, respectively (Supplemental Table S5). The top neuronal factors, BRD2, POU3F3, AATYK, SALL1, SOX2, and SOX10, are all known key neuronal regulators. For example, BRD2-deficient neuro-epithelial cells fail to differentiate into neurons (Tsume et al. 2012) and AATYK induces neuronal differentiation (Raghunath et al. 2000). ZIC3, ZIC2, SOX2, NANOG, and SALL1 are the top embryonic stem (ES) cell factors. ZIC2 (Luo et al. 2015) and ZIC3 (Declercq et al. 2013), for example, are required to maintain ES cell pluripotency. Similar observations can be made for the regulators with the highest median relative regulatory potentials in lymphoblastoid cells, PAX5, POU2AF1, MSC, and IKZF1. The relative regulatory potential is therefore a promising way of determining key tissue-specific regulators from H3K27ac ChIP-seq data.
Compendium of H3K27ac regulatory profiles predicts diverse gene expression responses
H3K27ac ChIP-seq profiles are shaped by a combination of biological and technical factors, including cell lineage and cell-type–specific transcription factor activity, immunoprecipitation efficiency, and DNA sequencing biases. We hypothesized that a compendium of diverse H3K27ac ChIP-seq profiles could be used to model gene expression changes in a variety of biological contexts. If true, this compendium would provide information about gene regulation in studies where ChIP-seq data are unavailable. To assess the possibility of using a set of 365 H3K27ac human ChIP-seq-defined regulatory potentials to model gene expression perturbations, we adopted a forward step-wise regression approach to identify informative samples. Termed MARGE-express, it defines a logistic regression model: . Here, yi is the indicator of whether a gene belongs to a given gene set (yi = 1) or not (yi = 0), pij is the transformed regulatory potential of gene i in sample j, and α is the vector of regression coefficients. At each step in the step-wise regression, the sample that maximizes the cross-validation AUC performance is added to the model. In preliminary analyses, we found that cross-validation performance plateaued before 10 H3K27ac samples, so we limit the maximum number of samples in the regression to 10 (Supplemental Fig. S10).
We tested MARGE-express on 671 molecular signature-based gene sets (MSigDB) (Liberzon et al. 2011) with over 200 genes, most of which were derived from either upward or downward differential expression between conditions. To rule out overfitting, for each gene set, we used genes in odd numbered chromosomes to train MARGE-express, then used the model to predict which genes on the even numbered chromosomes belong to the gene set. The proposed logistic regression model can indeed make accurate predictions for most gene sets (Fig. 3A), and in many cases the informative H3K27ac samples are closely related to the gene expression data set. The single ChIP-seq samples with the strongest predictive power for gene sets with keywords associated with breast, blood, liver, lung, prostate, neuron, or colon in their descriptions were most frequently derived from the relevant tissue type (Fig. 3B). For example, gene sets associated with breast were most frequently best predicted by breast H3K27ac ChIP-seq samples. The publicly available H3K27ac profiles therefore are of sufficient quality and variety to enable gene sets to be interpreted in a wide range of experiments. After the tissue-specific H3K27ac ChIP-seq samples that are often selected first in the step-wise regression, in later iterations MARGE-express frequently selects a diversity of cell lineages unrelated to the tissue of interest. Further work is needed to understand the signal in the MARGE-express models, for example whether information is derived from cell lineage or cell population, or whether some samples represent a generic background H3K27ac profile.
