
Pangolin genomes and the evolution of mammalian scales and immunity
- Siew Woh Choo1,2,3,22,
- Mike Rayko4,22,
- Tze King Tan1,2,
- Ranjeev Hari1,2,
- Aleksey Komissarov4,
- Wei Yee Wee1,2,
- Andrey A. Yurchenko4,
- Sergey Kliver4,
- Gaik Tamazian4,
- Agostinho Antunes5,6,
- Richard K. Wilson7,
- Wesley C. Warren7,
- Klaus-Peter Koepfli8,
- Patrick Minx7,
- Ksenia Krasheninnikova4,
- Antoinette Kotze9,10,
- Desire L. Dalton9,10,
- Elaine Vermaak9,
- Ian C. Paterson2,11,
- Pavel Dobrynin4,
- Frankie Thomas Sitam12,
- Jeffrine J. Rovie-Ryan12,
- Warren E. Johnson8,
- Aini Mohamed Yusoff1,2,
- Shu-Jin Luo13,
- Kayal Vizi Karuppannan12,
- Gang Fang14,
- Deyou Zheng15,
- Mark B. Gerstein16,17,18,
- Leonard Lipovich19,20,
- Stephen J. O'Brien4,21 and
- Guat Jah Wong1
- 1Genome Informatics Research Laboratory, Centre for Research in Biotechnology for Agriculture (CEBAR), University of Malaya, 50603 Kuala Lumpur, Malaysia;
- 2Department of Oral and Craniofacial Sciences, Faculty of Dentistry, University of Malaya, 50603 Kuala Lumpur, Malaysia;
- 3Genome Solutions Sdn Bhd, Research Management & Innovation Complex, University of Malaya, 50603 Kuala Lumpur, Malaysia;
- 4Theodosius Dobzhansky Center for Genome Bioinformatics, St. Petersburg State University, St. Petersburg, Russia 199004;
- 5CIIMAR/CIMAR, Interdisciplinary Centre of Marine and Environmental Research, University of Porto, 4050-123 Porto, Portugal;
- 6Department of Biology, Faculty of Sciences, University of Porto, 4169-007 Porto, Portugal;
- 7McDonnell Genome Institute, Washington University, St. Louis, Missouri 63108, USA;
- 8National Zoological Park, Smithsonian Conservation Biology Institute, Washington, DC 20008, USA;
- 9National Zoological Gardens of South Africa, Pretoria 0001, South Africa;
- 10Department of Genetics, University of the Free State, Bloemfontein, 9300, South Africa;
- 11Oral Cancer Research and Coordinating Centre, Faculty of Dentistry, University of Malaya, 50603 Kuala Lumpur, Malaysia;
- 12Ex-Situ Conservation Division, Department of Wildlife and National Parks, 56100 Kuala Lumpur, Malaysia;
- 13Peking-Tsinghua Center for Life Sciences, College of Life Sciences, Peking University, Beijing, China 100871;
- 14NYU Shanghai, Pudong, Shanghai, China 200122;
- 15Department of Neurology, Albert Einstein College of Medicine, Bronx, New York 10461, USA;
- 16Program in Computational Biology and Bioinformatics, Yale University, New Haven, Connecticut 06520, USA;
- 17Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut 06520, USA;
- 18Department of Computer Science, Yale University, New Haven, Connecticut 06520, USA;
- 19Center for Molecular Medicine and Genetics, Wayne State University, Detroit, Michigan 48201, USA;
- 20Department of Neurology, School of Medicine, Wayne State University, Detroit, Michigan 48201, USA;
- 21Oceanographic Center, Nova Southeastern University, Ft. Lauderdale, Florida 33004, USA
- Corresponding authors: l.choo{at}genomesolutions.com.my, lgdchief{at}gmail.com
-
↵22 These authors contributed equally to this work.
Abstract
Pangolins, unique mammals with scales over most of their body, no teeth, poor vision, and an acute olfactory system, comprise the only placental order (Pholidota) without a whole-genome map. To investigate pangolin biology and evolution, we developed genome assemblies of the Malayan (Manis javanica) and Chinese (M. pentadactyla) pangolins. Strikingly, we found that interferon epsilon (IFNE), exclusively expressed in epithelial cells and important in skin and mucosal immunity, is pseudogenized in all African and Asian pangolin species that we examined, perhaps impacting resistance to infection. We propose that scale development was an innovation that provided protection against injuries or stress and reduced pangolin vulnerability to infection. Further evidence of specialized adaptations was evident from positively selected genes involving immunity-related pathways, inflammation, energy storage and metabolism, muscular and nervous systems, and scale/hair development. Olfactory receptor gene families are significantly expanded in pangolins, reflecting their well-developed olfaction system. This study provides insights into mammalian adaptation and functional diversification, new research tools and questions, and perhaps a new natural IFNE-deficient animal model for studying mammalian immunity.
Pangolins (also known as scaly anteaters) are mammals of the order Pholidota. Their name is derived from the Malay word “pengguling,” meaning “something that rolls up.” Eight pangolin species are recognized, four from Asia (Manis javanica, M. pentadactyla, M. crassicaudata, and M. culionensis) and four from Africa (M. tricuspis, M. tetradactyla, M. gigantea, and M. temminckii) (Gaudin et al. 2009). Although pangolin morphology shares some similarity with South American anteaters and armadillos (superorder Xenarthra), they are phylogenetically distinct, with the pangolin order Pholidota and the order Carnivora being a sister group (Ferae) nested within the superorder Lauraisatheria (Murphy et al. 2001).
