
Pathoscope: Species identification and strain attribution with unassembled sequencing data
- Owen E. Francis1,
- Matthew Bendall2,
- Solaiappan Manimaran3,
- Changjin Hong3,
- Nathan L. Clement4,
- Eduardo Castro-Nallar5,
- Quinn Snell4,
- G. Bruce Schaalje1,
- Mark J. Clement4,
- Keith A. Crandall5,6 and
- W. Evan Johnson3,6
- 1Department of Statistics, Brigham Young University, Provo, Utah 84602, USA;
- 2Department of Biology, Brigham Young University, Provo, Utah 84602, USA;
- 3Division of Computational Biomedicine, Boston University School of Medicine, Boston, Massachusetts 02118, USA;
- 4Department of Computer Science, Brigham Young University, Provo, Utah 84602, USA;
- 5Computational Biology Institute, George Washington University, Ashburn, Virginia 20147, USA
Abstract
Emerging next-generation sequencing technologies have revolutionized the collection of genomic data for applications in bioforensics, biosurveillance, and for use in clinical settings. However, to make the most of these new data, new methodology needs to be developed that can accommodate large volumes of genetic data in a computationally efficient manner. We present a statistical framework to analyze raw next-generation sequence reads from purified or mixed environmental or targeted infected tissue samples for rapid species identification and strain attribution against a robust database of known biological agents. Our method, Pathoscope, capitalizes on a Bayesian statistical framework that accommodates information on sequence quality, mapping quality, and provides posterior probabilities of matches to a known database of target genomes. Importantly, our approach also incorporates the possibility that multiple species can be present in the sample and considers cases when the sample species/strain is not in the reference database. Furthermore, our approach can accurately discriminate between very closely related strains of the same species with very little coverage of the genome and without the need for multiple alignment steps, extensive homology searches, or genome assembly—which are time-consuming and labor-intensive steps. We demonstrate the utility of our approach on genomic data from purified and in silico “environmental” samples from known bacterial agents impacting human health for accuracy assessment and comparison with other approaches.
The accurate and rapid identification of species and strains of pathogens is an essential component of biosurveillance from both human health and biodefense perspectives (Vaidyanathan 2011). For example, misidentification was among the issues that resulted in a 3-wk delay in accurate diagnosis of the recent outbreak of hemorrhagic Escherichia coli being due to strain O104:H4, resulting in over 3800 infections across 13 countries in Europe with 54 deaths (Frank et al. 2011). The most accurate diagnostic information, necessary for species identification and strain attribution, comes from the most refined level of biological data—genomic DNA sequences (Eppinger et al. 2011). Advances in DNA-sequencing technologies allows for the rapid collection of extraordinary amounts of genomic data, yet robust approaches to analyze this volume of data are just developing, from both statistical and algorithmic perspectives.
Next-generation sequencing approaches have revolutionized the way we collect DNA sequence data, including for applications in pathology, bioforensics, and biosurveillance. Given a particular clinical or metagenomic sample, our goal is to identify the specific species, strains, or substrains present in the sample, as well as accurately estimate the proportions of DNA originating from each source genome in the sample. Current approaches for next-gen sequencing usually have read lengths between 25 and 1000 bp; however, these sequencing technologies include error rates that vary by approach and by samples. Such variation is typically less important for species identification given the relatively larger genetic divergences among species than among individuals within species. But for strain attribution, sequencing error has the potential to swamp out discriminatory signal in a data set, necessitating highly sensitive and refined computational models and a robust database for both species identification and strain attribution.
Current methods for classifying metagenomic samples rely on one or more of three general approaches: composition or pattern matching (McHardy et al. 2007; Brady and Salzberg 2009; Segata et al. 2012), taxonomic mapping (Huson et al. 2007; Meyer et al. 2008; Monzoorul Haque et al. 2009; Gerlach and Stoye 2011; Patil et al. 2012; Segata et al. 2012), and whole-genome assembly (Kostic et al. 2011; Bhaduri et al. 2012). Composition and pattern-matching algorithms use predetermined patterns in the data, such as taxonomic clade markers (Segata et al. 2012), k-mer frequency, or GC content, often coupled with sophisticated classification algorithms such as support vector machines (McHardy et al. 2007; Patil et al. 2012) or interpolated Markov Models (Brady and Salzberg 2009) to classify reads to the species of interest. These approaches require intensive preprocessing of the genomic database before application. In addition, the classification rule and results can often change dramatically depending on the size and composition of the genome database.
Taxonomy-based approaches typically rely on a “lowest common ancestor” approach (Huson et al. 2007), meaning that they identify the most specific taxonomic group for each read. If a read originates from a genomic region that shares homology with other organisms in the database, the read is assigned to the lowest taxonomic group that contains all of the genomes that share the homologous region. These methods are typically highly accurate for higher-level taxonomic levels (e.g., phylum and family), but experience reduced accuracy at lower levels (e.g., species and strain) (Gerlach and Stoye 2011). Furthermore, these approaches are not informative when the reads originate from one or more species or strains that are closely related to each other or different organisms in the database. In these cases, all of the reads can be reassigned to higher-level taxonomies, thus failing to identify the specific species or strains contained in the sample.