MARGE-express modeling of differential expression gene sets using H3K27ac regulatory potentials in diverse samples. (A) An analysis of 671 gene sets from MSigDB shows, using independent training and testing data, that this approach is highly predictive of most gene sets. (B) Heat map of the proportion, by tissue type, of H3K27ac samples that are most predictive of the tissue type-associated gene sets. Gene sets with descriptions that include the keyword liver, for example, are most often predicted by liver-derived H3K27ac ChIP-seq samples. In this example, the fraction of times that liver-derived H3K27ac samples are selected first in the step-wise regression analysis is represented in the liver-associated row of this heat map. (C) MARGE-express prediction of differentially expressed noncoding RNAs based on coding RNA data. A gene set based on the upward trending protein-coding genes in a time course of keratinocyte differentiation was used as input data. MARGE-express predicted scores for noncoding RefSeq genes. These scores are compared between upward and downward trending noncoding RNAs observed in a separate keratinocyte differentiation experiment. TINCR is a strongly up-regulated lncRNA in the differentiated state and is especially important to keratinocyte development.
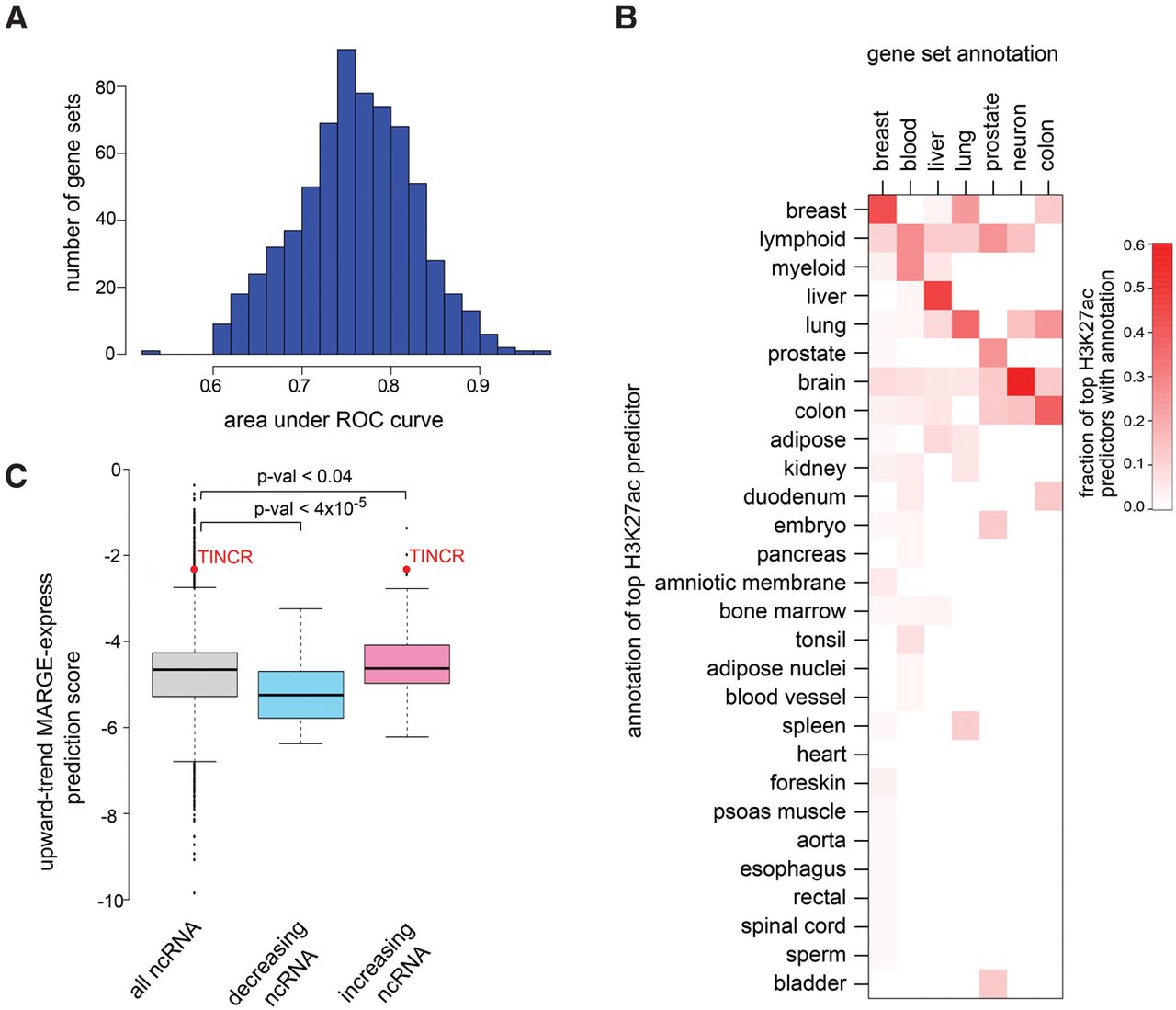
MARGE-express can predict, using data from one gene-expression-profiling platform, differential gene expression for genes that are not represented on that platform. Many DNA microarray platforms, for example, do not include probes for most noncoding RNAs (ncRNAs). As an application of MARGE-express, we tested the prediction of noncoding RNAs using DNA microarray data that were limited to coding genes. We obtained processed DNA microarray and RNA-seq data reported by two studies of keratinocyte development (Kretz et al. 2013; Lopez-Pajares et al. 2015). One study reported a set of protein coding genes with upward-trending mRNAs that are transcribed more rapidly over the time-course (Lopez-Pajares et al. 2015). The other reported ncRNAs that are differentially expressed (Kretz et al. 2013), including sets of ncRNAs that increase or decrease over the time course. We used MARGE-express to identify a model for increasing transcription using the upward-trend protein coding gene set and calculated scores for noncoding RefSeq genes based on this model. Please see Supplemental Methods for further details of this analysis. The MARGE-express prediction scores for the ncRNA set that was observed to follow an upward-trend are higher than all ncRNAs (P-val < 0.04) and the same prediction scores for the observed decreasing ncRNA set are much lower (P-val < 4 × 10−5) (Fig. 3C). TINCR, terminal differentiation-induced lncRNA, which plays an important role in this developmental process and is the focus of the Kretz et al. (2013) study, is correctly predicted by MARGE-express to be among the most highly up-regulated ncRNAs in keratinocyte development.
Semisupervised learning approach accurately infers cis-regulatory regions from genome-wide H3K27ac profiles
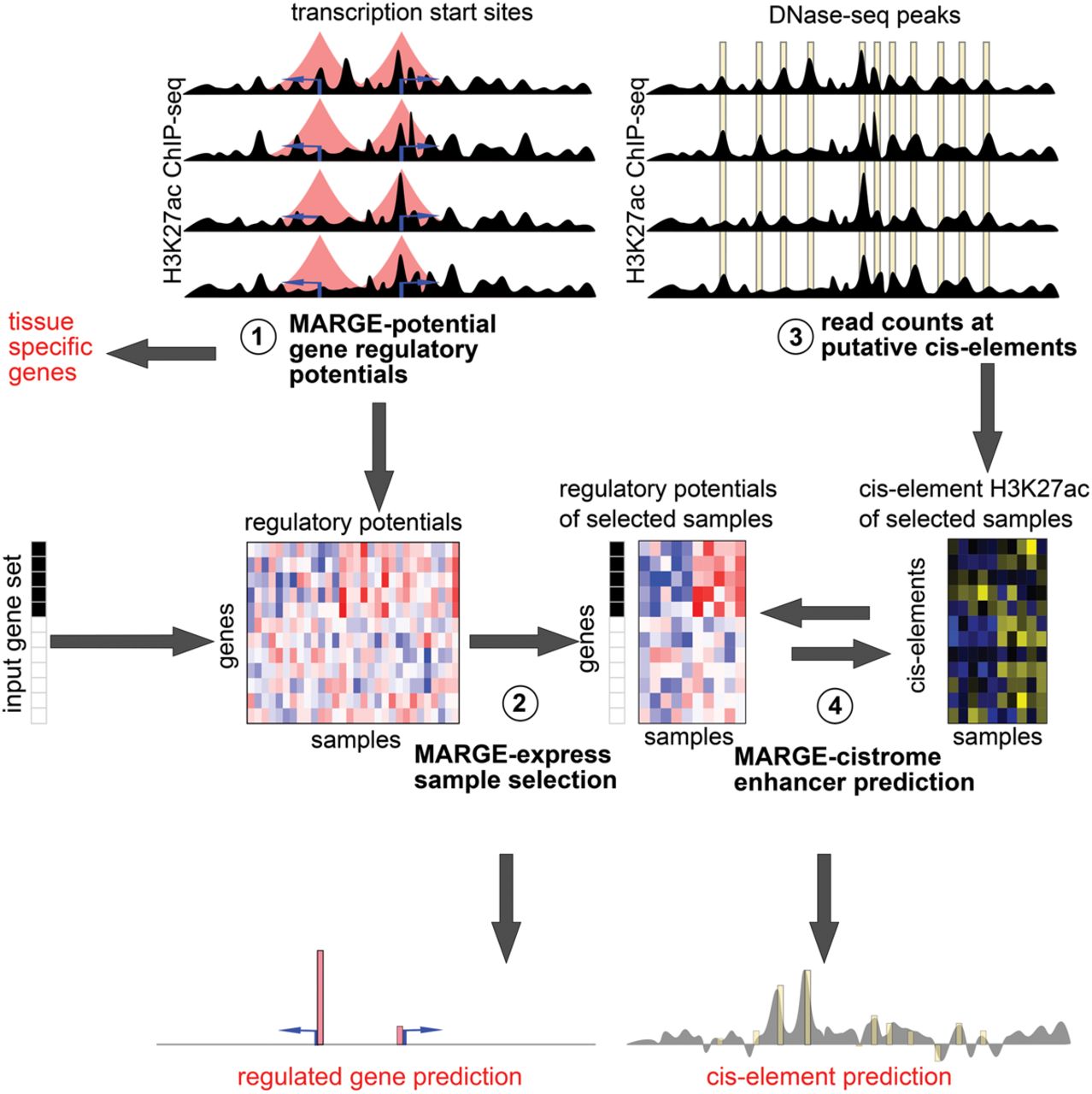
Understanding the cis-regulatory mechanisms underlying gene expression patterns is one of the key questions in modern biology. Although ChIP-seq, DNase-seq, and other chromatin profiling technologies can be highly informative, scarce or low-quality biological material from many systems is unsuitable for such experiments. To address this problem, we propose to determine cistromes associated with perturbed gene expression patterns using a compendium of H3K27ac ChIP-seq data. We hypothesize that the same H3K27ac ChIP-seq data that define regulatory potentials (Fig. 4, circle 1) predictive of gene expression perturbations can also predict the cistrome of regulating cis-elements. In our conceptual model, the perturbations in regulatory potentials that produce gene expression perturbations are in turn produced by correlated shifts in activity at individual cis-elements. MARGE assumes that the cis-elements are a subset of a union of DNase-seq peaks that serve to define the full repertoire of cis-elements in the genome (Fig. 4, circle 3). These sets of DNase I-hypersensitive regions are derived from 458 human and 116 mouse DNase-seq profiles. The unions of DNase I hypersensitive regions from these public DNase-seq profiles (Neph et al. 2012a; Thurman et al. 2012; Stergachis et al. 2014) include approximately 2.7 million and 1.5 million regions in human and mouse, respectively (more details can be found in the Supplemental Methods section). The H3K27ac ChIP-seq read counts across 1-kb genomic intervals centered on each UDHS region are summarized (Fig. 4, circle 3). Then, MARGE-cistrome (Fig. 4, circle 4) predicts cis-elements by comparing these H3K27ac signals at the UDHS level with H3K27ac summarized as regulatory potentials, without using DNA sequence information for predictions.
