
Unbalanced translocations arise from diverse mutational mechanisms including chromothripsis
- Corresponding author: katie.rudd{at}emory.edu
Abstract
Unbalanced translocations are a relatively common type of copy number variation and a major contributor to neurodevelopmental disorders. We analyzed the breakpoints of 57 unique unbalanced translocations to investigate the mechanisms of how they form. Fifty-one are simple unbalanced translocations between two different chromosome ends, and six rearrangements have more than three breakpoints involving two to five chromosomes. Sequencing 37 breakpoint junctions revealed that simple translocations have between 0 and 4 base pairs (bp) of microhomology (n = 26), short inserted sequences (n = 8), or paralogous repeats (n = 3) at the junctions, indicating that translocations do not arise primarily from nonallelic homologous recombination but instead form most often via nonhomologous end joining or microhomology-mediated break-induced replication. Three simple translocations fuse genes that are predicted to produce in-frame transcripts of SIRPG-WWOX, SMOC2-PROX1, and PIEZO2-MTA1, which may lead to gain of function. Three complex translocations have inversions, insertions, and multiple breakpoint junctions between only two chromosomes. Whole-genome sequencing and fluorescence in situ hybridization analysis of two de novo translocations revealed at least 18 and 33 breakpoints involving five different chromosomes. Breakpoint sequencing of one maternally inherited translocation involving four chromosomes uncovered multiple breakpoints with inversions and insertions. All of these breakpoint junctions had 0–4 bp of microhomology consistent with chromothripsis, and both de novo events occurred on paternal alleles. Together with other studies, these data suggest that germline chromothripsis arises in the paternal genome and may be transmitted maternally. Breakpoint sequencing of our large collection of chromosome rearrangements provides a comprehensive analysis of the molecular mechanisms behind translocation formation.
Translocation is one of the most common structural chromosome abnormalities found in humans, with a de novo frequency of 1 in 2000 (Warburton 1991). Unbalanced translocations lead to monosomy and trisomy for segments of different chromosomes and account for ∼1% of cases of developmental delay and intellectual disability (Ravnan et al. 2006; Ballif et al. 2007; Shao et al. 2008). The initial exchange of genetic material between two nonhomologous chromosomes can occur during premeiotic mitoses, meiotic recombination in the parental germline, or post-zygotic mitoses in the early embryo (Vanneste et al. 2009; Robberecht et al. 2013). Unbalanced translocations detected in affected children may be inherited from a parent who carries the balanced form of the rearrangement or may arise de novo.
Recurrent translocations may be mediated by nonallelic homologous recombination (NAHR) between segmental duplications (Giglio et al. 2002; Ou et al. 2011) or paralogous interspersed repeats (Luo et al. 2011; Hermetz et al. 2012; Robberecht et al. 2013). Palindromic AT-rich repeats on Chromosomes 3, 8, 11, 17, and 22 also generate recurrent translocations, the most common of which is the recurrent t(11;22) that causes Emanuel syndrome (Edelmann et al. 2001; Kurahashi et al. 2003; Gotter et al. 2007; Kato et al. 2012, 2014). Most germline translocations, however, are not recurrent, and sequencing of translocation breakpoints has revealed features of nonhomologous end-joining (NHEJ) and microhomology-mediated break-induced replication (MMBIR) at more than 60 unique translocation junctions (Chen et al. 2008; Higgins et al. 2008; Sobreira et al. 2011; Chiang et al. 2012; Robberecht et al. 2013). Recently, a study of 12 de novo unbalanced translocations, nine of which were sequenced, concluded that NAHR between paralogous repeats is the predominant mechanism of de novo unbalanced translocation formation (Robberecht et al. 2013).
Sequencing translocation breakpoints can also identify physically disrupted and fused genes. In the case of balanced translocations, genes altered at translocation breakpoints are strong candidates to explain neurodevelopmental phenotypes (Baptista et al. 2005; Higgins et al. 2008; Backx et al. 2011). On the other hand, the clinical features of individuals with unbalanced translocations may be explained by copy number changes in genes within monosomic and trisomic segments. In either case, identifying unique gene fusions is important for understanding the consequences of translocations.
Sequence analysis of breakpoint junctions can also reveal more complex rearrangement structures than expected from copy number studies alone (Luo et al. 2011; Chiang et al. 2012; Carvalho et al. 2013; Brand et al. 2014; Newman et al. 2015). Though most germline translocations involve only two chromosomes, some are the product of many breakpoints on three to five different chromosomes. Originally seen in cancer (Stephens et al. 2011), chromosome shattering or chromothripsis is now recognized as a cause of some germline translocations (Kloosterman et al. 2011, 2012; Chiang et al. 2012; Nazaryan et al. 2014; Pellestor et al. 2014; de Pagter et al. 2015).