Assembly-based algorithms can often lead to the most accurate strain identification. However, these methods also require the assembly of a whole genome from a sample, which is a computationally difficult and time-consuming process that requires large numbers of reads to achieve an adequate accuracy—often on the order of 50–100× coverage of the target genome (Schatz et al. 2010). Given current sequencing depths, obtaining this level of coverage is usually possible for purified samples, but coverage levels may not be sufficient for mixed samples or in multiplexed sequencing runs. Assembly approaches are further complicated by the fact that data collection at a crime scene or hospital might include additional environmental components in the biological sample (host genome or naturally occurring bacterial and viral species), thus requiring multiple filtering and alignment steps in order to obtain reads specific to the pathogen of interest.
Here we describe an accurate and efficient approach to analyze next-generation sequence data for species identification and strain attribution that capitalizes on a Bayesian statistical framework implemented in the new software package Pathoscope v1.0. Our approach accommodates information on sequence quality, mapping quality, and provides posterior probabilities of matches to a known database of reference genomes. Importantly, our approach incorporates the possibility that multiple species can be present in the sample or that the target strain is not even contained within the reference database. It also accurately discriminates between very closely related strains of the same species with much less than 1× coverage of the genome and without the need for sequence assembly or complex preprocessing of the database or taxonomy. No other method in the literature can identify species or substrains in such a direct and automatic manner and without the need for large numbers of reads. We demonstrate our approach through application to next-generation DNA sequence data from a recent outbreak of the hemorrhagic E. coli (O104:H4) strain in Europe (Frank et al. 2011; Rohde et al. 2011; Turner 2011) and on purified and in silico mixed samples from several other known bacterial agents that impact human health. Software and data examples for our approach are freely available for download at https://sourceforge.net/projects/pathoscope/.
Results
Overview of the identification approach
For the purposes of this demonstration, we constructed a reference database of bacterial genomes obtained from GenBank, chosen based on their phylogenetic affinity to eight bacterial agents from the “CDC Category A and B lists of bioterrorism agents/diseases” (http://www.bt.cdc.gov/agent/agentlist-category.asp). The query next-gen sequencing reads were independently aligned to the reference genomes using three different aligners, BLAST (Altschul et al. 1997), GNUMAP (Clement et al. 2010), and Bowtie 2 (Langmead and Salzberg 2012) (exact parameters used are given in the Methods section below). Reads with a single or unique alignment to only one organism in the database were denoted as uniquely mapped reads, or unique reads in short. However, since our database contains many closely related species and strains, many of the sequence reads map to multiple genomes in the database. These reads are denoted as non-unique reads. Reads that do not match any genome in the database are only utilized to help determine whether the source species is present in the database. From data examples presented in the sections below, we observed that between 6.4% and 99.9% of the reads map to multiple organisms, depending on the number of closely related strains in the database (see Fig. 1).
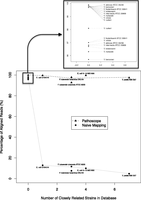
Impact of the closely related strains on the read alignment proportions. The genomes in the database were aligned to each other using an all-against-all BLASTN approach (Agren et al. 2012), and strains of the same species that were >98% similar using this metric were considered “closely related” strains. As the number of closely related strains increases, the naïve algorithm was not able to definitively identify the origin species. However, Pathoscope performed consistently well independent of the number of closely related strains.
When reads align to multiple genomes due to their sequence similarity, the reads are less likely to be assigned to the correct source genome. For example, in the E. coli K12 MG1655 example described below, >99.9% of the reads aligned to multiple genomes due to the presence of multiple related substrains in the database. In this case the correct genome received the same proportions of the reads as a closely related, but incorrect, substrain due to non-uniqueness. This leads to the inability to conclusively identify the correct substrain—especially for methods based only on the alignment, context matching, homology searching, or genome assembly. However, by reassigning the ambiguous reads, we show below that it is possible to remove reads assigned to genomes that are less likely to be the source of the reads, and reassign them to the source template of the reads.
Through an iterative process, our novel Bayesian read reassignment method is capable of identifying the genomes that are the most likely source of the reads. However, even though a set of reads could have originated from the DNA from multiple organisms, each individual read was derived from one template DNA strand that came from a single organism. To correctly and precisely identify the species present in the sample, the non-unique read probabilities must be reassigned to the correct template genome of origin. To address this need, we have formulated a Bayesian missing data mixture modeling approach (where the template genome of origin is the “missing data”) that integrates information contained within the read (mapping probability) with information obtained by borrowing strength across all reads from the sample (e.g., proportions of unique reads or imbalances in non-unique probabilities across all reads). This approach is superior to a naïve mapping approach that assigns reads based on information contained solely in the reads. Using this additional information helps to overcome mistakes in mapping caused by sequencing errors or low-quality bases.
