
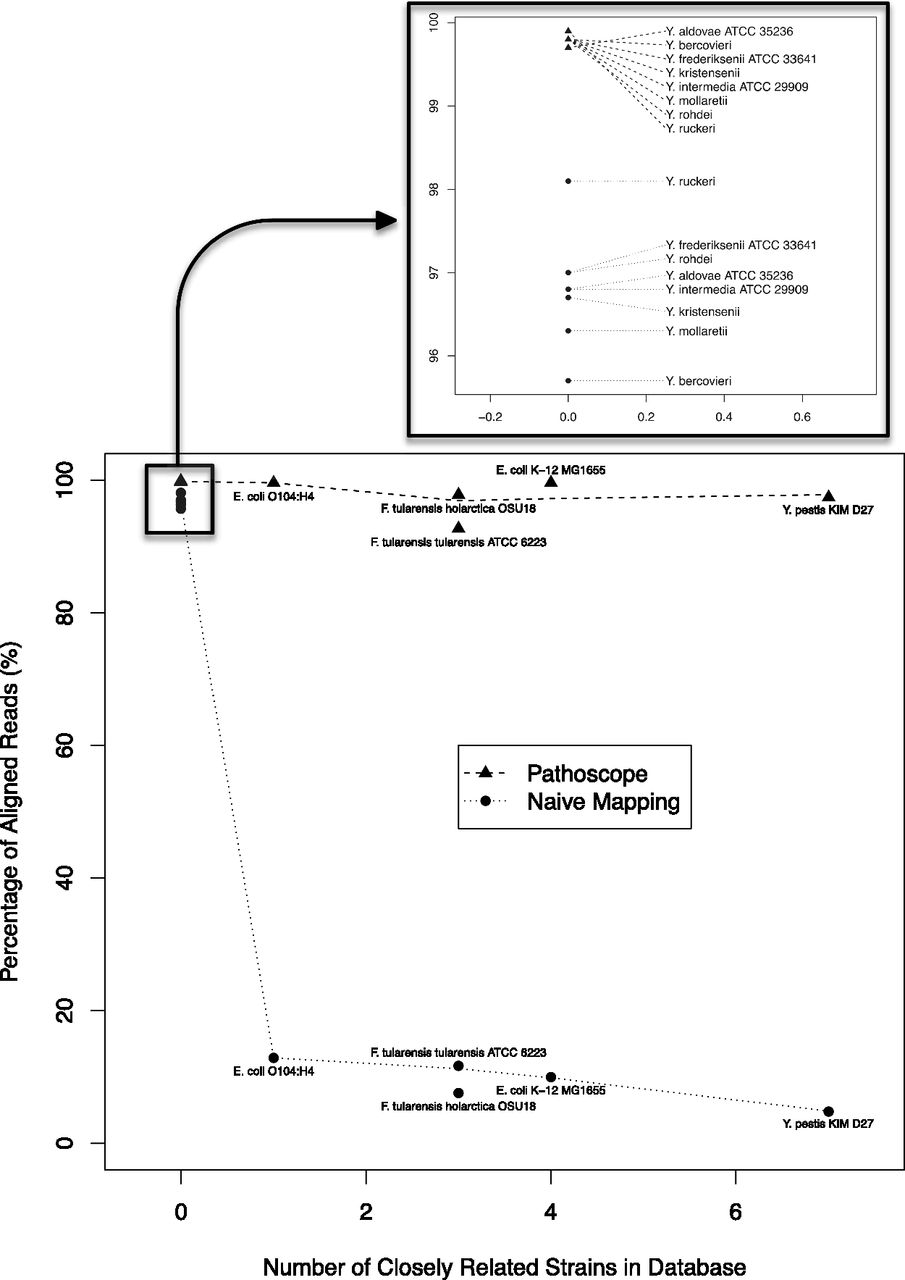
Figure 1.
Impact of the closely related strains on the read alignment proportions. The genomes in the database were aligned to each other using an all-against-all BLASTN approach (Agren et al. 2012), and strains of the same species that were >98% similar using this metric were considered “closely related” strains. As the number of closely related strains increases, the naïve algorithm was not able to definitively identify the origin species. However, Pathoscope performed consistently well independent of the number of closely related strains.