Schematic of MARGE framework. (1) MARGE-potential computes regulatory potentials from a compendium of H3K27ac ChIP-seq profiles. (2) MARGE-express uses stepwise regression to select a subset of informative H3K27ac ChIP-seq samples for the prediction of a user-provided input gene set. This regression selects columns (samples) from the matrix of normalized and centered regulatory potentials, represented as a blue-red heat map. MARGE produces a prediction of regulated genes that may include information on transcripts not included in the original gene expression study. (3) H3K27ac read counts in 1-kb regions centered on a list of DNase I-hypersensitive sites are extracted from the selected samples and assembled as a matrix of normalized and centered values, represented as a blue-yellow heat map. (4) MARGE-cistrome uses a semisupervised method to infer transcription factor binding sites from H3K27ac read counts at DNase I-hypersensitive sites (blue-yellow heat map), regulatory potentials (blue-red heat map), and the input gene set. MARGE-cistrome produces predictions of the cistrome of TFs that are responsible for the regulation of the gene set.
If the relevant TF binding data were available to define the cis-elements, we could directly use logistic regression to determine coefficients in a linear model that predicts cis-elements from H3K27ac ChIP-seq data. However, we are proposing to infer cis-elements without such TF binding data, and regression cannot be used to directly estimate the coefficients. Instead, we assume that a linear model that optimally classifies cis-elements using H3K27ac signal in UDHS regions will be similar to the one determined by MARGE-express to classify a gene set using H3K27ac regulatory potentials. Although changes in H3K27ac levels at enhancer sites produce changes in H3K27ac regulatory potentials, the two are not equivalent, as regulatory potentials are based on aggregates of regions of which only a fraction is likely to include enhancers with dynamic H3K27ac levels. In addition, the normalization of regulatory potentials is different from normalization at the level of individual cis-elements.
To infer cis-elements, MARGE-cistrome starts with the set of informative H3K27ac ChIP-seq data sets identified using MARGE-express (Fig. 4, circle 2). MARGE-cistrome (Fig. 4, circle 4) then generates a matrix of square-root H3K27ac signals, U′, in UDHS regions. The rows in this matrix correspond to all UDHS genomic loci and the columns correspond to the 10 regression-selected H3K27ac samples. Matrix U′ is normalized to U by row and column centering (details in Supplemental Methods). In a similar way, MARGE-cistrome generates a matrix of transformed normalized regulatory potentials, P, with columns corresponding to the same samples as in U, in the same order. A naive way of predicting TF binding is to transfer the parameters, α, estimated by logistic regression in MARGE-express (Fig. 5A, top left), directly to the TF binding inference problem, using α as coefficients of the H3K27ac signal at UDHS regions (Fig. 5A, top right). In this approach, the predictive score, , for a cis-element, i, is calculated as , where ui ∈ ℝ10 is row i of matrix U. Instead, MARGE-cistrome uses an alternative novel semisupervised learning method (Chapelle and Schölkopf 2006). This method assumes that the cis-elements associated with the regulation of the gene expression perturbation constitute a subset of the overall genome-wide cis-element repertoire and exhibit correlated H3K27ac signal patterns across the informative samples. The dominant H3K27ac signal patterns in the cis-elements are identified using k-means (K = 7) to cluster genomic loci by normalized H3K27ac signal, U, obtaining centroids λ1, …, λK (Fig. 5A, bottom right) for each cluster. In preliminary analysis, we tested alternative cluster numbers and did not find the algorithm to be sensitive to this choice, so we set this number as 7. Since we are clustering the same small number of samples (10), we do not expect the number of clusters to vary broadly. Moreover, the method does not rely on an optimal partitioning of UDHS sites into clusters; clustering merely serves as a guide to the distribution of the data.
MARGE-cistrome prediction of cis-regulatory regions from gene sets and H3K27ac ChIP-seq data. (A) Schematic of cis-regulatory region prediction through the direct transfer of MARGE-express coefficients from the H3K27ac regulatory potential domain to the domain of H3K27ac signal at UDHS regions (top). In this illustration, we represent a hypothetical case in which two samples are selected to predict gene sets and cis-regulatory regions. Using a supervised classification method (top left), such as logistic regression, we can identify the normal (red arrow) of a hyperplane that best separates genes in the gene set (red dots) from the rest (gray dots). Applied to the union of DNase-seq peaks (top right), this normal may not be the optimal classifier to separate transcription factor binding sites from the remainder of the candidate regions. Schematic of semisupervised learning for cis-regulatory element identification (bottom). Using k-means clustering (bottom right), using only two clusters for illustrative purposes, we can identify the centroids (orange and cyan arrows) of sets of putative cis-regulatory regions that have similar H3K27ac read count patterns across samples. Using gene sets (bottom left), we determine which of the centroid-derived normal vectors (orange arrow) is most predictive of the gene set. The optimal centroid derived vector (orange arrow) is then used to classify TF binding sites associated with the gene set (bottom right). (B) Applied to systems that are regulated chiefly by the respective transcription factors: the androgen receptor, the estrogen receptor, the glucocorticoid receptor, NOTCH, PPARG, and POU5F1, we find the centroids of the k-means clusters (left), predict gene sets (AUCGX , middle) with AUC performance that is highly correlated with AUC performance for the prediction of transcription factor binding sites (AUCTF , right). In these examples, 10 selected samples, S1 … S10, were clustered into seven clusters, C1 … C7. In each system, the samples with the greatest absolute positive and negative regression coefficients are as follows. AR: (S1) dihydrotestosterone-stimulated LNCaP cells, (S2) unstimulated LNCaP cells; ESR1: (S7) estradiol-stimulated MCF-7 cells, (S8) unstimulated MCF-7 cells; GR: (S1) dexamethasone-stimulated A549 cells, (S2) unstimulated A549 cells; NOTCH: (S1) CUTLL1 cells, (S2) γ secretase-inhibited CUTLL1 cells; PPARG: (S1) adipocytes, (S2) expanded memory T-cells; POU5F1: (S1) embryonic stem cells, (S2) embryonic stem cell-derived foregut. (C) In the prediction of TF binding sites from gene sets, the classifiers derived through semisupervised analysis perform better than those derived using the naive direct coefficient transfer approach in four examples, and almost as well as classifiers based on the direct application of logistic regression to transcription factor binding data.