Unlike other placental mammals, pangolin skin is covered by large and overlapping keratinized scales (Meyer et al. 2013). The selective forces underlying the origin of this unique mammalian trait remain a mystery, although observations of the eight modern species suggest a defensive armor function against predators. Furthermore, pangolins are edentulous or toothless, eating mostly ants and termites captured using long and muscular tongues. They also have a well-developed muscular system for fossoriality or arboreality and a remarkable olfactory system. Pangolins are the most poached and trafficked mammal in the world due to the huge demand of their meat as a delicacy and their scales for use in traditional medicines.
The Malayan pangolin and the Chinese pangolin are classified as critically endangered by the IUCN Red List of Threatened Species (http://www.iucnredlist.org/details/12763/0 and http://www.iucnredlist.org/details/12764/0). The Malayan pangolin is mainly found in Southeast Asia, whereas Chinese pangolins inhabit China, Taiwan, and some northern areas of Southeast Asia (Supplemental Fig. S1.1). Here, we present the first whole-genome sequencing and comparative analyses of these two unique species of the Pholidota, filling an important gap in our understanding of mammalian genome evolution and providing fundamental knowledge for further research in pangolin biology and conservation.
Results
Pangolin genome, divergence, and heterozygosity
A female Malayan pangolin derived from a wild specimen from Malaysia and a female Chinese pangolin from Taiwan were sequenced using whole-genome shotgun sequencing strategies to a coverage ∼145× and ∼59× based on the estimated genome size of 2.5 gigabases (Gbp) and 2.7 Gbp, respectively (Supplemental Information 1.0). We identified 23,446 and 20,298 protein-coding genes in the Malayan and Chinese pangolin genomes, respectively, using the MAKER annotation pipeline based on several sources of evidence from ab initio gene prediction, transcriptomic data, and protein evidence from the Canis familiaris reference genome (Broad CanFam3.1/canFam3) and transcriptome (Lindblad-Toh et al. 2005; Cantarel et al. 2008). The assemblies of both Malayan and Chinese pangolins capture at least 99.7% and 94.8% of the expressed genes assembled from RNA-seq data generated from eight different organs, respectively (Supplemental Information 2.1). The lower overall mapping rate of the Chinese pangolin is partially explained by the divergence of Malayan and Chinese pangolin, although it could also be impacted by the different sequencing depths. Furthermore, Core Eukaryotic Genes Mapping Approaches (CEGMA) and genome comparison analyses indicated that these genomes are good candidates for genome annotation (Supplemental Information 2.2, 2.3, 3.0).
Coalescent dating analysis suggests that pangolins diverged from their closest relatives, the Carnivora, ∼56.8–67.1 million years ago (MYA) (Supplemental Information 4.0) and that Malayan and Chinese pangolin species diverged from each other ∼4–17.3 MYA. Malayan pangolin has a more than three times higher heterozygosity rate than Chinese pangolin (1.55 × 10−3 and 0.4 × 10−3, respectively), which likely reflects the demographic impacts of a founder population and smaller effective population in the latter species (Supplemental Information 5.0). Interestingly, analysis of the trajectory of historical effective population size of the two pangolins using the pairwise sequential Markovian coalescent model showed an inverted pattern during the Middle Pleistocene, reflecting the influence of climate and sea level on both pangolin species (Supplemental Fig. S6.1).
Pangolin-specific phenotypes
To gain insight into possible genomic patterns linked with the unique traits of pangolins, we first searched for pseudogenized (presumably loss-of-function) genes. Gene loss through pseudogenization is increasingly recognized as an important factor in lineage and adaptive phenotypic diversification (Wang et al. 2006; Albalat and Canestro 2016). Given that pangolins are edentulous mammals, we screened 107 tooth development-related genes and found three pseudogenized candidate genes in pangolins. Most notable of these was ENAM, the largest protein of the enamel matrix and essential for normal tooth development (Meredith et al. 2009). ENAM mutations can lead to enamel defects (Hart et al. 2003). We identified several frameshift indels and premature stop codons in the ENAM genes of both the Malayan and Chinese pangolins (confirmed by Sanger sequencing; sample size = 8) (Fig. 1A; Supplemental Information 7.0; Meredith et al. 2009). ENAM pseudogenization has also been reported in the African tree pangolin (M. tricuspis) (Meredith et al. 2009). We also identified two other pseudogenized genes that have been linked with normal tooth development, amelogenin (AMELX) and ameloblastin (AMBN). These genes shared common mutations in both Asian species and all four African species (M. tricuspis, M. tetradactyla, M. gigantea, and M. temminckii) that we examined, suggesting that pseudogenization occurred early in the evolutionary history of the Pholidota lineage (Supplemental Figs. S7.1, S7.2). Loss of function of these genes through pseudogenization has also been reported in other edentulous vertebrates such as toothless baleen whales, birds, and turtles (Meredith et al. 2014).
