
Distinct global shifts in genomic binding profiles of limb malformation-associated HOXD13 mutations
- Daniel M. Ibrahim1,2,3,
- Peter Hansen1,4,
- Christian Rödelsperger1,6,
- Asita C. Stiege2,
- Sandra C. Doelken4,
- Denise Horn4,
- Marten Jäger1,4,
- Catrin Janetzki1,
- Peter Krawitz4,
- Gundula Leschik1,
- Florian Wagner5,
- Till Scheuer1,2,
- Mareen Schmidt-von Kegler1,
- Petra Seemann1,3,
- Bernd Timmermann2,
- Peter N. Robinson1,2,4,
- Stefan Mundlos1,2,3,4 and
- Jochen Hecht1,2,7
- 1Berlin-Brandenburg Center for Regenerative Therapies (BCRT), Charité Universitätsmedizin Berlin, 13353 Berlin, Germany;
- 2Max Planck Institute for Molecular Genetics, 14195 Berlin, Germany;
- 3Berlin-Brandenburg School for Regenerative Therapies (BSRT), Charité Universitätsmedizin Berlin, 13353 Berlin, Germany;
- 4Institute for Medical and Human Genetics, Charité Universitätsmedizin Berlin, 13353 Berlin, Germany;
- 5Atlas Biolabs GmbH, 10117 Berlin, Germany
Abstract
Gene regulation by transcription factors (TFs) determines developmental programs and cell identity. Consequently, mutations in TFs can lead to dramatic phenotypes in humans by disrupting gene regulation. To date, the molecular mechanisms that actually cause these phenotypes have been difficult to address experimentally. ChIP-seq, which couples chromatin immunoprecipitation with high-throughput sequencing, allows TF function to be investigated on a genome-wide scale, enabling new approaches for the investigation of gene regulation. Here, we present the application of ChIP-seq to explore the effect of missense mutations in TFs on their genome-wide binding profile. Using a retroviral expression system in chicken mesenchymal stem cells, we elucidated the mechanism underlying a novel missense mutation in HOXD13 (Q317K) associated with a complex hand and foot malformation phenotype. The mutated glutamine (Q) is conserved in most homeodomains, a notable exception being bicoid-type homeodomains that have lysine (K) at this position. Our results show that the mutation results in a shift in the binding profile of the mutant toward a bicoid/PITX1 motif. Gene expression analysis and functional assays using in vivo overexpression studies confirm that the mutation results in a partial conversion of HOXD13 into a TF with bicoid/PITX1 properties. A similar shift was not observed with another mutation, Q317R, which is associated with brachysyndactyly, suggesting that the bicoid/PITX1-shift observed for Q317K might be related to the severe clinical phenotype. The methodology described can be used to investigate a wide spectrum of TFs and mutations that have not previously been amenable to ChIP-seq experiments.
Transcription factors (TFs) determine cell type-specific gene expression and thereby govern developmental programs at all stages of development. TF binding to cis-regulatory elements regulates the tissue- and developmental stage-appropriate expression of target genes, and correspondingly, mutations in genes encoding TFs can lead to dramatic phenotypes in humans by disrupting developmental gene regulation programs. For instance, mutations in the TFs HOXD13, RUNX2, and PITX2 are associated with brachydactyly, cleidocranial dysplasia, and Axenfeld-Rieger syndrome (Kozlowski and Walter 2000; Otto et al. 2002; Johnson et al. 2003). However, the molecular mechanisms that cause these developmental phenotypes have been difficult to address experimentally, particularly for mutations that do not result in a simple loss of function. In these cases the phenotypes can be mutation specific, as it has been reported for, e.g., MSX2, FOXC1, PITX2, or HOXD13 (Kozlowski and Walter 2000; Wilkie et al. 2000; Johnson et al. 2003; Saleem et al. 2003). How these mutations exert their effect remains unclear, but it is likely that the mutant protein has altered binding specificity or affinity, resulting in abnormal targets or activity (Caronia et al. 2003; Saleem et al. 2003; Zhao et al. 2007).
The development of ChIP-seq technology, which couples chromatin immunoprecipitation (ChIP) with high-throughput sequencing (seq), has enabled researchers to investigate TF binding on a genome-wide scale, opening new approaches to exploring transcriptional control mechanisms (Valouev et al. 2008; Barski and Zhao 2009; Park 2009). To date, however, ChIP-seq has largely been used to investigate the binding of wild-type TFs in cell lines, and also in tissue samples where relatively large amounts of cells are readily available (Robertson et al. 2008; Ouyang et al. 2009; Welboren et al. 2009; Wang et al. 2011; Junion et al. 2012). In order to harness ChIP-seq methodologies to investigate mutant TFs involved in hereditary diseases, a number of technical hurdles must be overcome.
Current ChIP-seq methodologies are not well suited to investigating the effects of mutations in TFs, because antibodies typically do not distinguish between wild-type and mutant protein. Additionally, TFs often belong to gene families that consist of several close paralogs that in many cases are coexpressed. A further hindrance for analysis of TFs in developmental processes is that they are active during specific developmental time points that are difficult to recapitulate in cell culture systems and for which only very small amounts of tissue would be available in mouse models. This makes investigation of such factors essentially impossible with current ChIP-seq protocols that are based on antibodies specific to the protein of interest and that use cell culture or primary tissue samples. These difficulties apply especially to Hox genes, a family of highly conserved TFs. Arguably the most studied TFs in limb development are the 5′ Hoxa and Hoxd (Hoxa/d9-13) genes that pattern the proximo-distal axis and regulate growth and shape of the digits. Despite this, little is known about the targets of HOX proteins in limbs (Svingen and Tonissen 2006; Salsi et al. 2008; Villavicencio-Lorini et al. 2010). Genetic analyses have demonstrated that they are involved in limb patterning in the early phase of limb bud growth and later directly influence the size and shape of the skeletal elements (Muragaki et al. 1996; Zakany et al. 2004; Kuss et al. 2009; Delpretti et al. 2012).
Mutations in HOXD13, the most 5′ gene of the HOXD cluster, include polyalanine expansions and frame-shift or missense mutations, and are associated with severe digit malformations in humans (Debeer et al. 2002; Caronia et al. 2003; Johnson et al. 2003). Intriguingly, polyalanine expansions and frame-shift mutations are predominantly associated with the formation of extra digits and webbing between digits, collectively called synpolydactyly, while missense substitutions have been reported to cause distinctive shortening of the hands/feet (brachydactyly) (for a review, see Goodman 2002). The fact that these two classes of HOXD13 mutations are associated with two distinct phenotypes suggests that the underlying patho-mechanism is also distinct. Polyalanine expansions, which lead to aggregation of mutant HOXD13 in the cytoplasm (Albrecht et al. 2004), and HOXD13 frame-shift mutations are likely to represent loss-of-function mutations. Because of the different phenotype observed with HOXD13 missense mutations, we hypothesized that they represent gain-of-function mutations, whereby mutant proteins acquire altered genomic binding profiles. In this case, the phenotype would result not only from a lack of activation of target genes, but also from the activation of an abnormal set of target genes.