Application to the European E. coli outbreak of 2011
The recent outbreak of Escherichia coli (E. coli) O104:H4 in Europe resulted in a number of deaths that may have been prevented by an early identification of the affecting pathogen. We obtained 92,370 sequencing reads from an O104:H4 sequencing run generated at the BGI, using the Ion Torrent sequencing technology (Guilford, CT) (SRR227300; Li et al. 2011). Most of the reads in the data set (94.1%) ranged in length from 80 to 120 bp. We used BLAST, Bowtie 2, and GNUMAP to independently align these query reads to our reference database, which included the genomes of 30 strains of E. coli—many of which were closely related to the O104:H4 strain. The Pathoscope results from the BLAST, Bowtie 2, and GNUMAP alignments were nearly identical (<1% different), so we only report the results from the BLAST alignment below.
In addition to Pathoscope, we compared several other approaches for inferring the genomic source of sequencing reads. These included a naïve mapping strategy, where we aligned reads to the database and generated a posterior probability of alignment based on the read's alignment score for each genome. The read probabilities are then summed for each genome, resulting in the total (probabilistic) portion of the reads mapped to each specific genome. We also compared with PhymmBL (Brady and Salzberg 2009), MEGAN4 (Huson et al. 2007), PhyloPhythiaS (Patil et al. 2012), and MetaPhlAn (Segata et al. 2012). Finally, we applied an alignment approach using the Trinity assembler (Grabherr et al. 2011) to assemble high-quality contiguous sequences (contigs) from the reads, followed by the probabilistic alignment of the contigs to the database (see Methods for specific parameter settings for each algorithm).
For this example, we used the full data set of 92,370 reads, representing 1.3× coverage of the reference O104:H4 genome, as well as reduced data sets using 1000 random subsamples of reads for each of the following sample sizes: 9237 (0.13×), 924 (0.01×), and 92 (0.001×). For the smaller subsets (92, 924, 9237), we compared the average accuracy and range across samples for each method. These smaller sets were designed to evaluate algorithmic performance when the reads are generated using multiplexed sequencing runs or when they originated from contaminated samples that may be dominated by other genomic sources. However, we note that for MEGAN (graphical user interface), PhyloPhythiaS (manual webserver), and the assembly approach, we did not use 1000 random data sets; rather, we used a single random sample of each data set size, as they would either require thousands of manual submissions or an excessive amount of computation time. Table 1 contains the average accuracy and range across samples for each algorithm.
Results from the application of several species identification approaches to subsets of the 92,370 sequencing reads from the first O104:H4 Ion Torrent sequencing run
Naïve alignment, PhymmBL, and MetaPhlAn
The naïve algorithm consistently assigned around 12.9% of the read probability to the O104:H4 strain independent of the number of reads used. However, on average, between 7.4% and 9.4% of the read probability was assigned to the E. coli 55989, which is the closest fully sequenced genome to the O104:H4 strain (Rohde et al. 2011; Turner 2011). Several other E. coli strains received 1%–3% of the reads, and several species in the Shigella genus also received 1%–2% of the reads. In all, ∼93% of the read probabilities were assigned to an E. coli strain. The PhymmBL algorithm assigned 14.7% on average to the O104:H4 strain and exhibited similar profiles of false mapping to other strains and species. Overall, the performance of PhymmBL was only slightly better than the naïve approach. The MetaPhlAn algorithm aligns reads to taxonomic clade-specific markers, which in its current implementation can only identify DNA templates at the species level—and therefore cannot distinguish between strains or substrains of the same species. In addition, because it only uses short clade markers, merely 815 (0.9%) of the reads were assigned by MetaPhlAn. Of these reads, only 90.0% were aligned to E. coli, whereas 9.6% were incorrectly assigned to S. dysenteriae. The method gave inconsistent results for the subsamples of 9237, and most of the time failed to assign any reads to E. coli for the subsamples of 92 and 924. From these approaches, it is clear that an E. coli strain is present in the sample, and the naïve and PhymmBL approaches point to O104:H4 as the most likely source, but all results are ambiguous as to whether there are multiple E. coli strains or other species present in the sample.
Genome assembly approach
For the assembly approach, no contigs were generated from the 92 and 924 read data sets. For the data set with 9237 reads, only five contigs were generated, ranging in length from 221 bases to 442 bases in length (N50 = 409; N90 = 221). Although these five contigs best matched to the O104:H4 strain, they also aligned to several other (incorrect) genomes in the database. Finally, on the complete sequencing run representing 1.3× coverage of the genome, the assembler constructed 3637 short contigs (N50 = 292; N90 = 216) with only 21.5% of the contig mapping probability being assigned to the correct strain. Therefore, although this approach is a slight improvement over the naïve approach or context mapping, it is clear that a single sequencing run for a purified (single source) sample is not sufficient for strain attribution using an assembly-based approach.
Pathoscope reassignment
In contrast, as shown in Table 1, Pathoscope reassigned, on average, 99.4% of the read probability directly to the O104:H4 strain for the data sets with 92 reads and averaged 99.6% of the reads correctly for the larger data sets. These results imply that Pathoscope is a substantial improvement over naïve mapping, context mapping, and assembly-based methods for species identification and strain attribution.