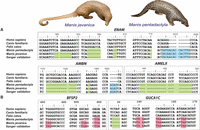
Case studies of pseudogenized genes in pangolins. (A) Three tooth development-related genes were pseudogenized and may be related to the lack of teeth in pangolins. There was a frameshift deletion in positions c1311–c1324, a single base pair deletion in c1476, and an insertion of AGAT at position c1621, resulting in another premature stop codon in the AMBN gene. The AMELX gene contains a large deletion at position c438–c497. (B) Two genes were pseudogenized and may be related to the poor vision of pangolins. Blue = insertion, green = deletion, pink = stop codon.
Pangolins are mostly nocturnal (the only exception being the long-tailed pangolin) and are thought to have poor vision (Soewu and Ayodele 2009). We screened 217 vision-related genes and identified several candidate genes that were pseudogenized (Fig. 1B). One of these, BFSP2, encodes a lens-specific intermediate filament-like protein that is a unique cytoskeletal element in the ocular lens of vertebrates (Sandilands et al. 2003). Interestingly, one frameshift insertion and three premature stop codons were consistently identified in the BFSP2 genes of both pangolin species (confirmed by Sanger sequencing; sample size = 8), suggesting possible loss of optical clarity, which may lead to progressive cataracts (Alizadeh et al. 2004). Another candidate gene, guanylate cyclase activator 1C (GUCA1C), expresses proteins that stimulate photoreceptors GC1 and GC2 at low concentrations of free calcium but inhibit GCs when the concentrations of free calcium ions are elevated, which is important for regulating the recovery of the dark state of rod photoreceptors following light exposure (Haeseleer et al. 1999). We found identical frameshift mutations and premature stop codons in GUCA1C in both pangolin species (confirmed by Sanger sequencing; sample size = 8), suggesting reduced stimulation of GC1 and GC2 and reduced rates of phototransduction in pangolins (Stephen et al. 2006). The GUCA1C gene was also pseudogenized in all African species with identical mutations (Supplemental Fig. S7.3). Interestingly, the pseudogenization of BFSP2 and GUCA1C have been reported in mice with reduced vision, supporting the hypothesis that the two genes are likely associated with the poor vision of pangolins (Imanishi et al. 2002; Sandilands et al. 2004; Song et al. 2009).
As an evolutionary consequence of being covered by scales, it is plausible that the pangolin immune system evolved differently than in other mammals. For example, genes such as IFNE, a unique interferon exclusively expressed in skin epithelial cells and inner mucosa-protected tisssues (e.g., lung, intestines, and reproductive tissues), establish a first line of defense against pathogens in other placental mammals (Day et al. 2008; Ponten et al. 2008; Xi et al. 2012; Fung et al. 2013; Demers et al. 2014; Uhlen et al. 2015). Interferons (IFNs) are a cluster of highly conserved gene families that encode for cytokines expressed by host cells for communication between cells, leading to the activation of the immune system in the presence of pathogens (De Andrea et al. 2002; Fensterl and Sen 2009). Strikingly, the single copy intronless IFNE gene is pseudogenized in both pangolin species (confirmed by Sanger sequencing; sample size = 8) but is intact in 71 other mammalian species (Fig. 2A). We found an insertion from positions 195 to position 219 and a point mutation at position c235C > T causing a premature stop codon. Other frameshift deletions at positions 264 and 540–546 would cause the loss of function. Although an alternate splicing pattern might avoid the premature stop codon and preserve the protein, high-throughput RNA sequencing of lung, cerebellum, cerebrum, and skin transcriptomes failed to detect IFNE expression (fragments per kilobase million [FPKM] = 0.0), although it is expressed in these organs in other mammals (Uhlen et al. 2005; Demers et al. 2014). We were also unable to identify any sequencing reads that mapped to the remainder of the gene after the premature stop codon, which is consistent with the hypothesis of a loss of function even if the stop codon were rescued. Interestingly, many putative functional domains or key signatures are deleted in the pangolin IFNE protein due to the premature stop codon (Fig. 2B). This suggests that the IFNE protein is unlikely to function properly if expressed, indicating that resistance to infection may be impacted in pangolins. We also examined other IFN families (IFNB, IFNK, IFNA, IFNG, and IFNL) and found that all families have intact gene copies in both pangolin species. It should be noted that the IFNA family consists of a cluster of 13 functional genes in human. Although we found that several intact genes from the IFN family in both pangolin species were retained, the number of IFNA genes is relatively low compared to human, suggesting diminished functions of this gene family in pangolins.

Multiple sequence alignment of all mammalian IFNE genes. Seventy-three mammalian species have available IFNE sequences, used for alignment. (A) Nucleotide sequence alignment of IFNE genes across different mammalian species. Blue = insertion, green = deletion, pink = stop codon. Protein sequence alignment of IFNE genes across different mammalian species is also shown. We identified an insertion (blue) in IFNE starting from nucleotide position 195 but not in other mammalian species, indicating that this insertion is specific to pangolins and possibly a marker to differentiate pangolins and other mammalian species. A premature stop codon or frameshift (orange) in the IFNE gene was consistently detected in both pangolin genomes. These mutations were validated by Sanger sequencing in eight Malayan pangolins. Protein sequences highlighted in orange are the frameshift mutations and premature stop codon. (B) Comparison between pangolin and human (reference) IFNE protein sequences. Pangolins have a short putative protein sequence because of a premature stop codon. Predicted functional domains and signatures in the human IFNE gene are represented by colored boxes. Yellow and pink boxes represent the predicted binding residues to IFNAR2 and IFNAR1, respectively, and which are bounded by interferons, which we obtained from a previosuly published paper (Fung et al. 2013). IFabd (SM00076) is a conserved functional domain in known interferons. The main conserved structural feature of interferons is a disulfide bond. INTERFERONAB (PR00266) is a three-element fingerprint that provides a signature for alpha, beta, and omega interferons. The elements 1 and 3 contain Cys residues involved in disulfide bond formation.