In this work, we apply a ChIP-seq methodology designed to investigate mutations in TFs to characterize a HOXD13 missense mutation identified in a patient with a severe form of brachydactyly/oligodactyly. The mutation, Q317K, alters the glutamine residue at position 50 of the homeodomain to a lysine residue, which is characteristic of bicoid-type homeodomain proteins including PITX1. We show that the Q317K mutation shifts the HOXD13 binding profile toward that of PITX1. The induced gene expression patterns and the phenotypic effects following injection of the constructs in chicken embryos provide further evidence for a global shift in the regulatory properties of the mutant HOXD13 toward that of PITX1. As a control, we investigated a second HOXD13 missense mutation that is associated with a distinct clinical phenotype, and found a strikingly different shift in the binding profile, suggesting that distinct molecular effects of mutations in TFs may underlie some of the clinical variability that can be seen with different mutations in the same TF. The methodology described here can be used to investigate a wide spectrum of TFs and mutations that have not previously been amenable to ChIP-seq experiments.
Results
The HOXD13Q317K mutation causes a complex brachydactyly/oligodactyly phenotype
We identified an individual with a complex brachydactyly/oligodactyly phenotype. She was born with normal measurements, and had normal psychomotor development and no family history of congenital malformations. The girl's hands had only four fingers each as well as severe shortening of the digits and camptodactyly. The terminal phalanges and the nails of the thumbs were absent (Fig. 1A). The left foot had only three, shortened toes, whereby the nail of the left great toe was absent. Partial cutaneous webbing (syndactyly) was seen between the other toes, and the fibular toe was deviated to the tibial side. The right foot had only four, shortened toes with partial syndactyly of two toes, and the nail of the great toe was absent (Fig. 1A).

A HOXD13 p.Q317K mutation causes a severe limb phenotype and alters the DNA binding specificity. (A) Photographs and radiographs of patient with HOXD13Q317K mutation. Missing digits (oligodactyly) and severe shortening of the digits (brachydactyly), as well as absent distal phalanges and nails are apparent. Radiographs show missing and deformed metacarpals, metatarsals, and rudimentary phalangeal bones. (B) The HOXD13Q317 mutations alter the binding motif at the 5′ end. The primary motifs found by de novo motif analysis for HOXD13wt (top), HOXD3Q317R (middle), and HOXD13Q317K (bottom). Both Q317 mutant proteins show an altered 5′ end of the sequence as compared with HOXD13wt. (C) In vitro binding of wild-type and mutant HOXD13 homeodomains to the HOXD13 motif. EMSA of purified wild-type and mutant HOXD13 homeodomains was performed using fluorescent dye-labeled double-stranded oligonucleotides containing a binding sequence for HOXD13 (CCAATAAAA). Oligonucleotides were incubated with purified wild-type and mutant HOXD13 homeodomains. Unlabeled competitor oligonucleotides reduced binding to the labeled oligonucleotide, whereas a mutant competitor did not, indicating sequence-specific binding. The HOXD13wt motif is bound by HOXD13wt and (weaker) HOXD13Q317R but not by HOXD13Q317K.
The clinical and radiological manifestations of this girl did not follow any known malformation patterns. Because of the absence of terminal phalanges, ROR2 mutations were initially sought and excluded. Mutation analysis of HOXD13 revealed a heterozygous mutation that was not found in either parent and had thus occurred de novo. The mutation is not present in the dbSNP database and has not been reported elsewhere. The severe brachydatyly/oligodactly phenotype described here has not been reported as part of the polyalanine expansion mutation spectrum, nor has it been described for other HOXD13 mutations. This c.949C>A substitution, in exon 2 of the HOXD13 gene (NM_000523.2), converts the glutamine residue at position 317 to a lysine residue (p.Q317K). At this position, a different point mutation (Q317R) has been reported previously in a family with a very different phenotype consisting of a form of brachysyndactyly (type V) (Zhao et al. 2007). The glutamine residue affected by the Q317 mutations corresponds to position 50 (Q50) of the homeodomain, which has been shown to be crucial for the binding site specificity of homeodomain proteins (Percival-Smith et al. 1990; Fraenkel et al. 1998; Chaney et al. 2005).
The Q317 mutations in Hoxd13 lead to distinct DNA binding properties of wild-type and mutant HOXD13 proteins
To overcome the hurdles for ChIP-seq analysis of TF-variants, we developed a cell culture-based system using mesenchymal limb bud cells and retroviral overexpression and applied it to compare Hoxd13wt to the Hoxd13Q317K and Hoxd13Q317R mutants (Supplemental Fig. S1). We used the chicken limb bud micromass system (chMM) for these experiments. Cells for chMM are taken from embryonic limb bud mesenchyme where HOXD13 is expressed physiologically. In culture, the cells undergo a chondrogenic differentiation process that can be inhibited by virally expressed Hoxd13 (Kuss et al. 2009).
To identify binding sites, we applied a targeting system in which a protein of interest is fused to a 3xFLAG-tag epitope in an RCASBP virus. This avian-specific retrovirus (RCAS) achieves high transfection levels while inserting only one copy of the virus per cell. An important motivation for the choice of the RCASBP system is the relatively mild degree of overexpression which is achieved. Using absolute quantification of transcripts, we found virally expressed Hoxd13 transcript levels from infected chMM to be approximately three to five times higher than endogenous HOXD13 expression in embryonic limb buds (Supplemental Methods; Supplemental Fig. S1). Cross-linked chromatin from chMM cultures infected with tagged murine Hoxd13wt, Hoxd13Q317K, or Hoxd13Q317R were used for ChIP-seq. For each experiment, we performed two replicates that were subjected to quality control standards as defined by The ENCODE Project Consortium (The ENCODE Project Consortium 2011; Landt et al. 2012).
We identified 34,267 peaks for HOXD13wt. To test the specificity of the identified binding sites, we conducted motif analysis using DREME (Bailey 2011). The primary motif inferred for HOXD13wt showed a characteristic HOX binding motif with an AT[A/T]AAA core sequence preceded 5′ by a somewhat more loosely defined [C/T][A/C], which is largely identical to the HOXD13 motif previously identified by protein-binding microarray analysis (Fig. 1B; Berger et al. 2008).