Identification of the nearest genome
The results from the MEGAN and PhyloPythiaS analyses were not included in the previous section because the annotation tables used by these approaches do not contain the O104:H4 strain (and cannot be manually added by the user). For this reason, we removed the O104:H4 strain from our reference database and reanalyzed the query reads using the naïve mapping and Pathoscope reassignment. In addition, we note that the PhyloPythiaS web server only allowed for a maximum of 10,000 reads for each submission, so the results presented here were based on random sets of 92, 924, and 9237 only (and not the full data set).
For the naïve mapping with O104:H4 removed, most of the aligned reads (99.8%) mapped to at least one strain of E. coli, thus rapidly and clearly identifying the species of origin. However, 96.1% of these reads aligned ambiguously to multiple E. coli strains. The 55989 strain received the largest proportion of the aligned reads (9.5%), followed by the O103:H2 strain (3.2%), the B7A, O26:H11, E24377A, and the E22 strains (3.1%), then the SE11 and IAI1 strains (3.0%). Therefore, although the correct species was easily identified using a naïve mapping strategy, the identification of the correct strain within the species proves to be more difficult, and a simple mapping strategy leaves much uncertainty in the process of identifying the strain most similar to the origin strain. This uncertainty can prove to be important for E. coli—which contains both benign and harmful strains—as the misclassification of the origin or nearest strain might lead to negative economic and human health consequences.
In contrast, the lowest common ancestor approach utilized by MEGAN assigned 80.2% of the reads to the family taxonomic level or higher. The remaining reads were assigned at the genus level; 19.7% of the total reads were assigned to the Escherichia genus and 0.2% of the reads were incorrectly assigned to the Shigella genus. MEGAN did not assign any reads at the species or strain level for any of the data sets. PhyloPhythiaS also performed poorly on this example: Overall, >84% of the reads were assigned to the family level or above, and <50% of all the reads were correctly assigned E. coli taxonomy levels. Furthermore, 32 incorrect genera received more reads than Escherichia, and five incorrect species received more reads than E. coli.
After application of the Pathoscope reassignment, 89.5% of the reads were reassigned to the 55989 strain. The genomes with the next highest read proportions were the O157:H7 strain (3.2%) and the O103:H2 strain (1.1%). Therefore, even though our approach did not completely converge on one genome (as it should not, because in this analysis the origin strain was not present in the database), it is clear that Pathoscope can clearly and definitively identify the closest fully sequenced neighboring strain with high confidence.
To evaluate whether the lack of sensitivity for MEGAN and PhyloPythiaS is due to the missing O104:H4 annotation, we applied MEGAN and PhyloPhythiaS to our analysis of reads from the E. coli K-12 MG1655 substrain (described in detail below), which is contained in the annotation. For MEGAN, the result was similar, in that all of the reads were assigned at the genus level or higher. For PhyloPythiaS, 98.5% of the reads were assigned to the genus level or above, and 34.7% of the reads were assigned to incorrect taxonomies. The E. coli species only received 1.4% of the reads, and no reads were assigned at the strain or substrain level. Therefore, these methods can fail to identify substrains, even when they are present in the annotation.
Computational time
MetaPhlAn was by far the fastest algorithm (Table 1), requiring only 1 min to complete because it aligns the reads to a set of small clade markers; however, the approach assigned <1% of the reads in this example. The naïve approach required 38 min for a BLAST alignment, 21 min for GNUMAP, and 13 min for Bowtie 2. Pathoscope and MEGAN used the naïve alignments and required an additional 7 min and 3 min, respectively. PylopythiaS required a total of 7 min to assign 9237 reads. PhymmBL required ∼36 h of database preprocessing, and then ∼2 h to assign the reads. Finally, the assembly approach required 30 min to complete.
NCBI sequence read archive data sets
To further evaluate the effectiveness of our method in different scenarios, we obtained sets of reads from 12 different bacterial species/strains from the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra), all of which were sequenced using the 454 platform (Roche). The reads from each sample were aligned to our full database of genomes and identified as though the true source of the reads were unknown. These data sets consisted of between 28,221 and 1,504,985 reads, with read lengths typically ranging from 77 to 277 bp. Overall, these data sets amounted to only 1.2× to 31.2× coverage of the target genomes. For more than half of these purified sample data sets, the read coverage is not sufficient to fully assemble a genome (Schatz et al. 2010).
Our Pathoscope strain attribution method worked extremely well on all of these samples (Table 2). Before reassignment, the read probability assigned to the correct genome ranged between 4.8% and 98.1%. To further evaluate this phenomenon, we plotted the naïve alignment probabilities (along with the Pathoscope reassignments) versus the number of closely related strains contained in the database (Fig. 1). Clearly, the accuracy of the naïve approach relies heavily on the number of similar genomes in the database, and to distinguish between closely related strains and substrains, a Pathoscope reassignment is absolutely necessary for proper identification. After reassignment using Pathoscope, the read probability for the correct genome ranged between 92.7% and 99.9%, showing very strong evidence for the correct genome of interest. In nine cases Pathoscope reassigned >99% of the reads to the correct genome. In the three sets where reassignment led to <99%, all had special circumstances and are discussed below. These examples clearly show the benefit of our pathogen detection approach and its ability to reliably identify the correct genome under a diverse set of conditions, not only to species, but also to strain level.