To test whether IFNE was also pseudogenized in the African pangolins, we sequenced the gene in all African species using the primers that we designed based on the genome sequence of Malayan pangolin. Our data showed that the IFNE gene was also pseudogenized in all African species and shared ancestral mutations with the Asian pangolin species (Supplemental Fig. S7.4). Therefore, we suggest that the pseudogenization of IFNE in pangolins most likely occurred before the divergence of the African and Asian pangolins, or at least 19–26.9 MYA, based on prior estimates (Bininda-Emonds et al. 2007; Fritz et al. 2009; Meredith et al. 2011).
Comparative analyses among pangolins and mammals
To further explore genetic differences between pangolins and their closest relatives, we compared the overlap of gene families among the two pangolin species and dog, cat, and giant panda. Our analysis identified 8325 ancestral gene families common to all five species (Fig. 3A). Interestingly, many gene families were unique to either Malayan pangolin (4958) or Chinese pangolin (3465), confirming the high level of divergence within pangolins, likely resulting from evolutionary adaptations to different ecological environments. We also found 1152 pangolin-specific gene families, of which 61% are expressed in at least one of the eight organs assessed, further suggesting that at least these genes are not assembly or annotation artifacts (Fig. 3B). Functional enrichment analyses suggest that the pangolin-specific genes are significantly overrepresented in signal transduction (496 genes; GO:0007165; FDR P-value = 2.53 × 10−9), neurological system processes (262 genes; GO:0050877; FDR P-value = 2.51 × 10−20), cytoskeleton organization (99 genes; GO:0007010; FDR P-value = 1.53 × 10−3), and cell junction organization (28 genes; GO:0034330; FDR P-value = 1.32 × 10−2) (Fig. 3C). The large number of pangolin-specific genes significantly enriched in signal transduction and neurological system processes suggests that pangolins evolved complex and unique signal transduction and neurological system networks. The large number of cytoskeleton organization-associated genes may be linked with the assembly, formation, and maintenance of the cytoskeletal elements involved in cell shape, structural integrity, and motility of the pangolin scales.
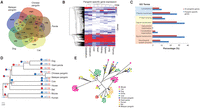
Comparative analysis between pangolins and mammals. (A) Venn diagram showing the unique and shared gene families among pangolins and their closest relatives (cat, dog, and giant panda). (B) Heat map showing the expression level of pangolin-specific genes across different pangolin organs, represented by FPKM values. Any FPKM values >5 were set to 5 in the heat map for visualization purposes. Only genes expressed (FPKM ≥ 0.3) in at least one organ were shown. (C) GO enrichment analysis of 1152 pangolin-specific genes. Significantly enriched GO terms are shown for the categories of cellular compartment (blue), molecular function (yellow), and biological process (light orange). (D) Phylogenetic tree and gene family expansion and contraction. Expanded gene families are indicated in blue, whereas contracted gene families are indicated in red. The proportion of expanded and contracted gene families is also shown in pie charts. (E) Phylogenetic tree showing significant contraction of the interferon gene family in pangolins.
Gene family analysis identified 147 families that are significantly expanded and 18 that are contracted in the pangolin lineage, indicating that gene expansion and contraction events played a major role in the functional diversification of pangolins (Fig. 3D; Supplemental Information 8.0). Notably, pangolins have a highly reduced number of interferon genes, which have a major role in responding to infections, inflammation, and healing of skin (Xi et al. 2012; Fung et al. 2013). Of the 10 genes in the interferon family, only three were found in Malayan pangolin and two in Chinese pangolin (Fig. 3E). The heat shock gene family also contracted significantly, possibly contributing to the sensitivity of pangolins to stress (Hua et al. 2015). However, we also document gene expansion in several important families, including ribosomal genes (17 families), olfactory receptor (OR) genes (six families), cathepsin genes (one family), and septin genes (one family), which were recently shown to deter microbial pathogens, preventing them from invading other cells (Supplemental Information 8.0; Mascarelli 2011). The significantly expanded OR gene families suggest that pangolins have an enhanced sense of smell, possibly helping locate prey and counterbalancing poor vision (Supplemental Fig. S8.2). Strikingly, functional enrichment analysis of the 147 expanded gene families reveal these genes are significantly overrepresented in neurological system processes (including neuromuscular processes and sensory perception) (207 genes; GO:0050877; FDR P-value = 6.43 × 10−14), which is consistent with our finding in the functional analysis of pangolin-specific genes (Supplemental Fig. S8.3). The gene families associated with symbiosis and host-parasite interactions were also overrepresented (82 genes; GO:0044403; FDR P-value = 2.67 × 10−13).