Here we investigate translocation structure, genes fused at breakpoints, and rearrangement mechanisms by analyzing a group of 57 unbalanced translocations, the largest cohort to date. Using a combination of array comparative genomic hybridization (CGH), fluorescence in situ hybridization (FISH), SureSelect sequence capture, and whole-genome sequencing (WGS), we provide a comprehensive sequence analysis of unbalanced translocations.
Results
Sequencing unbalanced translocation junctions
We recruited subjects with developmental delays, autism, intellectual disability (ID), and/or congenital anomalies after routine cytogenetics testing at Emory Genetics Laboratory (EGL). For 57 unrelated individuals with a previous diagnosis of an unbalanced translocation, we extracted DNA from peripheral blood for further study. In this cohort, translocation breakpoints are spread across all of the autosomes and the X Chromosome (Supplemental Table S1). From the 57 subjects, 51 rearrangements are simple unbalanced translocations with one derivative chromosome that fuses two chromosome breakpoints; six rearrangements have more than one breakpoint junction that joins multiple segments from two or more chromosomes. Subjects EGL312 and EGL356 have complex translocations involving two chromosomes, whereas EGL302, EGL305, and EGL321 have complex translocations between four or five chromosomes. EGL826 has one simple balanced translocation between Chromosomes 1 and 3 and a complex unbalanced translocation between Chromosomes 10 and 17.
To fine-map breakpoints, we designed custom oligonucleotide microarrays with dense probe coverage in 1-megabase (Mb) windows centered around the breakpoints determined by diagnostic chromosomal microarray analysis (CMA). High-density arrays resolve unbalanced translocation breakpoints to 200–1000 bp but do not detect copy-neutral structural variation. Next, we attempted SureSelect Target Enrichment to capture 40-kilobase (kb) regions surrounding 44 fine-mapped translocations (40 simple and four complex) (Supplemental Tables S1, S2). Since none of the breakpoints were shared between individuals, we pooled genomic DNA from five to seven subjects per SureSelect library and separated subject-specific junctions after next-generation sequencing (NGS) using Illumina HiSeq. We sequenced 100-bp paired-end reads and analyzed discordant reads where paired-ends map to different chromosomes, map too close together, or map too far apart relative to the GRCh37/hg19 reference genome.
Discordant reads spanned 19 of 40 simple translocations and two of four complex translocations targeted by SureSelect and Illumina HiSeq (Supplemental Tables S1, S2). To confirm NGS results, we PCR-amplified translocation junctions predicted by discordant reads and Sanger sequenced amplicons. We confirmed 18/19 of simple translocations supported by discordant reads. One translocation junction that failed PCR confirmation (EGL313) was supported by discordant reads between unique sequence and a segmental duplication. For the 21/40 simple translocations where SureSelect plus Illumina HiSeq did not yield discordant reads, we attempted long-range PCR using breakpoint estimates from high-resolution array CGH and successfully sequenced 12. We PCR-amplified and sequenced an additional seven simple translocations without attempting SureSelect, leading to a total of 37 simple translocation junctions confirmed by Sanger sequencing.
SureSelect followed by Illumina HiSeq successfully captured some breakpoint junctions for complex translocations in EGL305 and EGL321; however, for most complex translocations, we performed Complete Genomics WGS or Nextera mate-pair sequencing to capture multiple junctions in one experiment (see below).
Simple unbalanced translocations
We confirmed the junctions of 37 simple unbalanced translocations by Sanger sequencing (Table 1). Six junctions had blunt ends, and 20 junctions had 1–4 base pairs (bp) of microhomology shared between the two sides of the translocation. Eight translocations had short insertions or inversions at the breakpoint junction, ranging in length from 2 to 209 bp (Table 1). In four translocations, the inserted sequence is a copy of adjacent sequence, indicating DNA slippage (Viguera et al. 2001). Like other DNA replication-based rearrangements (Lee et al. 2007; Zhang et al. 2009; Conrad et al. 2010; Luo et al. 2011; Newman et al. 2015), two of these local duplications are in an inverted orientation relative to the reference genome, and two are in direct orientation. Insertions in LM219, EGL366, and EGL087 map to regions 210 bp, 1.5 kb, and 56 kb from the breakpoint, respectively (Supplemental Table S1). The origin of EGL089's 7-bp insertion is unknown.
Features of sequenced breakpoint junctions in simple, complex, and chromothripsis translocations
Three translocations have at least 335 bp of perfect homology shared between the two sides of the junction, consistent with NAHR. EGL051's translocation occurs between segmental duplications on Chromosomes 5 and 14 that are 95% identical over 1.5 kb. In EGL080, the translocation breakpoint spans a L1PA2 on Chromosome 8 and a L1PA3 on Chromosome 1 that are 93% identical across the 6.0-kb repeats. EGL083's junction lies in HERV-H elements on Chromosomes 8 and 12 that are 92% identical across the 3.2-kb and 3.0-kb repeats. In each of these translocations, recombination occurred at paralogous sites within repeats and created a hybrid repeat element at the breakpoint junction. Breakpoints in LM219's unbalanced translocation fall in AluSx and AluSx1 repeats; however, the junction does not lie in homologous parts of the Alus.