Results from the application of our species identification method on 12 data sets from the NCBI Sequence Read Archive (SRA, reference website)
Closely related strains in the database
There were 30 different strains and substrains of E. coli present in the genome database, three of which were substrains of the K-12 strain. Notably, the K-12 MG1655 and the K-12 W3110 substrains have >99.9% sequence similarity between the genomes; in fact, a recent study identified only 23 sites with point mutations to differentiate between these genomes (Hayashi et al. 2006). This created difficulty for strain attribution for the naïve mapping strategy: For example, when we attempted to assign reads from the K-12 MG1655 substrain, we observed that only 10.0% of the read probability mapped to K-12 MG1655, 10.0% to K-12 W3110, and 9.4% mapped to K-12 DH10B. Clearly, this shows the failure of a naïve mapping strategy and points to the need for a highly sensitive mapping strategy with greater differentiation among substrains. Our reassignment method, Pathoscope, was able to confidently reassign the reads to the correct genome. Our method reassigned an impressive 99.6% of the reads to the E. coli K-12 MG1655 genome. The ability to differentiate at the substrain level will become increasingly important as databases of bacterial genomes are rapidly growing.
Unassembled genome
The Yersinia pestis KIM D27 data set provided an interesting scenario that further illustrates the performance of Pathoscope in cases where the genome is not fully contained in the reference database. In our database there were 21 different strains or substrains of Y. pestis, two of which were substrains of the KIM strain. The correct KIM D27 genome was contained in our database, but was not fully assembled. Specifically, our database contained only nine contigs of the D27 substrain, whereas the database contained the complete genome of the KIM 10 strain. The percentage of read probabilities assigned using a naïve approach mapped only 4.8% of the reads to Y. pestis KIM D27, which was closely followed by the KIM 10 substrain (4.4%), and then the Mediaevalis strain (4.4%). After reassignment, 97.3% of the reads were reassigned to the correct KIM D27 unassembled substrain. While impressive, this percentage is smaller than was observed with many of the other genome examples from the SRA read sources, primarily because if the genome database contains closely related species to the target genome, many of the reads from unassembled regions will align to these genomes, resulting in a small but significant read probability for incorrect genomes. The greater probability assigned to the other genomes is an effect of the increased uncertainty due to the incomplete target genome.
Genome not present in the database
As was the case with the European E. coli example described previously, to further test our approach in a scenario where the source genome is not present in the database, we focused our attention on the SRA sample from the Francisella tularensis ATCC 6223 substrain. This substrain was not contained in the reference database; however, 13 strains and substrains of F. tularensis, including five substrains of the F. t. tularensis subspecies (type A), were present in the genome database. In this example, only 19.8% of the 67,276 reads mapped to a genome in the database, and 99.4% of these mapped reads aligned to more than one genome. However, after reassigning the reads, 92.7% of the read probability was assigned to the F. tularensis WY96-3418 strain, and 4.8% of the read mass was assigned to the F. tularensis SCHU S4 strain, both of the F. t. tularensis subspecies. It is interesting to note that there are two strong indicators providing evidence that the identified genome is not the true source, but just a closely related substrain. The first indicator is that only a small proportion of the reads (19.8%) mapped to any genome in this example. In addition, after reallocation, the read probabilities assigned is less than what was observed in the 11 cases when the true genome was contained in the reference database. Therefore, these two quantities provide promising metrics for identifying whether the true genome is contained in the reference database.
Combination of multiple SRA data sets
We also generated a mixed read data set by combining reads originating from Y. pestis KIM D27 (SRR033501), E. coli K-12 MG1655 (SRR031601), and F. tularensis subsp. holarctica OSU18 (SRR032505). After alignment to our genome database, 462,996 of the reads aligned to at least one genome in the database with 67.8%, 31.0%, and 1.2% originating from the Y. pestis, E. coli, and F. tularensis, respectively. Using a naïve mapping strategy, only 4.7% of the read probability was assigned to the correct Y. pestis strain, 4.4% matched the E. coli strain, and the F. tularensis strain received only 0.2% of the reads. In fact, the F. tularensis strain received fewer reads than 49 (of 131) genomes in the database. This clearly shows the failure of a naïve mapping strategy on mixed samples. Once the reads were reassigned using Pathoscope, 67.7%, 31.0%, and 1.2%, of the read probability was assigned to the correct Y. pestis, E. coli, and F. tularensis strains, respectively. Thus, Pathoscope was able to recover genome proportions almost identical to the original mixing proportions, and the results were substantially better than the naïve approach.