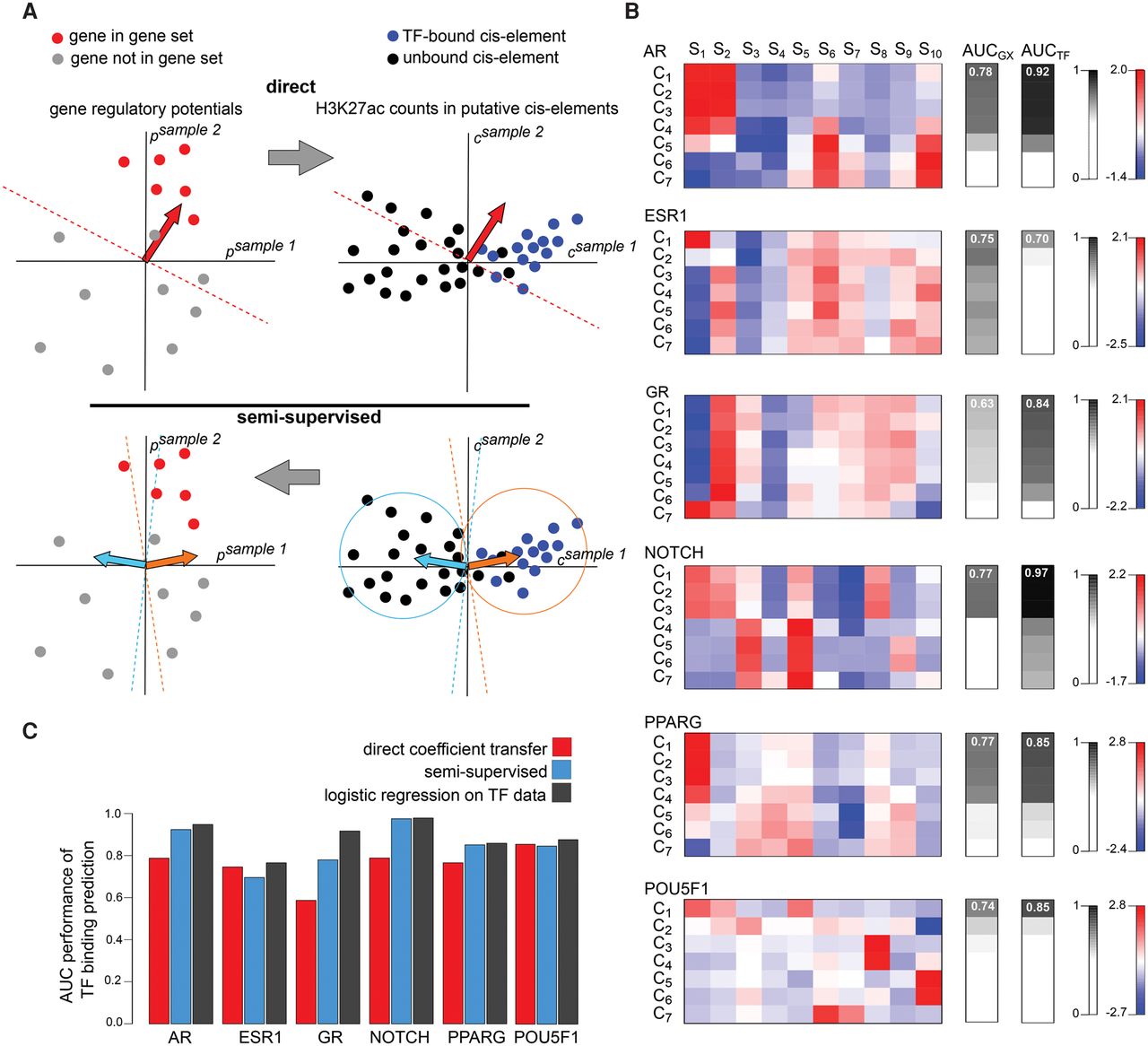
MARGE-cistrome then assesses which of these centroids is the most associated with the gene expression perturbation using a score for each gene j and cluster k, where pj is the jth row of matrix P. The AUC performance of sk in predicting the input gene set is then evaluated for each cluster k ∈ {1, …, K} (Fig. 5A, bottom left) to determine the cluster centroid λ* that best predicts the gene set. The MARGE-cistrome prediction score for cis-element i is calculated using this centroid: . MARGE-cistrome therefore predicts regulatory cis-elements by combining unsupervised and supervised methods to generate linear combinations of normalized H3K27ac read counts in each 1-kb cis-element ascribed region.
We tested MARGE-cistrome on six systems where gene expression changes are regulated by known transcription factors: the estrogen receptor ESR1 (Carroll et al. 2006); the androgen receptor AR (Wang et al. 2007); the glucocorticoid receptor NR3C1 (Muzikar et al. 2009); the peroxisome proliferator-activated receptor gamma (PPARG) (Mikkelsen et al. 2010); NOTCH1 (Wang et al. 2011); and POU5F1 (Kunarso et al. 2010). Details of the samples selected by MARGE-express in each case are described in Supplemental Table S6 (Supplemental Table S7 for MARGE-express predictions). As a gold standard for TF binding sites, we used ChIP-seq peaks for the TFs derived from relevant cellular contexts. In these test systems, we first checked the assumption that centroids that predicted gene expression well would also be good at predicting TF binding (Fig. 5B). This was indeed the case; the centroids that performed well in predicting gene sets (Fig. 5B, AUCGX) also performed well in predicting TF binding (Fig. 5B, AUCTF). We then compared the performance of the naive approach and the MARGE semisupervised method to an estimate of the attainable best performance in TF binding inference from H3K27ac ChIP-seq. The attainable performance was determined by applying logistic regression directly in the UDHS space on TF ChIP-seq data. We found that in all six cases the semisupervised approach was nearly as good as the attainable performance (Fig. 5C; Supplemental Table S8), whereas the direct approach performed worse in four cases. The semisupervised approach effectively up-weighs sample-specific H3K27ac signal that is associated with specific TF binding and down-weighs unrelated H3K27ac signal (Supplemental Fig. S11). These results show that MARGE-cistrome is a promising approach for predicting transcription factor binding sites associated with the cis-elements that regulate a user-provided gene set.