Complex translocations between two chromosomes
EGL312, EGL356, and EGL826 have complex translocations between two chromosomes. Though EGL826 has translocations involving four chromosomes, only two chromosomes form a complex rearrangement. According to array CGH, complex translocation breakpoints in EGL312 and EGL356 border repetitive regions, so we performed Nextera mate-pair sequencing (Illumina) of 5- to 7-kb inserts. This approach is ideal for junctions in repetitive DNA because mate pairs span repeats and map to unique sequence (Kloosterman et al. 2011; Talkowski et al. 2011, 2012; Hanscom and Talkowski 2014). We identified discordant reads for one of two junctions expected in EGL312 and for three of four junctions expected in EGL356. In EGL312's rearrangement, CMA and FISH analysis revealed an unbalanced translocation of two regions of Chromosome 9 to the short arm of Chromosome 13 (Fig. 1A). Mate-pair sequencing captured one inverted junction between the two translocated segments of Chromosome 9. This junction connects an L1PA3 repeat to a segmental duplication, so it is not surprising that we failed to capture this breakpoint by SureSelect. However, we did not sequence junction(s) that connect Chromosomes 9 and 13. EGL356 has an insertional translocation with three segments from Chromosome 13 translocated into the long arm of Chromosome 14 (Fig. 1B). We confirmed insertions by FISH, and CMA revealed a 1.4-Mb deletion at the insertion site on Chromosome 14. Mate-pair reads cross two translocation junctions between Chromosomes 13 and 14 and an inverted junction between two segments from Chromosome 13.
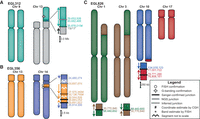
Models of the complex translocations from EGL312, EGL356, and EGL826. See legend for symbol definitions. Zoomed-in junctions point out those confirmed with PCR and Sanger sequencing, supported only by NGS reads, or inferred by FISH. Lighter-colored chromosome segments are deletions at breakpoints. Arrows indicate chromosomal orientation relative to the normal chromosome and are shown proximal to distal. (A) EGL312 has two regions of Chromosome 9 translocated onto the short arm of Chromosome 13. One NGS breakpoint junction (Nextera mate-pair sequencing) joins the two regions of Chromosome 9, and we infer a second breakpoint junction between Chromosome 9 and Chromosome 13. (B) EGL356's rearrangement is an insertional translocation of three regions of Chromosome 13 into the long arm of Chromosome 14. There is a 1.5-Mb deletion of Chromosome 14 at the insertion site. Nextera mate-pair sequencing revealed translocation junctions between Chromosomes 13 and 14, and we inferred one connection between two Chromosome 13 regions. (C) EGL826 has a maternally inherited balanced translocation between Chromosomes 1 and 3, in addition to a complex unbalanced translocation involving Chromosomes 10 and 17. At this translocation junction, there is an inverted triplication of a region of Chromosome 17. Breakpoint junctions were detected by WGS (Complete Genomics) and confirmed by PCR and Sanger sequencing.
We also used Complete Genomics WGS to sequence EGL826's two independent chromosome rearrangements (Fig. 1C). Her balanced translocation between Chromosomes 1 and 3 was maternally inherited, and her unbalanced translocation between Chromosomes 10 and 17 arose de novo. Whereas the balanced translocation has two simple translocation junctions, the unbalanced translocation has a 250-kb inverted triplication of Chromosome 17. Between the two rearrangements, we sequenced a total of four translocation junctions. There are blunt ends or up to 4 bp of microhomology at all breakpoint junctions analyzed in these translocations (Table 1).
Chromothripsis translocations
Chromosome banding and FISH analyses of EGL302, EGL305, and EGL321 revealed translocations involving four or five different chromosomes. Translocations between more than two chromosomes may be caused by germline chromothripsis (Kloosterman et al. 2011).
EGL305 has a four-way translocation that he inherited from his mother, who carries a more balanced form of the rearrangement (Fig. 2). We sequenced two junctions involving four different chromosomes by SureSelect followed by Illumina HiSeq. The derivative Chromosome 1 has a 530-kb deletion at the 1q21 junction that is connected to an inverted breakpoint on Chromosome 15q22. Since the segment of Chromosome 15 is inverted at the junction, there must be additional breakpoint(s) to account for the correct orientation of the end of the long arm of Chromosome 15. Junction sequencing of the derivative Chromosome 15 revealed an inverted segment of Chromosome 7 that lies between parts of Chromosomes 15 and 4. FISH analysis confirmed that EGL305's mother is balanced for the Chromosome 7 segment; she has a deletion of Chromosome 7 plus the derivative Chromosome 15 with the insertional translocation of Chromosome 7. EGL305 did not inherit the deleted Chromosome 7, so he has three copies of this 4.2-Mb region. DNA was depleted following targeted sequencing so we could not follow up with WGS to sequence additional breakpoints.