We then applied our chMM ChIP-seq system to compare HOXD13Q317K and HOXD13Q317R mutant proteins with HOXD13wt. After quality control and reproducibility testing, we identified 25,036 binding sites for HOXD13Q317K and 21,396 for HOXD13Q317R. Using these peaks, we first performed motif analysis for the mutant proteins. Strikingly, both the HOXD13Q317K and the HOXD13Q317R motif revealed a changed di-nucleotide at the 5′ terminus. The primary motif inferred for HOXD13Q317K has a guanine residue at the 5′ end that is followed by a variable base. The HOXD13Q317R motif also starts with a guanine, which is then followed by a thymine residue (Fig. 1B). Intriguingly, the AT-rich core remained largely unchanged in both motifs. Therefore, we infer that both Q317 mutations alter only the 5′ region of the DNA recognition sequence of HOXD13, while the 3′ part remains unchanged.
The HOXD13Q317K mutant, but not HOXD13Q317R, recognizes the PITX1-motif
We compared the HOXD13 homeodomain sequence to all other homeodomains carrying a lysine (K), like the mutant, at position 50. Sequence alignment between these homeodomains and the mutant HOXD13 homeodomain revealed the greatest similarity to the PITX-family (see Supplemental Fig. S2). Furthermore, the only homeodomain protein carrying a K at position 50 known to be involved in limb patterning is PITX1, one of the few TFs which is expressed in the hindlimb only and has been shown to confer hindlimb identity (Logan and Tabin 1999; Szeto et al. 1999). PITX1, like other K50-homeodomain TFs, binds a GGATTA motif (Tremblay et al. 1998). We hypothesized that the pathogenesis of HOXD13Q317K might be at least partially due to aberrant transactivation of genes that are normally regulated by PITX1. We therefore performed a series of experiments to test whether HOXD13Q317K, due to its altered sequence specificity, shares characteristics with PITX1.
As the motif analysis for HOXD13Q317K revealed a very similar but not identical motif to PITX1, we used electrophoretic mobility shift assays (EMSA) to test the binding of wild-type and mutant homeodomains with respect to characteristic HOXD13 and PITX1 target sequences. Wild-type HOXD13 specifically bound the wild-type HOXD13 motif, but HOXD13Q317K did not show appreciable binding activity. In contrast, the HOXD13Q317R mutant showed considerable residual binding to the wild-type HOXD13 motif (Fig. 1C). This binding affinity was also reflected in the de novo motif analysis, which identified a HOXD13wt-like motif as a secondary motif for HOXD13Q317R but not for HOXD13Q317K (data not shown). When we tested wild-type and mutant homeodomains for binding to the PITX1 target sequence, the Q317K mutant homeodomain efficiently bound the PITX1 motif, whereas the HOXD13wt and HOXD13Q317R homeodomains did not (Fig. 2B).
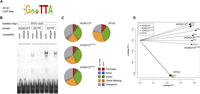
The HOXD13Q317K mutant binds the PITX1 binding site in vitro, and the ChIP-seq profile shifts toward PITX1. (A) The primary PITX1 motif identified from ChIP-seq. (B) In vitro binding of wild-type and mutant HOXD13 homeodomains to the PITX1 motif. EMSAs were performed as in Fig. 1C using oligonucleotides containing a binding sequence for PITX1 (GGATTA). HOXD13Q317K specifically binds the PITX1 motif, whereas it is not bound by HOXD13wt or HOXD13Q317R. (C) Genomic distribution of ChIP-seq peaks. The genome was divided into promoter (5 kb upstream of, 2 kb downstream from TSS), exon, intron, gene flanking (20 kb upstream, 20 kb downstream), and intergenic regions, each separated in conserved and nonconserved regions. (D) Principle component analysis for the coverage profiles of the uniquely mapped reads for all eight data sets (two biological replicates each for HOXD13wt, HOXD13Q317K, HOXD13Q317R, and PITX1).
The genome-wide binding pattern of the HOXD13Q317K mutant shifts toward a PITX1-like binding profile
We next wanted to see whether HOXD13Q317K or HOXD13Q317R were also able to recognize PITX1 binding sites in the genome. To identify PITX1 binding sites in the chicken genome, we used our established protocol to perform ChIP-seq from chMM-cultures infected with PITX1-expressing RCASBP-virus. We identified 40,881 peaks for PITX1 in chMM cultures. De novo motif analysis of ChIP-seq peaks readily identified the known PITX1 motif (Fig. 2A), indicating functionality of the protein.
The overall peak distribution of HOXD13wt, HOXD13Q317K, HOXD13Q317R, and PITX1 with respect to annotated genes was similar for all factors, but HOXD13wt peaks were more commonly found in conserved regions than PITX1 peaks (Fig. 2C). To compare the genome-wide distribution of protein binding we performed principle component analysis (PCA) on the read-count distribution in 500-bp windows for the eight data sets, representing two experiments each for HOXD13wt, HOXD13Q317K, HOXD13Q317R, and PITX1. The biological replicates for each condition clustered together and the coordinates for HOXD13Q317K were located at an intermediate position between those of HOXD13wt and PITX1 (Fig. 2D). In contrast, the coordinates for HOXD13Q317R remained closer to those of HOXD13wt.