Integration of public H3K27ac data to enhance in-house data on cis-element prediction
At times, to gain insight into cis-regulatory mechanisms, investigators conducting differential expression analyses augment the gene expression data with matching H3K27ac ChIP-seq profiles. We next investigated whether MARGE could utilize public H3K27ac data to enhance the analysis of matched H3K27ac ChIP-seq data. In the prostate cancer cell line LNCaP-abl, we conducted siRNA silencing of the transcription factors AR, E2F1, FOXA1, and FOXM1, lncRNA MALAT1, chromatin modifiers EZH2, KDM1A, and UTX, and the cohesin subunit RAD21. We then generated RNA-seq expression profiling and H3K27ac ChIP-seq data under control and the nine different knockdown conditions. Using public data alone, MARGE-express can retrieve the relevant H3K27ac profiles to model the down-regulated genes in each of the knockdown conditions with ROC AUC performances between 0.65 and 0.75 (Fig. 6A). Augmenting our 10 in-house LNCaP-abl H3K27ac data sets with the public H3K27ac ChIP-seq data, we only obtained subtle improvements in performance (Fig. 6A; Supplemental Table S10). Details of the samples selected in these analyses are described in Supplemental Table S9. This result indicates that having H3K27ac ChIP-seq data for the exact conditions is helpful but may not be required for studying the cis-regulation of gene expression in a cell system; public data alone may approach similar performance.
MARGE-cistrome prediction of cis-regulatory regions from knockdown gene expression and H3K27ac ChIP-seq data. (A) Down-regulated genes in LNCaP-abl prostate cancer cells on siRNA silencing of nine factors can be predicted from the compendium of H3K27ac ChIP-seq profiles. Augmentation of public data with H3K27ac ChIP-seq generated in LNCaP-abl samples improves prediction performance slightly. (B) Prediction of AR, E2F1, and FOXA1 binding sites using four methods: sample-specific H3K27ac ChIP-seq read count; difference of square root H3K27ac read counts between wild-type and knockdown samples; MARGE-cistrome based on public H3K27ac ChIP-seq data only; MARGE-cistrome based on public data augmented with H3K27ac ChIP-seq data in LNCaP-abl. (C) Example of predicted cis-regulatory loci with FOXA1 binding sites. The MARGE reweighted track is a linear combination of H3K27ac signal tracks with coefficients defined by MARGE-cistrome.
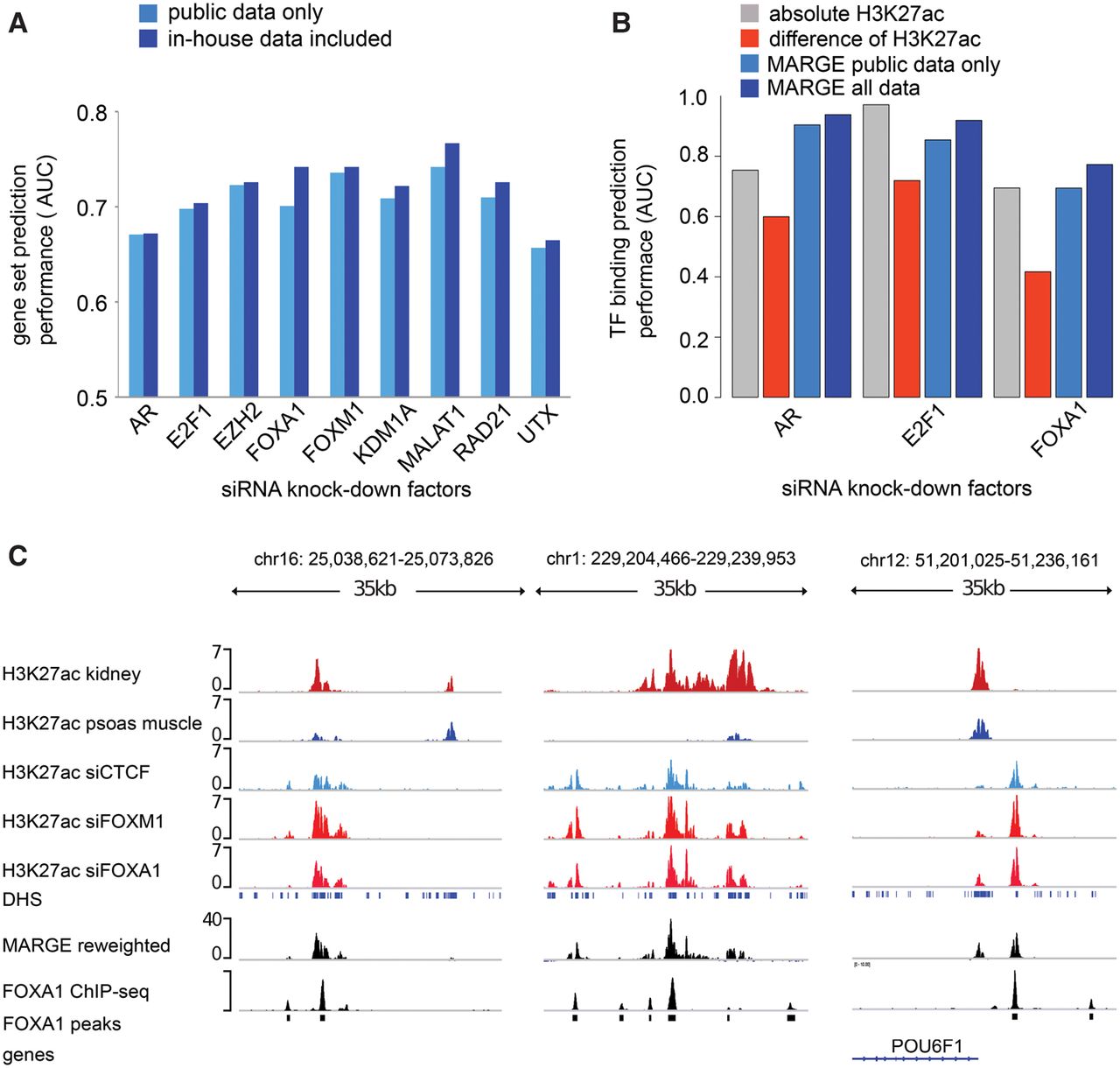
We then assessed the performance of MARGE-cistrome in the prediction of functional cis-regulatory regions in the siRNA knockdown experiments. Based on the assumption that the knockdown of a TF should have a direct effect on its genomic binding sites to dysregulate the target genes, we conducted ChIP-seq experiments to identify the binding sites of AR, E2F1, and FOXA1 to assess the performance of the prediction of cis-regulatory regions. We selected these factors as they interact directly with DNA and have high quality antibodies for ChIP. We compared the prediction performance of these binding sites (Fig. 6B; Supplemental Table S11) using the following methods: absolute H3K27ac read counts in UDHS regions; the normalized difference of square-root scaled H3K27ac read counts; MARGE-cistrome based on public H3K27ac data alone; and MARGE-cistrome based on public data and in-house LNCaP-abl-specific H3K27ac data. In the prediction of FOXA1 sites, MARGE-cistrome with public data alone has a similar performance as using the in-house data alone, and the integration of public and in-house data enabled MARGE-cistrome to improve performance from 0.70 to 0.77. In the case of AR, MARGE-cistrome on public data alone already outperforms the in-house data, and including the in-house data can further improve binding prediction. For E2F1, MARGE-cistrome with a combination of public and in-house data predicts E2F1 binding slightly less accurately than in-house H3K27ac data alone. Unlike AR and FOXA1, E2F1 tends to bind in promoter regions, suggesting that public data are more informative in predicting distal TF binding sites. The specific examples in Figure 6C illustrate how a linear combination of H3K27ac signal tracks, with MARGE-cistrome defined coefficients, can help to emphasize FOXA1 binding sites relative to other genomic regions. MARGE can therefore greatly enhance the analysis of investigator-generated H3K27ac ChIP-seq data by making use of a compendium of published data to improve the accuracy of target gene and distal cis-regulatory site prediction.