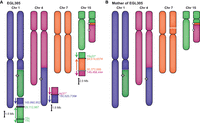
Maternal transmission of EGL305's chromothripsis. (A) A combination of G-banding and FISH revealed EGL305's four-way translocation between Chromosomes 1, 4, 7, and 15. SureSelect and Illumina HiSeq targeted to the Chromosome 1 deletion and Chromosome 7 duplication captured two junctions, and we inferred additional breakpoints. (B) EGL305's mother carries a more balanced form of the same four-way translocation.
We sequenced complex rearrangements in EGL302 and EGL321 via Complete Genomics WGS. In the original cytogenetic characterization of EGL302, we detected translocations involving Chromosomes 8, 9, 11, and 13 by chromosome banding. CMA revealed a 2.8-Mb deletion of Chromosome 8 and a 6.6-Mb deletion of Chromosome 9 that correspond to translocation breakpoints. SureSelect targeted to the Chromosome 8 and 9 deletion regions did not capture any translocation junctions, but WGS revealed 11 breakpoint junctions between Chromosomes 3, 8, 9, 11, and 13 (Fig. 3). We infer at least two additional breakpoint junctions by FISH mapping translocated segments (Supplemental Fig. S1). Though all the translocations are de novo, they appear to have arisen as two separate events. The reciprocal translocation between Chromosomes 11 and 13 has simple breakpoints on each derivative chromosome. However, derivative Chromosomes 3, 8, and 9 are part of complex translocations with multiple breakpoints and inserted fragments. Aside from the megabase-sized deletions on Chromosome 8 and 9, other breakpoints have only deleted 0–70 bp, for a total of 99 bp deleted.
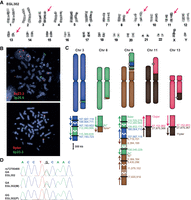
EGL302's chromothripsis translocations. (A) EGL302's karyotype; red arrows indicate translocation chromosomes. (B) FISH confirms the insertion of 8q23.3 (probe RP11-3A12) to the long arm of Chromosome 3 (3p26.6 control probe CTC-228K22) and the translocation of 9pter (probe CTB-41L13) to the long arm of Chromosome 8 (8p23.3 control probe RP11-410N18). (C) Model of the rearrangements in EGL302. The balanced translocation between Chromosomes 11 and 13 was confirmed by Sanger sequencing. Chromothripsis between Chromosomes 3, 8, and 9 results in many exchanges between the three chromosomes. (D) Example of parent-of-origin analysis for EGL302. The underlined guanine (G) at the breakpoint is derived from the paternal (P), not the maternal (M), allele.
EGL321 has a complex rearrangement involving Chromosomes 2, 3, 7, 10, and 11 (Fig. 4). We sequenced 23 breakpoint junctions in five derivative chromosomes using a combination of SureSelect and Complete Genomics WGS. According to FISH analysis, there are at least another six breakpoints (Supplemental Fig. S2). The translocation between Chromosomes 3 and 11 is restricted to those two chromosomes, and a portion of Chromosome 11 is inverted at both of the translocation junctions. Derivative Chromosomes 2 and 7 have swapped multiple segments of these two chromosomes, and the derivative Chromosome 10 has intermingled insertions of Chromosomes 2 and 7. Four breakpoints are completely balanced to the base pair, and the remaining breakpoints have 1- to 11-bp deletions. In addition to the 800-kb deletion of Chromosome 7 and the 2.2-Mb deletion of Chromosome 11, there are 55 total bp deleted at breakpoint junctions. The majority of breakpoint junctions in EGL302, EGL305, and EGL321 had no homology, and a few have short insertions (Table 1). No breakpoint junctions had more than 4 bp of microhomology.
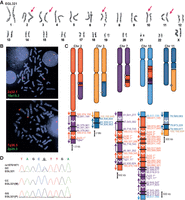
EGL321's chromothripsis translocations. (A) Karyotype of EGL321; red arrows indicate translocation chromosomes. (B) FISH confirms the translocation of the long arm of Chromosome 2 (probe RP11-89P7) to the long arm of Chromosome 10 (10p15.3 control probe CTB-23B11) and the translocation of the long arm of Chromosome 7 (probe RP11-3K23) to the long arm of Chromosome 2 (2p25.3 control probe RP11-71M21). (C) Model of EGL321's rearrangements. Zoomed-in translocation junctions show breakpoints on the derivative chromosomes. (D) Example of parent-of-origin analysis for EGL321. Underlined G is adjacent to a Chromosome 10 breakpoint and is derived from the paternal (P) allele.