To further evaluate Pathoscope on mixed samples, we generated 1000 mixtures of ∼5770 reads (based on the size of the smaller F. tularensis data set) with random proportions of each species. The naïve approach produced extremely biased results by consistently underestimating the correct read proportions, whereas Pathoscope closely estimated the read proportions with average absolute differences of 0.0008 for Y. pestis, 0.0092 for E. coli, and 0.0038 for F. tularensis (Table 3). In addition, the naïve approach consistently ranked genomes in the sample lower than many genomes that were not in the sample. For example, the average rank of Y. pestis across the 1000 simulations was 13.1 for the naïve approach, and for 627 samples Y. pestis was not ranked among the top 10 genomes. Alternatively, after Pathoscope, Y. pestis was ranked among the top three (there were three genomes in the mixture) in all but four of the mixtures and in the top five for all of the mixtures. In these simulations, Pathoscope did fail to rank the proper E. coli substrain in the top three for 67 of the samples, in which cases Pathoscope either selected a different E. coli K12 substrain or split the reads among the three K12 substrains in the database. For these 67 samples, we observed that the average number of E. coli reads was ∼700, representing ∼2.5% (0.025×) coverage of the E. coli genome. This points out that 2.5% coverage is not sufficient for Pathoscope to distinguish between substrains with 99.9% sequence identity (Hayashi et al. 2006), although Pathoscope did perform well in distinguishing between these substrains when coverage percentages ranged from 5% to 20%.
Results from 1000 random mixtures of ∼5770 reads from the Y. pestis KIM D27 (SRR033501), E. coli K-12 MG1655 (SRR031601), and F. tularensis subsp. holarctica OSU18 (SRR032505) data sets
Discussion
Here we present an accurate and sophisticated computational approach for species identification and strain attribution. Our approach relies on the construction of a genome database containing multiple strains or species that are possible source genomes for the sample and utilizes a probabilistic mapping approach to align the reads to the genome. Reads that map to multiple genomes are then reassigned to the most likely source genome using a Bayesian statistical framework that accommodates information on sequence quality and mapping quality. We attribute the increased accuracy of Pathoscope compared with other methods to the fact that Pathoscope considers all the reads jointly when reassigning reads to source genomes, whereas most other approaches only look at one read at a time. We show in multiple real data examples that our method is highly accurate in identifying the source genome or genomes for a biological sample. We show that in many cases, we can identify the source species or strain with only a small number of reads that represent only fractional coverage of the genome. In addition, we show that our approach is able to accurately identify the proper origin genome, even when several closely related strains or substrains are present within the database. We also show the failure of other approaches to assign reads and identify source genomes at the species, strain, and substrain level.
We demonstrate the performance of Pathoscope on purified samples and for “environmental” samples mixed in silico. In theory, this approach can also be applied to a variety of other scenarios including host-dominated clinical samples, unpurified environmental samples, and other types of community sequencing data. However, the performance and utility of Pathoscope in these contexts are yet to be determined. However, we believe that our approach will play an important role in future applications in pathology, bioforensics, and biosurveillance.
Methods
Genome database construction
Central to our approach is a robust database against which to map the query sequencing reads. For the purposes of this demonstration, we gathered a database of 170 complete bacterial chromosomes obtained from 131 distinct strains (610 Mbp) (see the Supplemental Material for accession numbers for the genomes included in this reference database). The database was intended to aid in the identification of eight bacterial agents of bioterrorism identified by the CDC: Bacillus anthracis, Burkholderia mallei, Burkholderia pseudomallei, Brucella sp., Clostridium botulinum, Escherichia coli O157:H7, Francisella tularensis, and Yersinia pestis.
In order to differentiate closely related strains and species (often nonpathogenic) from target strains of interest, we wanted to include in our reference database genomes from any closely related strains/species. Therefore, closely related species/strains were identified by phylogenetic analysis of the 16S ribosomal RNA genes. 16S sequences for all eight pathogens of interest were obtained from GenBank and used to query the nr database utilizing BLASTN (Altschul et al. 1997) using default parameters (Word Size: 28, Expect Value: 10, Match/Mismatch Scores: 1, −2, Gap Costs: Linear). We identified 3206 sequences corresponding to 1050 named species or subspecies with multiple sequences represented within a number of these taxonomic groups using a partial or full match with BLASTN. We then estimated phylogenetic relationships amongst these sequences and our target species. From this phylogeny, we selected 131 completed genome sequences, 332 fully sequenced plasmids, and 207 whole-genome shotgun sequencing projects to serve as our reference database (see the Supplemental Material for details). Although this study uses the entire genome database, any subset of these sequence types could be used for reference material. The genetic distances for Figure 1 were calculated by performing an all-against-all BLAST as implemented previously (Agren et al. 2012). Strains of the same species that were >98% similar using this metric were considered “closely related” strains for Figure 1.