As mentioned above, we identified 34,267 HOXD13wt-bound regions, 25,036 for HOXD13Q317K, 21,396 for HOXD13Q317R, and 40,881 for PITX1. Based on our definition of shared binding events, most peaks are TF specific, and only 45 sites are bound by all four factors. The overlap with HOXD13wt or PITX1 peaks was clearly different for HOXD13Q317K and HOXD13Q317R. HOXD13Q317R shared 27.5% (5887) of its binding sites with HOXD13wt, whereas this figure was only 6.0% (1492) for HOXD13Q317K (Fig. 3C). In contrast, HOXD13Q317K shared 6.6% (1671) of its binding sites with PITX1, but HOXD13Q317R shared only 2.9% (633). HOXD13wt shared only 1.1% (383) of its binding sites with PITX1 (Fig. 3C)
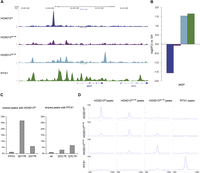
HOXD13Q317K binding, but not HOXD13Q317R, acquires PITX1-like properties. (A) Coverage profiles for HOXD13wt, HOXD13Q317R, HOXD13Q317K, and PITX1 in a 20-kb region covering the MGP gene show the similarity in binding between HOXD13Q317K and PITX1. (B) MGP is threefold up-regulated in PITX1- and Hoxd13Q317K-infected chMM cultures and threefold down-regulated in Hoxd13wt cultures (log2 fold changes compared to mock-infected cultures). (C) Peak overlap with respect to HOXD13wt and PITX1 peaks. (Left) PITX1 shares 1.0% (383/40,881) of its peaks with HOXD13wt; 27.5% (5887/21,396) with HOXD13Q317R; and 6.0% (1492/25,036) with HOXD13Q317K. (Right) HOXD13wt shares 1.1% (383/34,267) of its binding sites with PITX1; 3.0% (633/21,396) with HOXD13Q317R; and 6.7% (1671/25,036) with HOXD13Q317K. (D) HOXD13Q317K shares binding properties with both HOXD13wt and PITX1. Mean read densities centered (±3 kb) around the peak summits for HOXD13wt, HOXD13Q317R, HOXD13Q317K, and PITX1 (left to right). HOXD13wt (164.1 tags/50 bp; 93.5 tags/50 bp; 43.2 tags/50 bp; 23.6 tags/50 bp), HOXD13Q317R (49.7 tags/50 bp; 101.2 tags/50 bp; 49.8 tags/50 bp; 19.5 tags/50 bp), HOXD13Q317K (24.9 tags/50 bp; 51.9 tags/50 bp; 109.1 tags/50 bp; 25.6 tags/50 bp), and PITX1 (15.1 tags/50 bp; 24.2 tags/50 bp; 38.9 tags/50 bp; 191.2 tags/50 bp).
This shows that the fraction of PITX1 sites bound by HOXD13Q317K is considerably larger than the overlap observed for either HOXD13Q317R or HOXD13wt. Additionally, HOXD13Q317K and HOXD13Q317R share a large set of target sites (3094) that are not bound by HOXD13wt or PITX1.
Since enrichments at genomic regions are not always detected as peaks, we analyzed the distribution of reads from HOXD13wt, HOXD13Q317K, HOXD13Q317R, and PITX1 ChIP-seq around the identified binding sites using seqMINER (Fig. 3D; Ye et al. 2011). There was no enrichment of reads at the position of PITX1 peaks in HOXD13wt or HOXD13Q317R ChIP-seq experiments, and the same was true vice versa. In contrast, the HOXD13Q317K ChIP-seq displayed a moderate enrichment of reads at the site of PITX1 peaks, indicating HOXD13Q317K-binding at PITX1 peaks. We found a similar moderate enrichment of HOXD13Q317K around HOXD13wt peaks. Consistent with the peak overlap, HOXD13Q317K reads were enriched around HOXD13Q317R peaks and vice versa. Again, the genome-wide binding of the HOXD13Q317K mutant shifted toward PITX1 sites to an extent that was not found for HOXD13Q317R or HOXD13wt.
As an example for the changed binding behavior, Figure 3A shows the binding profiles of the four TFs at the genomic region of the matrix-Gla protein (MGP) gene. MGP encodes the matrix-Gla protein, which inhibits calcification in proliferating chondrocytes and blood vessels (Luo et al. 1997; Yao et al. 2011). HOXD13wt, HOXD13Q317K, and PITX1 bind to various sequences in the region upstream of MGP, while HOXD13Q317R shows only weak binding throughout this region. HOXD13wt binds a unique site. HOXD13Q317K and PITX1 share three binding sites with additional unique binding sites for each factor.
RNA-seq shows similar expression profiles induced by HOXD13Q317K and PITX1
To investigate whether similarities in genomic binding profiles are reflected in gene expression patterns, we performed RNA-seq expression analysis for chMM cultures infected with Hoxd13wt, Hoxd13Q317K, Hoxd13Q317R, or PITX1. In order to identify differentially expressed genes, expression levels were filtered according to a minimal expression of 2 RPKM in at least one experiment and at least twofold or higher differential expression in any of the cultures as compared to mock-infected cultures.
This left a total of 3118 transcripts belonging to 2506 genes that were differentially expressed in at least one of the chMM cultures (Supplemental Table 2). PCA analysis of the expression values for all differentially expressed transcripts demonstrated a similar profile as seen for ChIP-seq, with Hoxd13Q317K being close to PITX1 and Hoxd13Q317R close to Hoxd13wt (Fig. 4A). Similarly, hierarchical clustering of transcript levels clustered Hoxd13Q317K with PITX1-infected cultures, and Hoxd13Q317R with Hoxd13wt (Fig. 4B). For each factor, a comparable number of genes (between 845 and 1102) were differentially expressed. When we compared the genes regulated by mutant or wild-type HOXD13 with PITX1-regulated genes, we identified 436 genes coregulated by HOXD13Q317K and PITX1, whereas only 262 genes were coregulated by HOXD13wt and PITX1 and 237 genes by HOXD13Q317R and PITX1 (Supplemental Table 2).
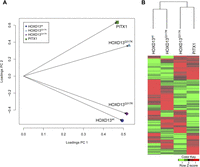
Gene regulation by HOXD13Q317K is more similar to PITX1 than to HOXD13wt or HOXD13Q317R. Global expression analysis of genes regulated by HOXD13wt, HOXD13Q317R, HOXD13Q317K, or PITX1 in chMM cultures. RNA-seq expression analysis found 3118 transcripts (2506 genes) to be at least twofold differentially regulated in Hoxd13wt, Hoxd13Q317R, Hoxd13Q317K, or PITX1 chMM compared to mock-infected cultures (RPKM cutoff 2). (A) Principle component analysis (PCA) of the expression values (normalized to expression values in mock-infected cultures) of these genes shows a similar clustering as seen in the ChIP-seq PCA. (B) Hierarchical clustering of the expression values (RPKM) clusters Hoxd13Q317K with PITX1 and Hoxd13Q317R with Hoxd13wt. Red indicates up-regulation and green indicates down-regulation, as compared to mock-infected cultures.
To identify the genes that might contribute to the similarity of HOXD13Q317K to PITX1 found in PCA and hierarchical clustering analysis, we focused on the 436 HOXD13Q317K/PITX1 coregulated genes. To characterize this group of genes, we performed GO analysis on the 305 co-down-regulated and 131 co-up-regulated genes. For the down-regulated genes, GO terms related to cartilage differentiation and limb development were overrepresented, which were also overrepresented in the sets of down-regulated genes for each individual factor (Supplemental Fig. S3). Among the HOXD13Q317K/PITX1 co-up-regulated genes, genes annotated to several GO categories related to muscle development as well as blood vessel development were overrepresented. These and related GO terms were specific for HOXD13Q317K and PITX1 up-regulated genes and were not similarly enriched in the HOXD13wt or HOXD13Q317R up- or down-regulated genes (Supplemental Fig. S3).