To determine the parental origin of de novo translocations in EGL302 and EGL321, we genotyped family trios for heterozygous SNPs adjacent to chromosome breakpoints. We isolated SNPs from derivative chromosomes by sequencing junctions in the probands and then determined the parental origin of the SNP at the breakpoint. Of the seven informative SNPs in EGL302 and six informative SNPs in EGL321, all were derived from paternal alleles (Figs. 3D, 4D; Supplemental Table S3).
Disrupted and fused genes at translocation junctions
Translocations may disrupt genes at breakpoints, leading to loss-of-function, or fuse genes that acquire a new function. Fusion genes are common in chromosome rearrangements in leukemia but are rarely reported in germline rearrangements (Backx et al. 2011; Rippey et al. 2013; Boone et al. 2014; van Heesch et al. 2014; Newman et al. 2015). In the 51 simple translocations with 102 sequenced or fine-mapped breakpoints, 44 (43%) of the breakpoints lie in a gene. Thirteen translocations do not disrupt a gene at either chromosome breakpoint, and 32 translocations disrupt a gene at one but not both breakpoints. In six simple translocations, both breakpoints lie in the open reading frame of genes. Genes juxtaposed by EGL064's and EGL352's translocations are not transcribed in the same direction, and EGL086's fusion gene is predicted to be out-of-frame (Supplemental Table S1). Translocations in EGL002, EGL019, and EGL308, however, are poised to create in-frame fusion transcripts (Fig. 5).
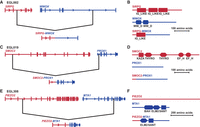
Predicted in-frame fusion genes at sequenced translocation junctions. Black lines indicate translocation breakpoints in genes (not drawn to scale). (A,B) Fusion of SIRPG and WWOX in EGL002. (C,D) EGL019's SMOC2-PROX1 fusion. (E,F) Fusion of PIEZO2 and MTA1 in EGL308.
EGL002's translocation between Chromosomes 16 and 20 joins SIRPG exons 1–2 to WWOX exon 5. The resulting SIRPG-WWOX fusion protein is predicted to retain a SIRPG immunoglobulin domain but lack WWOX WW domains. In EGL019, SMOC2 exon 1 is joined to PROX1 exons 2–5, but the fusion protein is not predicted to retain SMOC2’s functional domains. EGL308's translocation results in a truncated version of MTA1, with exons 8–21 fused to noncoding exons 1–2 of PIEZO1 upstream. Based on exon phase, all three of these fusion genes are predicted to be in-frame. However, RNA was not available, so we could not confirm the presence of fusion transcripts.
Complex translocations also have the potential to create fusion genes. Sequenced breakpoints in EGL305 and EGL312 do not disrupt genes. In EGL356's rearrangement, a deletion in Chromosome 14 interrupts DHRS4L1, and translocations interrupt MTUS2, ALG5, and POSTN on segments of Chromosome 13. (Supplemental Table S2). EGL826's translocation between Chromosomes 10 and 17 joins C1QTNF1 and STK32C genes in the same orientation, but fusion transcripts are predicted to be out-of-frame. Breakpoints in EGL302's rearrangements disrupt two genes, both on the derivative Chromosome 9. Three different breakpoints interrupt PTPRD, and one breakpoint disrupts SH3GL2. EGL321's breakpoints interrupt GRM3, KPNA1, DLG2, CACNA2D1, GULP1, COL5A2, KCNH7, PCLO, and TRRAP. In both EGL302 and EGL321, functional fusion genes are not predicted due to the fragmentation and orientation of the genes.
Discussion
Unbalanced translocation mechanisms
We analyzed translocations from 57 individuals with unique chromosome rearrangements and found that most junctions have little or no sequence homology. For the 37 simple unbalanced translocations we sequenced, 70% have 0–4 bp of microhomology, 22% have insertions or inversions, and only 8% have long stretches of homology shared between translocating segments (Table 1), suggesting that NHEJ and MMBIR are the predominant mechanisms of translocation formation (Hastings et al. 2009; Zhang et al. 2009). Recently, Robberecht et al. sequenced the junctions of nine de novo unbalanced translocations and found that six were mediated by NAHR between LINEs, HERVs, or segmental duplications (Robberecht et al. 2013). They concluded that NAHR between these longer repeats drives de novo unbalanced translocation formation. We determined translocation inheritance in 20 trios and found that eight were de novo, seven were maternally inherited, and five were paternally inherited. Similar to the 30% observed by Robberecht et al. (2013), 40% of our unbalanced translocations were de novo; however, only two out of eight de novo unbalanced translocations in our study were mediated by NAHR. As in Robberecht et al. (2013), these two junctions lie in homologous LINE or HERV repeats. Nonetheless, most de novo translocations in our study lack extensive sequence homology at junctions. Like other structural variation in the human genome (Conrad et al. 2010; Luo et al. 2011; Chiang et al. 2012; Newman et al. 2015), most de novo unbalanced translocations are the product of NHEJ or MMBIR.