Discussion
We have shown that MARGE-potential is more accurate than the ROSE superenhancer approach at predicting genes that respond to BET-inhibition and in the identification of key tissue-specific genes. While the emergence of superenhancer-like cis-regulatory regions through cooperation between cis-regulatory elements may be important in gene regulation, our proposed statistical framework does not make “superenhancer” calls or even peak calls. Our results support the idea that genes are typically regulated by multiple cis-regulatory elements. Quantitative modeling combined with chromatin profiling and high-throughput cis-regulatory knockout experiments will be required to understand how TFs act synergistically to create phenomena such as superenhancers. We used the H3K27ac signal within 100 kb (upstream and downstream) of the TSS to calculate the regulatory potential. This is consistent with the average size of topological associating domains in the chromatin measured by Hi-C. Explicit inclusion of TAD domain information does not have a significant impact on performance. Due to the exponential decay nature of the distance weighting factors, it makes little difference in the actual regulatory potential value between slightly different boundary locations. We observed that the relative regulatory potential is more predictive of genes down-regulated by BET-inhibition than the absolute regulatory potential and that genes with high median regulatory potentials tend to have CpG-rich promoters. This is consistent with previous work that describes the tendency for genes with CpG-rich promoters to be broadly expressed across cell types and those with CpG-poor promoters to be more cell-type specific (Natarajan et al. 2012) and expressed at a lower level (Karlić et al. 2010).
We demonstrated the power of published H3K27ac ChIP-seq data in predicting the cis-regulation of gene expression. The 365 collected H3K27ac ChIP-seq data sets covered a large variety of human tissues and cell types, which made our predictive model comprehensive and robust. We found that the compendium of H3K27ac regulatory potentials could be used to define predictive models for the majority of 671 gene expression perturbations in MSigDB. The striking ability of MARGE-express to predict the response of ∼20,000 genes using regulatory potentials from 10 out of 365 samples cannot be attributed to model overspecification. The existence of parsimonious models that explain some of these changes shows that even a limited cohort of cis-regulatory profiles can provide useful insights on many gene expression perturbation studies. While in many cases the informative H3K27ac samples are directly relevant to the gene expression perturbations, in some cases they are not. One explanation for the inclusion of unexpected H3K27ac data sets is that the samples from which the gene expression data are derived are composed of heterogeneous populations of cell types. The metadata for the informative H3K27ac profiles determined by MARGE-express might prove useful in determining the nature of these cell populations. Alternatively, the inclusion of some H3K27ac samples is to compensate for technical sources of bias. Further work is needed to interpret the biological or technical nature of this predictive power. While we have focused on H3K27ac in this study, as this mark has been extensively profiled and is also indicative of active enhancers, using appropriate methods complementary chromatin profiles might be incorporated to improve prediction performance. Further research will be needed to determine how other chromatin data types can be effectively used in combination with this mark to improve prediction performance.
MARGE-cistrome makes use of the H3K27ac mark both as an indicator of the general cis-regulatory environment influencing a gene as well as an indicator of localized histone acetyltransferase activity associated with the binding of specific transcription factors. To use information derived from the regulatory potential domain to infer transcription factor binding, we developed a semisupervised learning algorithm. This approach is based on the assumption that H3K27ac at the regulatory set of TF binding sites tends to produce a pattern across the selected samples that forms a cluster of UDHS regions. In this way, MARGE-cistrome provides a useful strategy for identifying the cis-regulatory loci that regulate a differentially expressed set of genes. The success of this approach depends on the level at which the gene expression changes occur relative to the resolution of the H3K27ac compendium in terms of samples that represent the treatment and control conditions in the gene expression experiment. The predictive performance of MARGE-express and MARGE-cistrome will continue to improve as more H3K27ac ChIP-seq data become available in a greater variety of cell types and conditions.
Methods
MARGE-potential
MARGE-potential calculates the regulatory potential of each gene: , where , ti is the genomic position of the TSS of gene i, and sk is the MACS2 summary of H3K27ac ChIP signal at this position. The parameter µ, which determines the decay rate as a function of distance from the TSS, is set so that a H3K27ac read 10 kb from the TSS contributes one-half of that at the TSS. MARGE-potential also calculates the relative regulatory potential defined as the ratio of the regulatory potential in sample j to the median regulatory potential for that gene across all samples in the H3K27ac compendium: .
MARGE-express
MARGE-express generates a gene set prediction model from the H3K27ac ChIP-seq compendium. MARGE-express analyzes an input list of genes that are differentially expressed in a uniform direction as a result of some perturbation (e.g., gene knockdown, gene overexpression, differentiation, chemical or genetic perturbations). MARGE-express employs forward step-wise logistic regression to identify the 10 most informative samples from the H3K27ac ChIP-seq compendium. MARGE-express solves the regression model: , where yi is the indicator of whether a gene belongs to the given gene set (yi = 1) or not and (yi = 0) and . In each step of the forward step-wise regression, the sample that produces the highest average ROC-AUC value in fivefold cross-validation is selected. By default, MARGE selects 10 H3K27ac samples from the compendium. In the examples we used in this paper, DHT in LNCaP, E2 in MCF7, Dex in A549, GSI in CUTLL, adipose differentiation status, and POU5F1 (also known as OCT4) knockdown, the gene sets were defined setting FDR ≤ 0.01 and fold-change ≥ 2 as thresholds.