We then wanted to see whether the genes coregulated by HOXD13Q317K and PITX1 were also cobound by the factors. Therefore, we classified a gene as cobound when a HOXD13Q317K/PITX1 shared peak was found in the gene body or within 25 kb up- or downstream from the gene. Examples for cobinding of HOXD13Q317K and PITX1 at coregulated genes are shown in Supplemental Fig. S4. Using this classification, 13.1% (57) of the 436 coregulated genes were also cobound by HOXD13Q317K/PITX1, significantly more than expected by chance (Fisher's exact P = 1.295 × 10−3). An analogous analysis performed for the HOXD13Q317R mutant and PITX1 found 5.06% (12 out of 237) coregulated genes cobound (P = 0.1335) (Supplemental Fig. S5).
Overexpression of Hoxd13Q317K and PITX1 induces similar phenotypes
Chicken micromass cultures are derived from limb bud mesenchymal cells. Cultured at high density, they differentiate into various cell lineages, including chondrocytes which form cartilage nodules that undergo differentiation, mineralization, and finally bone formation (Ahrens et al. 1979). Viral expression of wild-type Hoxd13 strongly inhibits chondrogenic differentiation. Instead, cells in every part of the culture except the very center do not assemble into organized structures (Fig. 5). Overexpression of Hoxd13Q317R does not inhibit chondrocyte formation like the wild type does. The cultures, although a bit smaller in size than mock-infected chMM, appear to undergo the same chondrogenic differentiation process (Fig. 5). chMM infection with Hoxd13Q317K results in a completely different phenotype. Inhibition of chondrogenic differentiation is even stronger, but in contrast to Hoxd13wt, the cells form a multilayered fibrotic tissue which radially extends from the center of the culture (Fig. 5). This radially extending fibrotic tissue was also observed in PITX1-expressing chMM cultures, although inhibition of chondrogenesis was not as strong as in cultures infected with Hoxd13Q317K.

Some effects on cell differentiation and development are shared between Hoxd13Q317K and PITX1. (Upper row) Eosin-stained chMM cultures for Hoxd13wt, Hoxd13Q317R, Hoxd13Q317K, and PITX1. Both Hoxd13Q317K and PITX1 induced differentiation into thick, fibrotic tissue. (Lower row) Viral overexpression of Hoxd13wt, Hoxd13Q317R, Hoxd13Q317K, and PITX1 in chicken wing buds (HH36). Hoxd13wt and Hoxd13Q317R overexpression leads to an overall shortening of skeletal elements. Hoxd13Q317K-infected wing buds lack or have a reduced forelimb-specific patagium, have a straightened wrist, and develop a fourth digit (arrowheads). PITX1 overexpression leads to a partial hind- to forelimb transformation.
So far, our data showed that binding specificity, gene expression, and morphology of Hoxd13Q317K-infected chMM cultures shift from Hoxd13 to PITX1 characteristics. To test whether this shift in specificity can also be observed in vivo, we expressed Hoxd13wt, Hoxd13Q317R, Hoxd13Q317K, and PITX1 using the same FLAG-tagged constructs as used for ChIP-seq experiments in chicken limb buds. The effects of Hoxd13wt and PITX1 in this experiment have been described and are clearly distinguishable. As previously shown, expression of PITX1 in the developing chick wing bud leads to a partial fore-to-hindlimb transformation of the infected wing (Logan and Tabin 1999; Szeto et al. 1999). This manifests itself in three characteristic features. The infected wings show an absence of the patagium (the skin that will form the surface of the wing), demonstrate straightening of the posterior wrist flexure, and develop an additional digit (Fig. 5). In contrast, ectopic expression of Hoxd13wt leads to a reduction in bone size but has no effect on limb patterning (Fig. 5; Goff and Tabin 1997).
Like Hoxd13wt, viral infection of wing buds with Hoxd13Q317R mainly leads to an overall reduction of the limb structures (Fig. 5). We note that in a minority of cases, we observed reduced interdigital cell death or mispositioning of the wrist (Supplemental Fig. S6).
In Hoxd13Q317K-infected wing buds, we observed the lack or reduction of the patagium as well as a straightening of the posterior flexure of the wrist. Also, a fourth digit developed. However, the position of this digit was different from that observed in PITX1-infected wings. This extra digit extended from the first phalanx of digit 1, while with PITX1 overexpression, the additional digit was located completely apart from digit 1 (Fig. 5, arrowheads; Supplemental Fig. S6).
Taken together, the effects of Hoxd13Q317K expression in chMM cultures and limb bud injections are distinct from those induced by Hoxd13wt or Hoxd13Q317R and display a partial overlap with those of wild-type PITX1.
Discussion
TFs are frequently involved in the pathogenesis of disease, in particular, congenital malformation syndromes. In many instances, this is due to haploinsufficiency, i.e., the functional loss of one allele caused by deletions, nonsense mutations, frame-shifts, or other inactivating variants. Point mutations that result in amino acid alterations are, however, more difficult to interpret. Provided that a missense mutation does not simply destabilize the protein, it may induce aberrant function by mechanisms such as altering protein-protein or protein-DNA binding. Mutations in HOXD13 are associated with a broad spectrum of limb phenotypes including webbing of fingers (syndactyly), additional fingers (polydactyly), and shortening of fingers or of the entire hand/foot (brachydactyly). Polyalanine expansions in HOXD13 are associated with synpolydactyly, consisting of syndactyly between fingers III and IV and an additional finger located in between digits III and IV (Muragaki et al. 1996). Amino acid substitutions in the homeodomain have been described in cases with brachydactyly mainly affecting the distal thumb and/or the metacarpals (Johnson et al. 2003). Interestingly, a mutation at the identical position as the one described here but to a different amino acid, p.Q317R, causes yet another phenotype—a combination of synpolydactyly and brachydactyly (Zhao et al. 2007). The complex limb malformation that is associated with the Q317K mutation is different from the previously described phenotypes and thus extends the phenotypic spectrum associated with HOXD13 mutations even further. The presence of several distinctive phenotypes suggests mutation-specific disease mechanisms for each type of mutation. In the case of HOXD13 polyalanine expansions, the pathomechanism appears to be a combination of loss of function and gain of function caused by protein aggregation and the interaction with other alanine repeat containing TFs (Villavicencio-Lorini et al. 2010). For missense mutations, considering the drastic differences in phenotypic expression, a common pathomechanism for all mutations is a rather unlikely scenario, and specific effects, such as alterations in binding capacity, are more likely to explain this phenomenon.