Probabilistic alignment
We used the FastQC pipeline (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to assess the quality of the read data sets. For the data sets used in these examples, the qualities of the data sets were
generally acceptable by the standards set for the FASTQC pipeline. Once the quality of the data set was ascertained, we aligned
the reads utilizing a modification of BLAST (Altschul et al. 1997) alignment scores and the GNUMAP probabilistic alignment algorithm (Clement et al. 2010) because of their abilities to compute the likelihood that a read will be mapped to multiple locations in a reference database.
In these approaches, segments of size k bases (k-mers) from the reads are indexed into a genomic hash table that contains all k-mers and their location in the reference genomes from the database. Once a set of putative locations is identified using
the k-mer hash, the reads are then aligned to the full genomic sequences at these locations using a seed extension (Altschul et al. 1997) or a probabilistic Needleman-Wunsch algorithm (Clement et al. 2010). The latter approach incorporates the base-quality information provided for each nucleotide, allowing GNUMAP to rely more
on high-quality base calls and less on bases that are less certain. This approach improves mapping results from reads with
bases that may be low quality or miscalled by the sequencer and reduces the chance that the reads will be misaligned to an
incorrect genome. After alignment, the scores for each alignment location for both alignment algorithms (BLAST and GNUMAP)
are then converted to posterior probabilities. Given the alignment scores S1,S2,…, Sn the posterior probability assigned to the jth alignment, Pj, is computed as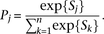
These posterior probabilities are interpreted as the probability that each alignment is the true source of the read. A probabilistic aligner performs significantly better than other alignment algorithms that discard non-unique reads or randomly assign non-unique reads to a single genome (Li et al. 2008, 2009; Langmead et al. 2009; Li and Durbin 2009). Alignment algorithms that discard non-unique reads lose much of the power to identify the correct genome. Unique reads often represent only a small fraction of the available reads and mapping algorithms that discard reads that occur in multiple locations are not able to discriminate between possible pathogens based on this limited amount of data. Randomly assigning the non-unique reads creates similar problems because it does not allow for read reassignment, leading to many reads being attributed to incorrect genomes.
Bayesian reassignment method
The Bayesian mixture model of Pathoscope assumes that reads are drawn from a small subset of unknown size from the pathogen genomes in the database. It assumes that each read is drawn from only one of the genomes in the subset. Parameters in the model represent the proportions of reads that originate from each genome as well as the proportion of the non-unique reads that are incorrectly assigned to each genome due to sequence similarity. Our Bayesian missing data mixture model reweights the read assignment probabilities using the mapping qualities and the parameters of the model. In practice, in the reassignment process the parameters are designed to penalize the value of non-unique reads in the presence of unique reads and reweight the non-unique reads based on overall mapping proportions when no reads map uniquely.
To formally describe our model, let i=1,…,R be the index of the reads and let j=1,…,G be the index of the genomes in the database. Let xi=(xi1,xi2,…,xiG)={xij} be a set of genome indicators for read i where xij=1 if the read originated from the jth genome and xij=0 if the read did not come from genome j. Note that by assumption, one and only one element in the vector xi can be equal to 1 (i.e., each read has only one template genome). We assume that xi follows a multinomial distribution, with probability of success π=(π1,π2,…,πG)={πj}, where πj is the proportion of the reads that originated from the jth genome.
For the unique reads, we know the template genome of interest or, in other words, we directly observe the genome indicator xi for these reads. In the case of the non-unique reads, the genome indicator xi is unobserved or missing data. For the non-unique reads, the observations are partial mapping qualities for each of the genomes. These mapping probabilities are provided as posterior probabilities, which are scaled mapping qualities or relative likelihood alignment scores obtained from the algorithm. More specifically, for the ith read we denote these mapping scores by qi=(qi1,qi2,…,qiG)={qij}. For unique reads, the qij values are equal to the xij values. For non-unique reads, these represent the uncertainty in mapping and need to be rescaled—or equivalently these reads need to be reassigned to the correct template genome of origin. In order to do this, we define a second set of parameters, θ=(θ1,θ2,…,θG)={θj} where θj is a reassignment parameter that represents the proportion of the non-unique reads that need to be reassigned to the jth genome.
In order to simplify the notation in the likelihood function, we define yi as the uniqueness indicator for read i, namely letting yi=1 if read i is unique and yi=0 otherwise. Under the modeling assumptions above, the complete data likelihood of the parameters (π, θ) given the observed data (reads, yi, unique xi) and the missing data (non-unique xi) is given by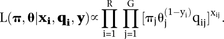
Although the reassigned reads (estimated xi) and reassignment parameters (estimated θ) are very informative, the quantities of interest from the modeling steps are the estimates for the genome read proportions (estimated π). These probabilities will identify the single or multiple organisms from the database that are present in the samples, based on the proportion of the reads that are assigned to the genome after the reads are reassigned.