Positive selection analysis
To identify signatures of natural selection, we used a set of 8250 protein-coding orthologs shared among the dog, cat, panda, cow, horse, mouse, human, and megabat genomes (Supplemental Information 9.0). Branch site tests identified evidence of positive selection along the pangolin lineage in 427 genes that had significant signals (P < 0.05 adjusted) (Supplemental Table S9.1).
We hypothesize that the reduced IFN-mediated immunity from the loss of IFNE and the contraction of the interferon gene family in pangolins imposed strong selective pressure on immunity-related genes. We identified a large proportion of genes under selection in the pangolin lineage that involve a wide range of immunity-related pathways, including hematopoietic cell lineage, cytosolic DNA-sensing pathway, complement and coagulation cascades, cytokine–cytokine receptor interaction, and the phagosome pathway (Supplemental Table S9.2). For instance, in the hematopoietic cell lineage, the colony stimulating factor 3 receptor (CSF3R) gene encodes a transmembrane receptor for a cytokine (granulocyte colony stimulating factor 3) (Fig. 4A). CSF3R proteins are present on precursor cells in the bone marrow, playing important roles in the proliferation and differentiation into mature neutrophilic granulocytes and macrophages which are essential for combating infection (Liongue and Ward 2014). As predicted by Protein Variation Effect Analyzer (PROVEAN), CSF3R protein contains a critical pangolin-specific mutation at position 247(G→H) in the functionally relevant Fibronection Type III domain that likely affects its protein function in pangolins (Fig. 4C). Another gene, integrin subunit alpha M (ITGAM), mediates leukocyte activation and migration, phagocytosis, and neutrophil apoptosis. It has been reported that ITGAM can migrate neutrophils across intestinal epithelium to maintain intestinal homeostasis and eliminate pathogens that have translocated across the single layer of mucosal epithelial cells (Parkos et al. 1991). We identified two pangolin-specific amino acid changes in the functional Integrin alpha-2 domain of ITGAM which likely affect protein function (Fig. 4C). In the cytosolic DNA-sensing pathway, transmembrane protein 173 (TMEM173), also known as Stimulator of Interferon Genes, plays an important role in innate immunity by sensing cytosolic foreign DNA and inducing type I interferon production when cells are infected with pathogens (Fig. 4B; Ran et al. 2014). We identified two critical amino acid changes in pangolins at positions 216(D→G) and 217(P→L) in the functionally relevant region, suggesting that they may have a functional impact on TMEM173 (Fig. 4C).
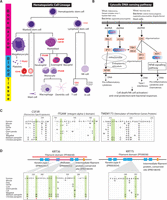
Evolution of immunity-related pathways and scale-related genes in the pangolin ancestor. Genes under positive selection in the pangolin lineage in hematopoietic cell lineage (A) and cytosolic DNA-sensing pathway (B) are highlighted in red. (C) Several critical pangolin-specific mutations were identified in the functionally relevant Fibronection Type III signatures (INTERPRO ID = IPR003961) of CSF3R, the Integrin alpha-2 signatures (IPR013649) of ITGAM, and the Stimulator of Interferon Genes Protein region (INTERPRO ID = IPR029158) of TMEM173. For the CSF3R, ITGAM, and TMEM173, P-values of the detection of positive selection were 1.58 × 10−2, 3.75 × 10−2, and 2.92 × 10−5, respectively. (D) Several critical pangolin-specific amino acid changes were detected in hair-/scale-related keratin proteins, KRT36 and KRT75, which are located in functionally relevant regions that may affect protein functions. For the KRT36 and KRT75, P-values of the detection of positive selection were 2.24 × 10−2 and 1.33 × 10−5, respectively. We also examined whether African species (M. tricuspis, M. tetradactyla, and M. temminckii) have the critical pangolin-specific amino acid changes that we observed in the Asian pangolins. Circles at the bottom indicate our preliminary results: Red circle = all African species that we examined have the identical amino acid change, yellow circle = not all African species that we examined have the identical amino acid change (but they all have amino changes/deletion that likely affect protein function as predicted by PROVEAN), brown circle = all African species that we examined have the same amino acids like the human reference sequence, white circle = data not available. The alignment results are shown in Supplemental Figure S7.5.
We further postulate that genetic changes during the evolution of hair-derived pangolin scales likely involved genes related to hair formation in general, and particularly keratins, which are essential components of scales and hair (Meyer et al. 2013). Among the positively selected candidate genes associated with hair formation are KRT36, KRT75, KRT82, and KRTAP3-1. For instance, type II keratin 75 (KRT75), a hair follicle–specific keratin, has an essential role in hair and nail integrity (Chen et al. 2008). Type I keratin 36 (KRT36), a hair or “hard” sulfur-rich keratin, is mainly responsible for the extraordinarily high degree of filamentous cross-linking by specialized keratin-associated proteins (Moll et al. 2008). Many critical pangolin-specific amino acid changes were identified in keratin genes in functionally relevant domains (Fig. 4D). These critical amino acid changes are also present in both KRT36 and KRT75 genes in the distantly related African pangolin species that we examined, suggesting that they potentially contributed to the development of pangolin scales (Fig. 4D; Supplemental Fig. S7.5). We also identified genes related to energy storage and metabolism (12 genes) and mitochondrial metabolism (11 genes), perhaps an adaptive response to reduce their metabolic rate given their large body size but low energy diet of ants and termites (da Fonseca et al. 2008). Other candidate genes are associated with the development of the robust muscular system of pangolins, which assists its fossorial lifestyle (10 genes), and the nervous system (10 genes) (Supplemental Table S9.2).