The identical Q to K substitution, as in the patient described here, has been introduced in the Drosophila fushi-tarazu (ftz) homeodomain. This changed the recognition motif in vitro from CCATTA to GGATTA, the motif of the homeodomain-protein bicoid that endogenously carries a K (lysine) at position 50 of the homeodomain (Percival-Smith et al. 1990; Schier and Gehring 1992). The effects of the homeodomain Q50K substitution have been investigated in Drosophila, and its altered regulatory capacity has been studied using reporter constructs (Schier and Gehring 1992; Zhao et al. 2000). Flies transgenic for an ftzQ50K allele develop normally and do not show any malformations, although reporter constructs respond to the altered binding of ftzQ50K (Schier and Gehring 1992). The ftzQ50K protein binds with high affinity to constructs carrying consensus bcd binding sites and activates them; however, it fails to activate natural enhancers harboring nonconsensus binding sites, demonstrating the importance of other homeodomain sequences and sequences outside the homeodomain that are needed to confer the full regulatory capacity of the TF (Zhao et al. 2000).
In the mouse genome, there are 25 K50 (bicoid-type) homeodomain TFs (20 in humans), among them the genes of the Otx, Obox, Rhox, Six, and Pitx families. The only bicoid-type TF in vertebrates that is known to play a major role in limb development is PITX1. Studies in mouse and chicken have shown that Pitx1, expressed in the developing hindlimb but not in the forelimb, is required for the formation of hindlimb characteristics and produces hindlimb-like morphologies when misexpressed in forelimbs (Logan and Tabin 1999; Szeto et al. 1999).
Our results show that, in comparison with HOXD13wt, HOXD13Q317K leads to a clearly different regulation of target genes, many of which are also recognized and regulated by PITX1. This is likely due to the fact that HOXD13Q317K is capable of binding to and regulating a substantially greater number of PITX1 targets than is observed for ftzQ50K and bcd. This is supported by the change in sequence specificity induced by HOXD13Q317R, which is similar but distinct, and the fact that only HOXD13Q317K can recognize the PITX1 binding site in vitro, indicating that the recognition of PITX1 sites is specific to HOXD13Q317K. Furthermore, the number of coregulated genes for PITX1 and HOXD13Q317K is much higher than for the other factors, and shared binding sites for PITX1 and HOXD13Q317K are enriched in the vicinity of the coregulated genes, also indicating shared properties of HOXD13Q317K and PITX1. However, it remains difficult to assign individual ChIP-seq peaks to target genes (for review, see MacQuarrie et al. 2011; Spitz and Furlong 2012). As in our results, developmental TFs have been found to bind throughout the genome, and many peaks are found in intergenic regions that cannot be easily attributed to the regulation of a specific target (Cao et al. 2010). Therefore, the absence of a nearby peak does not exclude the regulation of a certain gene. Neither does the presence of ChIP-seq peaks in promoters of differentially expressed genes prove a regulatory function of an individual peak. Thus, while the enrichment of shared peaks near coregulated genes links cobinding to coregulation, the list of candidate genes will neither be complete nor will all the genes be direct targets.
The adoption of a novel set of regulatory targets is only possible if the new binding sites in the genome are meaningful in the regulatory context of the cell. The cells in the embryonic limb bud are responsive to Pitx1 expression, as indicated by Pitx1 overexpression and knockout experiments in transgenic models. In the patient, the altered binding of HOXD13Q317K is therefore likely to disturb the regulatory balance during limb bud development by regulating PITX1 targets in the HOXD13 expression domain. PITX1 is expressed in the entire hindlimb at early stages and becomes more restricted to the middle part (zeugopod) in later stages, whereas HOXD13 is expressed in fore- and hindlimb, first in a small domain in the posterior limb bud and then in the entire distal limb (autopod). Thus, a PITX1-like activity in the HOXD13 expression domain can be expected to result in a major degree of misexpression. The hypothesis that normal HOXD13 function is partially replaced by PITX1 activity is supported by the effects of overexpressing Hoxd13Q317K and PITX1 in chMM cultures and in infected chicken wing buds, which leads to overlapping developmental consequences in both cases, suggesting a similar set of downstream effects. Our in vitro results demonstrate the mutant is able to activate PITX1 target genes. Furthermore, the in vivo experiments show that this effect is biologically relevant, as the mutant is able to induce PITX1-like morphological changes. The specificity of these effects is further corroborated by our investigations of the HOXD13Q317R mutant.
The phenotype in the HOXD13Q317K patient is different and more severe than that described for the HOXD13Q317R patients. The less severe phenotype of HOXD13Q317R is possibly due to two reasons. On the one hand, HOXD13Q317R still demonstrates considerable residual binding to the wild-type HOXD13 motif and may thus have remaining wild-type activity. On the other hand, the new binding sites do not seem to correspond to known TF target sites and may therefore have only minimal effects. Thus, our results show that a missense mutation in a TF can act as a gain-of-function mutation that shifts the binding profile of the relevant TF on a genome-wide scale. The individual consequences of such a shift, though, are dependent on the presence of compatible cis-regulatory elements/targets as well as the properties of protein domains involved in transactivation and protein-protein-interaction. This applies not only to pathogenic mutations but is also likely to be relevant in respect to TF evolution.
ChIP-seq has been used to characterize the genomic binding profiles of numerous TFs in many different experimental settings. Nevertheless, it has not yet been systematically used to investigate the effects of mutations in TFs associated with hereditary diseases. The main roadblocks consist of the difficulty in distinguishing between wild-type and mutant TFs with a primary antibody and in obtaining sufficient amounts of tissue of a relevant anatomical site and developmental stage for the analysis. We have concentrated our efforts on the chMM system in this work, but similar methodologies are likely to be useful for other organ systems that are amenable to infection with the RCASBP virus, including models for muscle, neuronal, and neural crest cell development (McCobb et al. 1990; Dorman and Johnson 1999; Etchevers 2011). While we chose the murine Hoxd13 for our viral constructs, the very high conservation between mouse and human HOXD13 (94% protein identity, 100% identity in the homeodomain) and strong conservation of TF binding specificities (Hecht et al. 2008; Schmidt et al. 2010) makes the findings directly translatable between mouse and human. The advantage of the chMM system over other cell culture systems is that the cultures undergo a physiological differentiation process, and thus are well suited to investigating the factors that influence this process. The RCASBP system leads to overexpression of the transduced TF which might lead to false-positive binding sites. However, for the comparison of mutated TFs vs. wild type, this is not likely to be relevant. Additionally, the overexpression levels in the case of Hoxd13 and PITX1 in chMM cultures are still in the range of endogenous expression levels in the developing limb.