It is possible that at least some of the 14 simple translocations that failed junction sequencing have repetitive DNA or cryptic complexity at the breakpoints that prevented SureSelect, NGS, or junction PCR. Even if all 14 translocations were the product of NAHR, junctions without significant sequence homology still outnumber those formed by NAHR. Translocations in EGL045 and EGL315 may be NAHR-mediated, since breakpoints determined by high-resolution array CGH map to homologous repeats (HERV-H and L1PA2/L1PA3, respectively). However, breakpoints of the remaining 12 translocations map to regions that lack homology between both sides of the junction. Furthermore, breakpoints that fine-map to homologous interspersed repeats are not guaranteed to be the product of NAHR. For example, array CGH mapped both breakpoints in EGL103's translocation to AluSx1 repeats, but sequencing revealed that breakpoints were outside of the repeats and the junction lacked significant sequence homology.
Forty-eight percent (86/179) of sequenced breakpoints from simple and complex translocations lie within repeats (Supplemental Tables S1, S2). This is not surprising since approximately half of the human genome is repetitive (Lander et al. 2001), and similar repeat content has been reported at other CNV breakpoints (Vissers et al. 2009; Bose et al. 2014). Translocation junctions of EGL051, EGL080, and EGL083 are located in paralogous segmental duplications, L1s, and HERV-H elements, respectively. Robberecht et al. (2013) found the same classes of repeats at breakpoint junctions of unbalanced translocations. These repeats are more than 1-kb long, are found only in primates, and are >92% identical. While recombination between Alus has been described for numerous interstitial deletions and duplications (Luo et al. 2011; Boone et al. 2014; Newman et al. 2015), Alu-Alu events rarely mediate germline translocations (Rouyer et al. 1987; Chen et al. 2008; Luo et al. 2011; Chiang et al. 2012; Fruhmesser et al. 2013; Robberecht et al. 2013). These data suggest that specific types of repeats may be favored in aberrant homologous recombination that gives rise to translocations.
We identified two breakpoints shared between our translocations and those described in Robberecht et al. (2013). Translocations in EGL083 and Robberecht Case 3 are mediated by NAHR and have a breakpoint on Chromosome 12 in the same HERV-H (hg19; Chr 12: 4,128,160–4,131,129). However, the translocation partners are different chromosomes. Recombination between HERV-H repeats has been implicated in other translocations and deletions (Hermetz et al. 2012; Shuvarikov et al. 2013; Campbell et al. 2014). Robberecht Case 7 has an unbalanced translocation likely mediated by NAHR between L1PA4 elements on Chromosomes 9 and 10. EGL319's translocation has a breakpoint in the same Chromosome 9 L1PA4 (hg19; Chr 9: 15,595,148–15,601,275), although the translocation partner is different and the junction has microhomology rather than features of NAHR. It is possible that this L1PA4 is a breakage hotspot that may be resolved by diverse DNA repair mechanisms.
Complex translocations and chromothripsis
We characterized six chromosome rearrangements with multiple breakpoints. Translocations in EGL312, EGL356, and EGL826 have more than one breakpoint and have inversions at the translocation junctions, but only two chromosomes are involved in the complex rearrangements. EGL302, EGL305, and EGL321 have translocations between at least four different chromosomes and many balanced insertions with altering orientations, all of which had blunt ends or microhomology at the junction. These features are hallmarks of chromothripsis (Kloosterman et al. 2011, 2012; Chiang et al. 2012; Pellestor et al. 2014).
Rearrangements in EGL305 were transmitted from his mother, who carried a more balanced form of the translocations. In addition to EGL305, maternal chromothripsis transmission has recently been observed in three other families (de Pagter et al. 2015). In both EGL302's and EGL321's de novo chromothripsis events, rearrangements occurred on paternal alleles. Though our sample size is too small to determine a parent-of-origin bias, these data are consistent with other studies that find an enrichment of paternally derived chromosome rearrangements (De Gregori et al. 2007; Grossmann et al. 2010; Thomas et al. 2010; Hehir-Kwa et al. 2011; Kloosterman et al. 2011, 2012; Liu et al. 2011).