Furthermore, we explored whether there was evidence that genes under selection were associated with any response to diseases, particularly those related with infection or skin/mucosal organs. We identified genes associated with inflammation (18), cancer or viral infection (25), bacterial infection (10), pneumonia and gastrointestinal disease (21), and skin diseases (15) (Supplemental Table S9.3). Interestingly, one gene, lactotransferrin (LTF), encodes a multifunctional immune protein found at mucosal surfaces, providing the first line of defense to the host against inflammation and infection (Ward et al. 2002). It has been shown that LTF has antimicrobial activities and induces both systematic and mucosal immune responses, for example, against lung and gut-related systemic infections (Debbabi et al. 1998). Two pangolin-specific amino acid changes were detected in the LTF protein sequence, potentially having an impact on LTF function (Supplemental Fig. S9.1). Together, evidence of selection on these genes provides possible links with the unique phenotypes and physiological requirements of pangolins. Importantly, the immunity- or inflammation/infection-related genes provide avenues to further explore the evolution of mammalian immunity.
Pangolins have a noncanonical repeat layout
Transposable elements accounted for approximately 28.9%–30.0% of the pangolin genomes, lower than in other Carnivora such as the giant panda (35.3%), tiger (36.7%), cat (37.2%), and dog (36.2%), suggesting divergence in genome architecture between pangolins and other placental mammals (Supplemental Fig. S10.1a). Pangolins also have relatively low proportions of SINE repeats (2.6%–2.8% vs. 8.2%–11%), mainly due to a relatively smaller number of tRNA SINEs (Supplemental Fig. S10.1a,b). A search using a de novo constructed repeat library supports the comparatively low proportion of this repeat family (Supplemental Table S10.2). Furthermore, both pangolin species have relatively low proportions of intronic repeats (SINEs, LINEs, and LTRs) (Supplemental Fig. S10.1c).
Discussion and conclusion
Our analyses have revealed that, similar to observations in other lineages (Wang et al. 2006; Meredith et al. 2014), gene loss through pseudogenization has had a significant impact on the evolution and diversification of pangolin genomes and their biology. We detected and validated pseudogenes in pathways related to multiple morphological or physiological functions including dentition, vision, and immunity. The pseudogenization of the IFNE gene in pangolins, otherwise functional in all other mammal genomes surveyed so far, is especially noteworthy. In mice, a deficiency of IFNE in epithelial cells of the female reproductive tract (FRT) can increase susceptibility to microbial infection (Fung et al. 2013). Therefore, the pseudogenization of this interferon gene in pangolins suggests that innate immunity may be compromised, resulting in an increased susceptibility to infection, particularly in the skin and mucosa-protected organs. Since pangolins may have intrinsically weak mucosal immunity, Burkholderia spp. might easily infect the lung by penetrating lung epithelial cells and the brain by penetrating nasal mucosa and migrating to the brain through olfactory nerves, which has previously been shown with other Burkholderia (Owen et al. 2009; Sim et al. 2009; Taylor et al. 2010; Dando et al. 2014; St John et al. 2014). Furthermore, captive pangolins are prone to frequently fatal gastrointestinal disease, pneumonia, and skin maladies (Clark et al. 2008; Hua et al. 2015). Pangolins are notoriously difficult to maintain in captivity, and the stress of captivity and/or poor husbandry might render pangolins even more vulnerable to infections or diseases by suppressing immune responses. Whether or not these afflict free-ranging pangolins where pathogen exposure is rampant remains unknown.
Our study raises the fundamental evolutionary question of how pangolin adaptations occurred given their seemingly increased risk to infection if their skin and mucosal surfaces are constantly being exposed to pathogens. Our analyses suggest several evolutionary changes in genes that may respond to or interact with pathogens, including immunity-related genes/pathways, the significant expansion of the septin gene family, and the enrichment of the genes in symbiosis. These changes may enhance the immunity system of pangolins, although we do not know the relative efficiency of these adaptations compared to interferon-mediated immunity. We propose that pangolin scales may be an important morphological innovation to compensate for the decrease in immunity normally provided by the skin. These hard and overlapping scales may act as defensive armor to protect pangolins against injuries (or stress) which would make pangolins even more vulnerable to infection or the invasion of pathogens. Moreover, pangolins curl into near impregnable balls, covering their scaleless abdomen using their well-developed neuromuscular system, during sleep or when threatened, which would also support the hypothesis that these adaptations serve to protect pangolins from skin injuries. It is noteworthy that besides the antimicrobial effects, plasmacytoid dendritic cell (pDC)-produced type 1 IFNA/B helps heal wounds through re-epithelization of injured skin (Gregorio et al. 2010). It is unknown whether keratinocyte-produced type-I IFNE is also involved in skin wound healing (e.g., recruiting pDCs to the wound). Pangolins have a marked contraction of interferon genes that likely facilitates wound healing. Future studies are needed to test the relationship between IFNE, skin healing, and scale development.