Bayesian prior distributions
We assume a priori that both π and θ follow Dirichlet distributions, the densities of which can be seen in the following equations: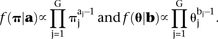
If aj=1 for all j=1,…,G, this is equivalent to adding one unique read for each of the G genomes, and aj=n would be the equivalent of adding n unique reads to the jth genome. Similarly, bj=n is the equivalent of adding n reads of non-unique read probabilities to the jth genome. However, the prior information for θ does not behave like true non-unique reads because it is not subject to reassignment. Prior information assigned to each genome will always be associated with that genome, but its effect is diminished as the number of reads increases. This can be seen clearly in the maximization formulas given in the following section. The prior information stabilizes the algorithm by preventing the estimates of π and θ from converging to the boundaries of 0 and 1. Inclusion of prior information will bias the results, possibly even leading to the identification of the wrong genome if the prior is not selected carefully. However, this would only happen in rare circumstances, and it would require initially favoring some genomes above others. To avoid this, each genome will usually receive the same values for its priors for a and b. If prior information is included, evidence of a read being present in the sample can be inferred based on whether or not the final read probability is statistically greater than the original prior information inserted. Note that noninformative priors can also be used for the experimental data by assigning zero unique reads and zero non-unique reads to each genome.
Read reassignment via the EM algorithm
Estimation of the model parameters and reassignment of the reads is accomplished using an expectation-maximization (EM) algorithm (Dempster et al. 1977). Each of the iterations of the EM algorithm consists of two simple steps. The first, called the expectation step or E-step, reassigns each read to its most likely or expected genome based on its mapping quality score and current estimates of the read proportion and non-unique misclassification model parameters. In the second step, called the maximization step or M-step, the model parameters are re-estimated using the new read assignment probabilities from the most recent E-step. These steps are repeated until the read assignments and proportion estimates converge to stable values between iterations. This algorithm is guaranteed to converge to a local maximum (Hastie et al. 2009) and in our data examples presented above, the parameter's posterior distributions appeared to be unimodal, and therefore the EM converged to a global maximum without issue.
To implement this algorithm, initial estimates of the parameters π and θ are proposed, usually πj=θj=1/G for all j. In the E-step, the expected value of xi is computed for each combination of i=1,…,R and j=1,…,G-based estimates of π and θ, as well as the observed data qi and y. In the E-step, the expected values of the elements of xi are estimated as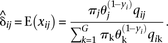
Next, the M-step calculates the new estimates of π and θ given qi, y and the current expected values 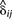 . The formulas for estimating π and θ provide the Bayesian maximum a posteriori (MAP) estimates; however, if the prior information aj and bj are set to 0 for all j genomes, these equations provide the maximum-likelihood estimates. Letting
. The formulas for estimating π and θ provide the Bayesian maximum a posteriori (MAP) estimates; however, if the prior information aj and bj are set to 0 for all j genomes, these equations provide the maximum-likelihood estimates. Letting 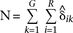 , these estimates are as follows:
, these estimates are as follows: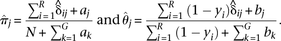
The E-step is then repeated using the updated estimates of π and θ, followed again by the M-step. These steps are repeated until the expected value of xi and the estimates of π and θ converge to stable values across iterations.
Parameters used in methods comparisons
For the O104:H4 example comparisons and SRA data sets, we used the following parameters:
Naïve alignment
(A) BLAST version 2.2.27+ at default parameters, (B) Bowtie 2 version 2.0.2 with: “-k 100”, and (C) GNUMAP version 3.0.2 with: “-m 16 -h 500 -a 0.8–print_all_sam”.
MEGAN
Version 4.70.4 with default parameters and the BLAST output file (described above).
PhymmBL
Version 4.0 at default parameters after manually adding O104:H4 to the database.
MetaPhlAn
Version 1.7.7 at default parameters and the BLAST marker database.
PhyloPhythiaS
Webserver: http://phylopythias.cs.uni-duesseldorf.de; Submission date: 3/2013.
Trinity Assembler
Release 2012-06-08 with “–seqType fa–JM 10G–single”.
Software availability
Software is available at https://sourceforge.net/projects/pathoscope/.
Acknowledgments
We gratefully acknowledge allocation of supercomputer resources from the Fulton Supercomputing Lab at Brigham Young University and the Linux Clusters for Genetic Analysis (LinGA) at the Boston University Medical Campus. This work was partially supported by funds from the National Institutes of Health (R01HG00569).
Author contributions: K.A.C. and W.E.J. conceived the study. W.E.J., Q.S., M.J.C., and G.B.S. developed the algorithm and provided direction for its implementation, and O.E.F., S.M., C.H., N.L.C., and W.E.J. implemented the algorithm and developed the software. M.B., E.C.N., and K.A.C. developed the target genome database and calculated genome similarity. O.E.F., S.M., and C.H. applied the software to the sequencing data sets. O.E.F., W.E.J., and K.A.C. wrote the paper.
Footnotes
-
↵6 Corresponding authors
E-mail wej{at}bu.edu
E-mail kcrandall{at}gwu.edu
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.150151.112.
Freely available online through the Genome Research Open Access option.
- Received October 1, 2012.
- Accepted June 25, 2013.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported), as described at http://creativecommons.org/licenses/by-nc/3.0/.