As more germline chromothripsis genomes are being sequenced, common features have begun to emerge. Though there are many breakpoints in chromothripsis, few are accompanied by large copy number changes. CGH, WGS, and FISH revealed that EGL302 has at least 18 breakpoints but only two large deletions of Chromosomes 8 (2.8 Mb) and 9 (6.6 Mb). EGL321 has at least 33 breakpoints, including two with large deletions of Chromosomes 7 (800 kb) and 11 (2.2 Mb). Other breakpoints have small deletions (up to 70 bp), insertions (1–7 bp), or inversions, but do not have duplications (Supplemental Table S2). Similar breakpoint junction characteristics and “mostly balanced” copy number have been described at other chromothripsis rearrangements (Kloosterman et al. 2011, 2012; Chiang et al. 2012; Macera et al. 2014; Nazaryan et al. 2014; Pellestor et al. 2014; de Pagter et al. 2015). In EGL302 and two other chromothripsis events in the literature, breakpoints disrupt the PTPRD gene on Chromosome 9 (Macera et al. 2014; de Pagter et al. 2015), suggesting that this locus may be a chromothripsis hotspot. Clinical features in individuals with germline chromothripsis may be due to loss of genes within deletions, or due to genes disrupted by copy-neutral rearrangements. Thus, copy number studies alone may not pinpoint the genes responsible for phenotypes.
Translocation annotation and technical limitations
Mapping translocation breakpoints at the nucleotide level required a tiered approach consisting of high-resolution array CGH, targeted sequence capture with NGS, WGS, and confirmation by junction PCR followed by Sanger sequencing. We successfully confirmed the breakpoints of 37/51 simple unbalanced translocations. Fourteen translocation junctions could not be verified by the above methods, and this is due to a combination of technical limitations, lack of genomic DNA, and the nature of the rearrangements.
FISH analysis revealed that copy number gains in EGL354, EGL357, and EGL358 were unbalanced translocations to the short arms of Chromosomes X, 21, and 22, respectively (Supplemental Table S1). However, we did not detect genomic losses of those chromosome arms by array CGH. This is consistent with small deletions of ends of the derivative chromosomes that may lie in segmental duplications or other repetitive DNA not included in microarray analysis (Rudd 2012). Though we targeted the breakpoints corresponding to the terminal gains of these unbalanced translocations, SureSelect plus Illumina HiSeq did not identify reads that cross the translocation breakpoints.
We fine-mapped 14 breakpoint regions from 12 translocations to LINEs and attempted to capture these loci by SureSelect. Discordant reads spanned the junction from LINE to unique sequence in only five of these breakpoints (EGL002, EGL064, EGL306, EGL317, and EGL319), which is consistent with the previously recognized limitation in LINE breakpoint sequencing (Talkowski et al. 2011). Surprisingly, our SureSelect approach was successful in mapping informative reads to three segmental duplications. Discordant reads and Sanger sequencing supported EGL051's junction between two 95% identical segmental duplications. EGL313's junction was supported by discordant reads that anchor the segmental duplication at the breakpoint to unique sequence; however, we were not able to confirm this junction by Sanger sequencing. EGL062's breakpoint failed SureSelect, but we sequenced this junction from segmental duplication to unique sequence by long-range PCR.
Though array CGH, FISH, and chromosome banding do not provide nucleotide resolution of breakpoints, they are essential to interpret CNV breakpoints from NGS data. Following WGS of complex translocations and chromothripsis genomes, we performed iterative rounds of FISH to place insertional translocations on the correct derivative chromosome (Supplemental Figs. S1, S2). Furthermore, initial FISH and/or chromosome banding studies are necessary to distinguish unbalanced translocations from terminal deletions and duplications detected by copy number assays (Rudd 2012). Thus, as NGS and WGS approaches become routine for CNV detection (Xi et al. 2011; Michaelson and Sebat 2012; English et al. 2015), techniques that visualize chromosomes will continue to be important for interpreting structural variation.
WGS identified many copy-neutral rearrangements that were missed by microarray analyses of EGL302 and EGL321. Though the copy number changes were relatively minor in these individuals, chromosome banding revealed multiple translocations, so we were not surprised to find additional breakpoints besides those detected by array CGH. On the other hand, WGS does not always reveal additional complexity at translocation junctions. WGS of EGL382's simple translocation and EGL826's complex translocation only identified the breakpoints we had already predicted by array CGH. Thus, it is unlikely that most translocations have cryptic complexity. Chromothripsis is estimated to occur in 2–4% of cancers (Forment et al. 2012; Pellestor et al. 2014), which is similar to the incidence of chromothripsis in germline chromosome rearrangements (Kloosterman et al. 2011; Chiang et al. 2012; Forment et al. 2012; Macera et al. 2014).
In this large-scale analysis of unbalanced translocations, we report a paucity of sequence homology at breakpoint junctions and predict three novel in-frame fusion genes. Our approach to combine SureSelect, Illumina HiSeq, mate-pair sequencing, and WGS uncovered a wide range of breakpoints in this diverse cohort. This comprehensive analysis revealed that most unbalanced translocations are simple and likely formed by NHEJ and MMBIR repair processes. Rarer translocations between four or five chromosomes proved to have tens of breakpoints, most of which were not recognized by standard cytogenetic methods. Combined with other complex chromosome rearrangement studies (Borg et al. 2005; Kloosterman et al. 2011, 2012; Chiang et al. 2012; Macera et al. 2014; Pellestor et al. 2014), these data suggest that translocations involving more than two chromosomes are likely to be the product of chromothripsis.