Our analyses showed that pangolin-specific genes were significantly overrepresented in the GO categories governing cytoskeleton organization, neurological system process, and signal transduction. Further, certain genes involved in the muscular and nervous systems showed evidence of positive selection. These could be linked to the unique adaptations and behaviors of pangolins, particularly those related with their highly sophisticated musculoskeletal system (Supplemental Information 12).
The Chinese pangolin had a significantly larger predicted genome size but significantly fewer genes than the Malayan pangolin, despite the lower sequencing coverage in the former species. We found segmental duplications were identified in a higher proportion in the Chinese pangolin compared to the Malayan pangolin, suggesting that the segmental duplications partly contribute to the larger genome size of the Chinese pangolin (Supplemental Information 3.4). It should be noted that a previous comparative chromosome map between the Malayan (2n = 38) and Chinese (2n = 36–42) pangolins revealed many chromosomal rearrangements between the two species (Nie et al. 2009). Besides that, we did not observe significant recent repeat family activity unique to Chinese pangolins, suggesting that the genome expansion is unlikely directly accounted by repeats (Supplemental Fig. S10.2). However, we cannot rule out the possibility that the recent segmental duplications and repeats were underestimated (e.g., some might collapse together due to high sequence similarity during assembly since we sequenced the pangolin genomes using the short-read Illumina technology), which would complicate the analyses.
Our data also showed that the number of gene families unique to each of the pangolin species is 3–4 times greater than the number of shared gene families. In this analysis, we faced the common problem of almost all draft genomes—the potential difference between the real and available gene set. We can expect a number of misannotated genes because of fragmented assemblies, and this can partly inflate the number of genes for each species. However, it should not affect subsequent analyses because we had a refined pangolin-specific gene set by orthology clustering as described in the Methods section. It is also worth noting here that the discrepancy in the unique gene family numbers indeed can be explained by biological reasons, such as a high rate of molecular evolution. It was previously shown that the chromosome complements of Malayan pangolin and Chinese pangolin differ by seven Robertsonian rearrangements (three centric fissions and four centric fusions), which could also affect the number of unique gene families for each species after their divergence from the common ancestor (Nie et al. 2009).
In conclusion, this study provides new insights into the biology, evolution, and diversification of pangolins. The annotated reference genomes will be invaluable for future studies addressing issues of species conservation and gene function and may provide a new natural animal model for understanding mammalian immunity.
Methods
Genome sequencing, assembly, and annotation
Genomic DNA from female pangolins was extracted and sequenced using the Illumina sequencing platform. Different insert sizes of short-read and mate-pair libraries ranging from ∼180 bp to 8 kbp were used. The genome sequence of the Malayan pangolin was assembled using CLC Assembly Cell 4.10, SGA 0.10.10, and SOAPdenovo2, and the best assembly was chosen for downstream analyses (Supplemental Information 1.3.1; Luo et al. 2012; Simpson and Durbin 2012). The genome of the Chinese pangolin was assembled using SOAPdenovo v1.0.5 (Supplemental Information 1.3.2). The Malayan pangolin assembly has a genomic length of ∼2.5 Gbp with an assembled N50 scaffold size of 204,525 bp, whereas the Chinese pangolin scaffold size was ∼2.2 Gbp with an assembled N50 scaffold size of 157,892 bp (Supplemental Information 1.3).
Protein-coding genes in the pangolin genomes were predicted using the MAKER annotation pipeline by combining de novo gene prediction, RNA-seq data, and homology-based methods (Supplemental Information 3.0; Cantarel et al. 2008). Identified protein-coding genes in the Malayan and Chinese pangolin genomes are supported by functional assignments from at least one biological database by the InterProScan 5 pipeline (Supplemental Information 3.0; Jones et al. 2014).
Genome size was estimated by k-mer analysis using short-insert sequencing reads (Supplemental Information 1.4). The genome size of the Malayan pangolin and Chinese pangolin was estimated to be 2,492,544,425 and 2,696,930,760 bp, respectively.
Gene family clustering
OrthoMCL (Li et al. 2003) was used to identify homologous protein sequences among the Malayan pangolin, the Chinese pangolin, cat, dog, and giant panda. BLASTALL (Pearson 2014) was first performed among the protein sequences. For each pair of gene members of a gene family/cluster, both local and global matched regions must be at least 40% of the longer gene protein sequence. The minimum score value of 50 and e-value of 1 × 10−8 in BLAST was used. After clustering the protein sequences into gene families, pangolin-specific genes were retrieved for downstream analysis using in-house Perl scripts.
Functional enrichment analysis
Enrichment analysis of pangolin-specific and the significantly expanded gene families were performed using Blast2GO with all pangolin genes used as background for comparisons (Conesa et al. 2005). To identify significantly overrepresented gene ontology categories (e.g., molecular function and biological processes), Fisher's exact test was used with a cut-off of a false discovery rate of 0.05 after multiple test correction.
Repetitive element analysis
Repetitive elements in the pangolin genomes and other Carnivora genomes (giant panda, tiger, cat, and dog) were identified using RepeatMasker open-4.0.5 (Smit et al. 2013–2015) against the Repbase TE database (version 2014-01-31) (Supplemental Information 6.0; Jurka 2000).