The chMM-ChIP-seq system enables a relatively quick functional characterization by comparing wild-type and mutant proteins in the genomic context and reveals not only where the mutant TF fails to bind but also if the protein changes its binding specificities. To date, the analytical tools to characterize missense mutations have been limited. Most frequently, nuclear localization of the protein is tested in transient transfection assays supplemented with gel shift assays and/or luciferase assays on reporter constructs. Failure to bind the consensus motif or to activate the reporter result in the characterization of the mutation as a loss of function (Kozlowski and Walter 2000; Saleem et al. 2003; Zhao et al. 2007). According to these criteria, the HOXD13Q317K mutation described here would qualify as a loss-of-function mutation. However, our data show that this is not the case. Our results suggest that missense mutations in TFs may have other effects such as the acquisition of novel functions via expansion and/or alterations of binding motifs.
Methods
Clinical analysis and Sanger sequencing
One family with brachydactyly was investigated. Mutation analysis in the ROR2 and HOXD13 genes was performed using standard Sanger sequencing to test both parents and the affected child. HOXD13 mutation nomenclature is according to RefSeq Accession NM_000523.2. Primer sequences are given in the Supplemental Methods. All patients provided signed, informed consent to this study. The study was approved by the local ethics committee.
Chicken micromass cultures
Chicken micromass cultures were prepared as described elsewhere (Solursh et al. 1978). In brief, cells were obtained from 4.5-d chicken embryos (HH24) and mixed with virus before seeding. chMM cultures were harvested after nine days incubation in chicken micromass medium consisting of DMEM/Ham's F12 (Biochrom) with 10% FBS (PAA), 0.2% chicken serum (Sigma), 1% L-glutamine (Lonza), and penicillin/streptomycin (Lonza). For harvesting, chMM cultures were digested with 0.1% collagenase (Sigma) in chMM-medium to obtain a roughly single-cell suspension and fixed for 10 min on ice with 1% formaldehyde in chMM-medium. Before chromatin extraction and ChIP, fixed cells were frozen in liquid N2 and stored at −80°C. For histological staining, cultures were fixed in the cell culture dish with 4% PFA in PBS overnight and stained with eosin solution (Sigma).
3xFLAG-tag vectors as a universal ChIP-seq tool
In order to generate fusion proteins for ChIP-seq that would be recognized by a single antibody under standardized conditions, we modified the pSlax-13 vector (Morgan and Fekete 1996) to allow fusion of an insert to an N-terminal triple FLAG-tag and directional cloning via ClaI and SpeI. We cloned the coding sequences for wild-type (wt) and mutant (Q317K, Q317R) murine Hoxd13 (NM_008275.2) as well as wild-type chicken PITX1 (NM_001167684) into a modified RCASBP(A)-vector using the ClaI and SpeI sites.
Retroviral infection of chicken limb buds
Infection of the chicken limb buds with the viruses described above was performed as described previously (Logan and Tabin 1998). Chicken embryos (VALO BioMedia) were infected by injecting virus into the presumptive wing field at HH 9-11 and incubated for the appropriate time. Embryos were fixed in 4% PFA overnight and analyzed using a dissecting microscope.
Electromobility shift assays
Cloning and expression of wild-type and mutant homeodomains in Escherichia coli were performed as described in the Supplemental Methods. EMSA was performed using 20 μl binding buffer (100 mM NaCl, 2 mM MgCl2, 0.1 mg/mL BSA, 4 mM spermidine, 25 mM HEPES, pH 7.5, 1× Roche complete protease inhibitor), and 1.5 μL poly(dIdC) (100 ng/μL) as well as 1 μL purified homeodomain (400 ng/μL) were added. Reactions were incubated on ice for 5 min, and then 2 μL of the Cy3-labeled oligonucleotide (1.5 pmol/μL) and—if appropriate—1 μL (15 pmol/μL) of the unlabeled competitor were added and incubated for 15 min at room temperature. Oligonucleotide sequences are given in the Supplemental Methods. Before loading of the sample onto the gel, 2 μL loading buffer (40% glycerol + 0.01% bromphenol blue) were added. Electrophoresis was performed on an 8% native polyacrylamide gel in 0.5× TBE and scanned on a FLA-5000 Scanner (Fuji).
Chromatin immunoprecipitation
ChIP was performed as described in Lee et al. (2006) with the following alterations. Cells were suspended in 1.5 mL L3 buffer, sonicated with a Diagenode Bioruptor for 45 cycles (30-sec pulse, 30-sec pause, HI power), and stored at −80°C before use. Chromatin concentration was determined by measuring the DNA content in a 150-μL sample of the sonicated chromatin. For each ChIP, 40 μL of magnetic Protein G beads (Invitrogen, #100.04D) were preincubated with 10 μg anti-FLAG M2 antibody (Sigma, F1804), and incubated with ∼35 μg chromatin overnight.
ChIP-seq
For ChIP-seq library preparation, ethanol-precipitated DNA from ChIP was redissolved in 46 μL of water and subjected to library preparation using the NEBNext ChIP-seq Library Prep Master Mix for Illumina (New England Biolabs), selecting for a fragment size range of 300–450 bp according to the manufacturer's instructions. Sequencing was performed on an Illumina GAIIx sequencer with 36-bp single reads using TruSeq v5 sequencing chemistry and TruSeq v5 cluster generation kits.
RNA-seq
Total RNA from chMM cultures was isolated using peqGOLD TriFast Reagent (PEQLAB) and subsequent purification with RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. mRNA for RNA-seq libraries was purified using the Oligotex mRNA Mini Kit (Qiagen) from 6 μg of total RNA. RNA-seq libraries were constructed using the NEBNext mRNA Library Prep Master Mix Set for Illumina according to the manufacturer's instructions, selecting for a fragment size range of 300–500 bp. Sequencing was performed on an Illumina GAIIx sequencer with 115-bp single reads using TruSeq v5 sequencing chemistry and TruSeq v5 cluster generation kits.
Computational analysis
ChIP-seq
Data
In this study, sequence data for a total of eight ChIP-seq experiments were analyzed (see Supplemental Table 1). Two libraries each were generated for ChIP and input samples of HOXD13wt, HOXD13Q317R, HOXD13Q317K, and PITX1 with an average of 27,193,329 nonredundant uniquely mappable reads.