Methods
Custom array CGH
This study was approved by the Institutional Review Board (IRB) at Emory University. Subjects had CMA testing with a version of the EmArray oligonucleotide array (Baldwin et al. 2008), followed by confirmation by chromosome banding or FISH. G-banding of chromosomes from peripheral blood has a resolution of 550–700 bands, and FISH was performed as described (Baldwin et al. 2008). For most subjects, DNA extracted from whole blood was used for all microarray and breakpoint sequencing experiments. We used DNA from lymphoblastoid cell lines for EGL302, EGL316, EGL321, EGL382, EGL826, and LM219. To fine-map unbalanced translocation breakpoints, we performed high-resolution array CGH. We designed custom 4×180K oligonucleotide arrays with ∼200-bp probe spacing using eArray from Agilent Technologies (https://earray.chem.agilent.com/earray/). The array design ID (AMADID) identifiers are 018181, 021634, 021635, 021636, 021637, 034386, 037387, 035709, 035730, 037646, 040718, and 063584. Each subject's array AMADID is listed in Supplemental Tables S1 and S2. Arrays were hybridized, scanned, and analyzed as previously described (Luo et al. 2011).
Sequencing unbalanced translocations
We used Agilent SureSelect Target Enrichment to pull down 40-kb regions around breakpoints fine-mapped by custom array CGH. SureSelect followed by Illumina HiSeq sequencing was performed at HudsonAlpha Genomic Services Laboratory, and sequence analysis was performed as described previously (Hermetz et al. 2014). Custom SureSelect library numbers (ELID) are listed in Supplemental Tables S1 and S2. Arrays and SureSelect libraries were designed using the GRCh37/hg19 genome build, and we kept genomic coordinates in this version so that the design IDs correspond to the coordinates in our tables. Providing genomic coordinates in this genome build does not affect our conclusions.
We performed long-range PCR and Sanger sequencing to confirm breakpoints (Supplemental Table S3). We used the Qiagen LongRange PCR kit (Catalog # 206403), following the manufacturer's protocol. Sanger sequencing was performed by Beckman Coulter Genomics, and the reads were aligned to the human genome reference assembly (GRC37/hg19) using the BLAT tool on the UCSC Genome Browser (http://genome.ucsc.edu/). Junction sequences are provided in Supplemental Table S4.
Whole-genome sequencing
WGS libraries for EGL312 and EGL356 were prepared using the Nextera Mate Pair Sample Prep kit (Catalog # FC-132-1001) according to the manufacturer's instructions. We used the Gel-Plus protocol to size-select 5- to 7-kb genomic fragments for sequencing. The two libraries were barcoded and sequenced on one lane of Illumina HiSeq, and the reads were analyzed as previously described (Hermetz et al. 2014).
WGS of genomic DNA from EGL382, EGL826, EGL302, and EGL321 was performed by Complete Genomics as described (Drmanac et al. 2010). Complete Genomics provided the individual reads, quality scores, and initial mappings to the GRCh37 reference genome in .tsv format. To identify discordant read pairs, we converted reads and mappings flagged as structural variant candidates to SAM format with the map2sam command in CGATools 1.7.1 (http://cgatools.sourceforge.net/). We used SAMtools (Li et al. 2009) to sort, index, and convert files to BAM. To account for intra-read gaps, we used a custom Perl script that extracts discordant read pairs that map aberrantly relative to the reference genome. We viewed discordant reads with Integrative Genomics Viewer (Robinson et al. 2011) to identify and interpret structural variation.
Fusion gene prediction
For breakpoints that interrupt genes oriented in the same direction, we predicted the reading frame of fusion genes. We used all gene isoforms included in the Ensembl release 75 gene transcript database (Flicek et al. 2014) to predict whether the reading frame was preserved following the rearrangement. Juxtaposed exons with the same phase were predicted to be in-frame. We predicted fusion protein motifs (Fig. 5) by analyzing cDNA sequence from Ensembl 75 with ScanProsite (http://prosite.expasy.org/scanprosite/).
Data access
Agilent array CGH data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE68019. Breakpoint junction sequences have been submitted to GenBank (http://www.ncbi.nlm.nih.gov/genbank/) under accession numbers KR072894–KR072971. Illumina sequencing data have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRP057518, and Complete Genomics whole genome sequencing data have been submitted to the database of Genotypes and Phenotypes (dbGaP; http://www.ncbi.nlm.nih.gov/gap) under accession number phs000845.v1.p1.
Acknowledgments
We thank Vanessa Jump, Jacob Sloan, and Lan Dang for FISH experiments and Cheryl Strauss for editorial assistance. We also thank families for participating in this project. This study was supported by a grant from the National Institutes of Health (MH092902 to M.K.R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.191247.115.
- Received February 16, 2015.
- Accepted May 15, 2015.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