RNA-seq expression analysis
The RNA-seq raw data were from our Malayan pangolin transcriptome project. Briefly, we sequenced the transcriptomes of cerebellum, cerebrum, lung, heart, kidney, liver, spleen, and thymus using the Illumina HiSeq technology platform (100-bp Paired End [PE] strategy). The data from each sample ranged from 41 to 53 million of PE reads or ∼8.2–10.6 Gbp. For each organ, the PE reads were mapped to the genome assembly using TopHat 2.0.11 (Trapnell et al. 2009). Properly mapped PE reads with the best match were sorted and indexed with SAMtools (Li et al. 2009). The expression of each gene was calculated and represented in fragments per kilobase of transcript per million mapped reads based on the coordinates of the MAKER-generated gene models. Heat maps showing expression of pangolin-specific genes across the eight different pangolin organs were generated using in-house R scripts. Using the same approach, we also calculated the expression level of the pseudogenized IFNE gene in the transcriptomes of three organs (lung, cerebrum, and cerebellum) of the Malayan pangolin, as well as a transcriptome of skin tissue (250-bp PE; # of PE reads = ∼17 million) which we generated in a separate project using Illumina MiSeq technology.
Functional impact of substitutions in positively selected genes
We assessed the potential functional impact of pangolin-specific amino acid changes in the positively selected genes of interest in the pangolin ancestor using PROVEAN (Choi and Chan 2015). Amino acid substitutions were considered “deleterious” if the PROVEAN score was ≤−2.5 and “neutral replacements” if the PROVEAN score was >−2.5.
Data access
The Malayan pangolin and Chinese pangolin BioProjects are accessible at the NCBI BioProject (BioProject: http://www.ncbi.nlm.nih.gov/bioproject/) under accession numbers PRJNA256023 and PRJNA20331, respectively. The assembled whole-genome sequences from this study have been submitted to GenBank (http://www.ncbi.nlm.nih.gov/genbank/) under accession numbers JSZB00000000.1 (Malayan pangolin) and JPTV00000000.1 (Chinese pangolin). The raw sequencing reads of Malayan pangolin and Chinese pangolin have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession numbers SRP078903 and SRX247981, respectively. All RNA-seq reads can be accessed at the NCBI SRA under accession number SRP064341. The assembled transcriptome sequences of Malayan pangolin have been submitted to Transcriptome Shotgun Assembly (TSA; http://www.ncbi.nlm.nih.gov/genbank/tsa/) under accession number GEUV00000000. All PCR trace data have been submitted to the NCBI Trace (Trace; http://www.ncbi.nlm.nih.gov/Traces/) under trace IDs (SID 271513, SID 271514, SID 271515, SID 271516, and SID 271517). In addition, the pangolin genome sequences, raw sequencing reads, and RNA-seq data of Malayan pangolin are also accessible at our pangolin genome hub (http://pangolin-genome.um.edu.my/).
Acknowledgments
We thank Kerstin Lindblad-Toh and Jessica Alföldi for generously providing guidance and sharing their advice. We also thank all members of the Genome Informatics Research Laboratory, University of Malaya in contributing to this research. Special thanks to Tan Shi Yang for providing IT and bioinformatics support in this project. This project was mainly supported by the University of Malaya and Ministry of Education, Malaysia, under the High Impact Research (HIR) grant UM.C/HIR/MOHE/08 and UMRG grant (grant number: RG541-13HTM) from the University of Malaya and Ministry of Education, Malaysia. This research was also supported in part by the Russian Ministry of Science (Mega-grant no.11.G34.31.0068; S.J.O., Principal Investigator), and the funding to R.K.W. was provided by NIH-NHGRI grant 5U54HG00307907.
Author contributions: The project is a part of the International Pangolin Research Consortium (IPaRC). The analyses were mainly conducted by members from the Genome Informatics Research Laboratory, University of Malaya and the Theodosius Dobzhansky Center for Genome Bioinformatics, St. Petersburg State University, Russia. For full details of author contributions, please see below:
S.W.C., S.J.O., M.R., G.J.W., W.E.J., and A.M.Y. conceived and coordinated this project. S.W.C., K.V.K., A.M.Y., G.J.W., J.J.R.R., S.J.L., and F.T.S. performed animal handling, sampling, and tagging. S.W.C., R.H., and I.C.P. coordinated and performed DNA extraction. S.W.C., R.K.W., and P.M. led and designed library construction and NGS experiments. S.W.C. and T.K.T. designed primers, PCR, and Sanger sequencing validation experiments. A.K., D.L.D., and E.V. performed Sanger sequencing validation experiments for African pangolins. S.W.C., M.R., T.K.T., R.H., W.Y.W., A.A., K.P.K., A.K., A.A.Y., M.R., S.K., P.D., R.K.W., P.M., D.Z., M.B.G., G.T., K.K., and G.F. performed data analyses, data interpretation, and oversaw various analyses. S.W.C., T.K.T., M.R., A.K., R.H., and G.J.W. wrote the manuscript. S.J.O., A.A., K.P.K., W.C.W., W.E.J., I.C.P., and L.L. revised the manuscript. S.W.C. and S.J.O. were the principal investigators of this project.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.203521.115.
- Received December 17, 2015.
- Accepted August 4, 2016.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