MARGE-cistrome
MARGE-cistrome infers cis-regulatory regions that are indicative of a pattern of transcription factor binding that induces either an increase or a decrease in gene expression for all genes in a gene set. The MARGE-cistrome procedure is as follows:
Use MARGE-express to identify 10 H3K27ac samples that best model the gene set.
Generate a matrix of square-root H3K27ac signals, U′, in UDHS regions. The rows in this matrix correspond to UDHS regions and the columns correspond to the 10 samples selected by MARGE-express.
Normalize matrix U′. For each column, subtract the column median from all elements in this column. For each row, subtract the row mean from each row element. The normalized matrix is U.
Generate a matrix P′ of the square root of regulatory potentials where each column of P′ is derived from the sample used to generate the corresponding column of U′ and the rows of P correspond to all nonredundant genes.
Normalize P′ using the same procedure that is used to normalize U′. Denote the normalized matrix P. Note that, although the matrices U and P are normalized using the same procedure, the column medians are, in general, not the same for both matrices.
Identify the dominant H3K27ac signal patterns in the cis-element matrix U using k-means (K = 7) to cluster genomic loci by normalized H3K27ac signal, obtaining cluster centroids λ1,…, λK, λk ∈ R10 for clusters 1, …, K.
Assess which of these centroids is most highly associated with the gene expression perturbation. Calculate a score for each gene j and cluster k, where pj is the jth row of matrix P. Measure the performance of this score in predicting the input gene set by evaluating the AUC for each cluster k ∈ {1, …, K}. Determine the cluster centroid λ* that produces the largest AUC.
Calculate a prediction score for cis-element i using this centroid: . The higher is, the more likely UDHS region i is to be bound by the factors that regulate the input gene set.
Gene expression microarray analysis
Affymetrix microarray gene expression data were normalized using the standard multichip average (RMA) package in R (Irizarry et al. 2003); differential expression analyses were performed with the linear model for microarray (LIMMA) (Smyth 2004). Please see Supplemental Methods for details.
Superenhancer and superenhancer-associated gene detection
Superenhancer analysis was carried out using ROSE (Lovén et al. 2013; Whyte et al. 2013) (https://bitbucket.org/young_computation/rose.git). Please see Supplemental Methods for details.
Performance evaluation
ROC and precision recall curves were generated using the R package ROCR (Sing et al. 2005).
Figures were plotted using R (R Core Team 2016).
ChIP-seq and DNase-seq analysis
MACS2 was used for DNase-seq peak calling. Signal summarization for H3K27ac ChIP-seq and DNase-seq was carried out using MACS2 (Zhang et al. 2008). Please see Supplemental Methods for details.
Data access
MARGE code is available in the Supplemental Material and at http://cistrome.org/MARGE/. LNCaP-abl ChIP-seq and RNA-seq data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE72467 and GSE72534, respectively.
Acknowledgments
This work was partially funded by the National Natural Science Foundation of China 31329003, the National Institute of General Medical Sciences of the National Institutes of Health 1U41 HG007000, U01 CA180980, and R01 GM099409 to X.S.L., a Dana-Farber Cancer Institute (DFCI) Department of Biostatistics and Computational Biology Research Scientist Fund to C.A.M., and a Leukemia and Lymphoma Society (LLS) fellow award to C.Z.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.201574.115.
References
- ↵Ahn Y, Mullan HE, Krumlauf R. 2014. Long-range regulation by shared retinoic acid response elements modulates dynamic expression of posterior Hoxb genes in CNS development. Dev Biol 388: 134–144.
- ↵Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837.
- ↵Bender MA, Ragoczy T, Lee J, Byron R, Telling A, Dean A, Groudine M. 2015. The hypersensitive sites of the murine β-globin locus control region act independently to affect nuclear localization and transcriptional elongation. Blood 119: 3820–3827.
- ↵Boyle AP, Song L, Lee B-K, London D, Keefe D, Birney E, Iyer VR, Crawford GE, Furey TS. 2011. High-resolution genome-wide in vivo footprinting of diverse transcription factors in human cells. Genome Res 21: 456–464.
- ↵Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218.
- ↵Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP, 2015. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527: 192–197.
- ↵Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, 2006. Genome-wide analysis of estrogen receptor binding sites. Nat Genet 38: 1289–1297.
- ↵Chapelle O, Schölkopf B. 2006. Semi-supervised learning. The MIT Press, Cambridge, MA.
- ↵Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MGM, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, 2013. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24: 777–790.
- ↵Crawford GE, Holt IE, Whittle J, Webb BD, Tai D, Davis S, Margulies EH, Chen Y, Bernat JA, Ginsburg D, 2006. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res 16: 123–131.
- ↵Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 107: 21931–21936.
- ↵De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, Helin K, Hornick JL, Mautner V, Kehrer-Sawatzki H, 2014. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 514: 247–251.
- ↵Declercq J, Sheshadri P, Verfaillie CM, Kumar A. 2013. Zic3 enhances the generation of mouse induced pluripotent stem cells. Stem Cells Dev 22: 2017–2025.
- ↵Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. 2003. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc Natl Acad Sci 100: 8758–8763.
- ↵Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380.
- ↵Ernst J, Kellis M. 2010. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol 28: 817–825.
- ↵Ernst J, Kellis M. 2015. Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat Biotechnol 33: 364–376.
- ↵Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49.
- ↵Frankel N, Davis GK, Vargas D, Wang S, Payre F, Stern DL. 2010. Phenotypic robustness conferred by apparently redundant transcriptional enhancers. Nature 466: 490–493.
- ↵Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan K-K, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, 2012. Architecture of the human regulatory network derived from ENCODE data. Nature 489: 91–100.
- ↵He HH, Meyer CA, Hu SS, Chen M-W, Zang C, Liu Y, Rao PK, Fei T, Xu H, Long H, 2014. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat Methods 11: 73–78.
- ↵Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318.
- ↵Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, 2009. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods 6: 283–289.
- ↵Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. 2013. Super-enhancers in the control of cell identity and disease. Cell 155: 934–947.
- ↵Hoffman MM, Ernst J, Wilder SP, Kundaje A, Harris RS, Libbrecht M, Giardine B, Ellenbogen PM, Bilmes JA, Birney E, 2013. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res 41: 827–841.
- ↵Hong J, Hendrix DA, Levine MS. 2008. Shadow enhancers as a source of evolutionary novelty. Science 231: 1314.
- ↵Irizarry RA, Hobbs B, Beazer-barclay YD, Antonellis KJ, Scherf UWE, Speed TP. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4: 249–264.