Quality filtering and read mapping
Reads with an average Phred score lower than 28 were discarded. Quality filtered reads were mapped against the reference genome galGal3 using the aligner BWA, allowing up to two mismatches (Version 0.5.9-r16) (Li and Durbin 2009). Non-uniquely mapped reads were discarded and redundant reads were removed using SAMtools rmdup (Li et al. 2009). The quality of ChIP-enrichment was validated using spp (Kharchenko et al. 2008; Landt et al. 2012).
Principle component analysis (ChIP-seq)
For principle component analysis (PCA) of the coverage profiles of the uniquely mapped reads, the galGal3 genome was divided into nonoverlapping 500-bp windows. For each sequence data set, the number of uniquely mapped reads for each window was determined, yielding count vectors of length 2,198,570. Q-mode PCA was then performed on the normalized counts of each of the eight experiments with the R function prcomp.
ChIP quality control and peak calling
Quality control and tests for reproducibility of ChIP-seq experiments were performed according to the guidelines suggested by The ENCODE Project Consortium (Li et al. 2011; Landt et al. 2012). Peak calling was performed according to Kundaje (2012) using the MACS2 peak caller (Zhang et al. 2008). Briefly, peaks for each replicate were called against pooled input controls of both biological replicates with low stringency (parameters: -g 1.0 × 109, –p 1 × 10−1,–too-large,–bw, as determined by spp in the quality control step). The final peak set was called from the pooled replicates, sorted according to P-value, and the threshold was determined by using the number of peaks above IDR 0.0025 from the pool-consistency IDR-analysis (see Supplemental Table 1).
Motif analysis
Motif analysis was conducted using DREME (MEME package, version 4.7.0) (Bailey 2011). Sequences of 150 bp surrounding the summits of the 1000 strongest peaks (sorted by P-value) were used as input (parameters: -e 0.05, -m 10, -g 100, -s 1, -v 2, -mink 3, -maxk 10).
Peaks in genomic regions
The galGal3 genome was subdivided into the following five classes of regions: promoter, exon, intron, gene flanking, and intergenic on the basis of annotation data from Ensembl BioMart (database: Ensemble Genes 66; data set: WASHUC2).
Regions between 5 kb upstream of and 2 kb downstream from the gene start position were defined as promoter regions. For genes shorter than 2 kb, the downstream part of the promoter was omitted. All exon and intron regions that do not overlap with promoter regions were defined as intron and exon regions. Regions between 25 kb and 5 kb upstream of the gene start position, as well as regions between the gene end position and 20 kb downstream, were defined as gene flanking regions. Regions not covered by any of the previously defined regions were defined as intergenic regions. All classes of regions were further subdivided into the subclasses, conserved and not conserved, using the most conserved track for vertebrates of UCSC (phastConsElements7way).
To count peaks in different genomic regions, summit files were intersected with the regions defined above using intersectBed of BEDTools (Quinlan and Hall 2010), yielding for all peaks which are not in intergenic regions at least one association with a gene. One summit may be assigned multiple genes because the regions are overlapping to some extent.
seqMINER analysis
We used seqMINER version 1.3.3 (Ye et al. 2011). The peak summits for HOXD13wt, HOXD13Q317K, HOXD13Q317R, and PITX1 were taken as reference coordinates. Subsequently, mean read densities relative to these locations were calculated for merged reads from the two biological replicates, and were calculated for each factor. The graph displays the distribution of mean read densities (tags/50 bp) centered on the peak summits (±3000 bp).
RNA-seq
The reference transcriptome for Gallus gallus was downloaded from the Ensembl release 64 database using BioMart 0.7 on October 7, 2011. The database is based on the WASHUC2/GALGAL3 genome assembly and contains 23,392 transcripts (exon models) for 17,934 genes. The reads for each data set/condition were separately mapped to the reference transcriptome using bowtie (release 0.12.7). Except for the seed length (l = 40) and the number of allowed mismatches in the seed region (m = 3), the default parameters were used for the alignment. A preceding quality filtering of the reads was skipped due to Bowtie's ability to use Phred quality scores of reads during the alignment step. Reads mapping to multiple transcripts were randomly assigned to one of them. To quantify the transcript expression and normalize between the data sets, the reads per kilobase of exon model per million mapped reads (RPKM) values for all exon models were calculated (Mortazavi et al. 2008) and used for further analysis.
Hierarchical clustering and principle component analysis of expression data
The analysis of expression data was performed for the five RNA-seq data sets with RPKM values for 3118 transcripts showing a minimum RPKM value of 2 in at least one of the experiments with the additional criterion of a fold change of at least two compared to the RPKM values obtained for the control RCASBP vector. Q-mode PCA and hierarchical clustering were applied to the base-2 logarithms of the RPKM fold-change values. The PCA was performed with the R function prcomp (retx = TRUE, center = TRUE, scale = TRUE). The hierarchical clustering and dendrogram calculation was done using the Matlab 2011 function clustergram with the Euclidean metric for pairwise comparison and unweighted average distance for the linkage. GO-analysis was performed using DAVID (Huang et al. 2008, 2009).
Cobinding of TF to differentially coregulated genes
For the 3118 differentially regulated transcripts, the RPKM values were summed up for each gene and condition. RPKM fold changes relative to control cultures were calculated as for transcripts. Genes with a twofold up-regulation for HOXD13Q317K and PITX1 or a twofold down-regulation for HOXD13Q317K and PITX1 were defined as HOXD13Q317K/PITX1 coregulated genes. HOXD13Q317R/PITX1 coregulated genes were defined accordingly.
Shared peaks for HOXD13Q317K and PITX1 and, accordingly, HOXD13Q317R and PITX1 were assigned to genes if they fall in the promoter, the gene body, or gene flanking regions of annotated ENSEMBL genes (see Peaks in Genomic Regions section above). Genes with at least one shared peak for HOXD13Q317K and PITX1 were defined as HOXD13Q317K-PITX1 cobound genes. HOXD13Q317R-PITX1 cobound genes were defined accordingly.
Data access
Data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE44799.
Acknowledgments
This study was supported by grants from the Bundesministerium für Bildung und Forschung (Förderkennziffer FKZ 1315848A) and the Deutsche Forschungsgemeinschaft (DFG) (grant numbers MU880/11-01, MU 880/8-1) to S.M., J.H., and P.N.R.
Footnotes
-
↵7 Corresponding author
E-mail hecht{at}molgen.mpg.de
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.157610.113.
- Received March 13, 2013.
- Accepted August 28, 2013.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported), as described at http://creativecommons.org/licenses/by-nc/3.0/.