- ↵Jang MK, Mochizuki K, Zhou M, Jeong H, Brady JN. 2005. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell 19: 523–534.
- ↵Ji H, Vokes SA, Wong WH. 2006. A comparative analysis of genome-wide chromatin immunoprecipitation data for mammalian transcription factors. Nucleic Acids Res 34: e146.
- ↵Jiang P, Freedman ML, Liu JS, Liu XS. 2015. Inference of transcriptional regulation in cancers. Proc Natl Acad Sci 112: 7731–7736.
- ↵Johnson D, Mortazavi A, Myers R, Wold B. 2007. Genome-wide mapping of in vivo protein-DNA interactions. Science 316: 1497–1502.
- ↵Karlić R, Chung H-R, Lasserre J, Vlahovicek K, Vingron M. 2010. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci 107: 2926–2931.
- ↵Köster J, Rahmann S. 2012. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 28: 2520–2522.
- ↵Kretz M, Siprashvili Z, Chu C, Webster DE, Zehnder A, Qu K, Lee CS, Flockhart RJ, Groff AF, Chow J, 2013. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 493: 231–235.
- ↵Kunarso G, Chia N-Y, Jeyakani J, Hwang C, Lu X, Chan Y-S, Ng H-H, Bourque G. 2010. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet 42: 631–634.
- ↵Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. 2011. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27: 1739–1740.
- ↵Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289–293.
- ↵Lopez-Pajares V, Qu K, Kretz M, Khavari PA, Zarnegar BJ, Boxer LD, Rios EJ, Tao S, Kretz M, Khavari PA. 2015. A lncRNA-MAF:MAFB transcription factor network regulates epidermal differentiation. Dev Cell 32: 693–706.
- ↵Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. 2013. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153: 320–334.
- ↵Luo Z, Gao X, Washburn MP, Shilatifard A, Lin C, Smith ER, Marshall SA, Swanson SK, Florens L. 2015. Zic2 is an enhancer-binding factor required for embryonic stem cell specification. Mol Cell 57: 685–694.
- ↵Meyer CA, Liu XS. 2014. Identifying and mitigating bias in next-generation sequencing methods for chromatin biology. Nat Rev Genet 15: 709–721.
- ↵Meyer MB, Benkusky NA, Pike JW. 2015. Selective distal enhancer control of the Mmp13 gene identified through clustered regularly interspaced short palindromic repeat (CRISPR) genomic deletions. J Biol Chem 290: 11093–11107.
- ↵Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-K, Koche RP, 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448: 553–560.
- ↵Mikkelsen TS, Xu Z, Zhang X, Wang L, Gimble JM, Lander ES, Rosen ED. 2010. Comparative epigenomic analysis of murine and human adipogenesis. Cell 143: 156–169.
- ↵Montavon T, Soshnikova N, Mascrez B, Joye E, Thevenet L, Splinter E, de Laat W, Spitz F, Duboule D. 2011. A regulatory archipelago controls Hox genes transcription in digits. Cell 147: 1132–1145.
- ↵Muzikar KA, Nickols NG, Dervan PB. 2009. Repression of DNA-binding dependent glucocorticoid receptor-mediated gene expression. Proc Natl Acad Sci 106: 16598–16603.
- ↵Natarajan A, Yardimci GG, Sheffield NC, Crawford GE, Ohler U. 2012. Predicting cell-type–specific gene expression from regions of open chromatin. Genome Res 22: 1711–1722.
- ↵Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, Stamatoyannopoulos JA. 2012a. Circuitry and dynamics of human transcription factor regulatory networks. Cell 150: 1274–1286.
- ↵Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK, 2012b. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489: 83–90.
- ↵Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, Rodig SJ, Kung AL, Bradner JE, Weinstock DM. 2012. BET bromodomain inhibition targets both c-MYC and IL7R in high-risk acute lymphoblastic leukemia. Blood 120: 2843–2852.
- ↵Ouyang Z, Zhou Q, Hung W. 2009. ChIP-Seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells. Proc Natl Acad Sci 106: 21521–21526.
- ↵Perry MW, Boettiger AN, Levine M. 2011. Multiple enhancers ensure precision of gap gene-expression patterns in the Drosophila embryo. Proc Natl Acad Sci 108: 13570–13575.
- ↵Picaud S, Wells C, Felletar I, Brotherton D, Martin S, Savitsky P. 2013. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc Natl Acad Sci 110: 19754–19759.
- ↵Price DH. 2000. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol 20: 2629–2634.
- ↵R Core Team. 2016. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/.
- ↵Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470: 279–283.
- ↵Raghunath M, Patti R, Bannerman P, Lee CM, Baker S, Sutton LN, Phillips PC, Reddy CD. 2000. A novel kinase, AATYK induces and promotes neuronal differentiation in a human neuroblastoma (SH-SY5Y) cell line. Brain Res Mol Brain Res 77: 151–162.
- ↵Roadmap Epigenomics Consortium. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330.
- ↵Sing T, Sander O, Beerenwinkel N, Lengauer T. 2005. ROCR: visualizing classifier performance in R. Bioinformatics 21: 3940–3941.
- ↵Smyth GK. 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3.
- ↵Spitz F, Furlong EEM. 2012. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13: 613–626.
- ↵Stergachis AB, Neph S, Sandstrom R, Haugen E, Reynolds AP, Zhang M, Byron R, Canfield T, Stelhing-Sun S, Lee K, 2014. Conservation of trans-acting circuitry during mammalian regulatory evolution. Nature 515: 365–370.
- ↵Tang Q, Chen Y, Meyer C, Geistlinger T, Lupien M, Wang Q, Liu T, Zhang Y, Brown M, Liu XS. 2011. A comprehensive view of nuclear receptor cancer cistromes. Cancer Res 71: 6940–6947.
- ↵Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, 2012. The accessible chromatin landscape of the human genome. Nature 489: 75–82.
- ↵Tsume M, Kimura-yoshida C, Mochida K, Shibukawa Y, Amazaki S. 2012. Brd2 is required for cell cycle exit and neuronal differentiation through the E2F1 pathway in mouse neuroepithelial cells. Biochem Biophys Res Commun 425: 762–768.
- ↵Wang Q, Li W, Liu XS, Carroll JS, Jänne OA, Keeton EK, Chinnaiyan AM, Pienta KJ, Brown M. 2007. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell 27: 380–392.
- ↵Wang H, Zou J, Zhao B, Johannsen E, Ashworth T, Wong H, Pear WS, Schug J, Blacklow SC, Arnett KL, 2011. Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc Natl Acad Sci 108: 14908–14913.
- ↵Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, Tang Q, Meyer CA, Zhang Y, Liu XS. 2013. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc 8: 2502–2515.
- ↵Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. 2013. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153: 307–319.
- ↵Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137.