
Abstract
A complex interplay between transcription factors (TFs) and the genome regulates transcription. However, connecting variation in genome sequence with variation in TF binding and gene expression is challenging due to environmental differences between individuals and cell types. To address this problem, we measured genome-wide differential allelic occupancy of 24 TFs and EP300 in a human lymphoblastoid cell line GM12878. Overall, 5% of human TF binding sites have an allelic imbalance in occupancy. At many sites, TFs clustered in TF-binding hubs on the same homolog in especially open chromatin. While genetic variation in core TF binding motifs generally resulted in large allelic differences in TF occupancy, most allelic differences in occupancy were subtle and associated with disruption of weak or noncanonical motifs. We also measured genome-wide differential allelic expression of genes with and without heterozygous exonic variants in the same cells. We found that genes with differential allelic expression were overall less expressed both in GM12878 cells and in unrelated human cell lines. Comparing TF occupancy with expression, we found strong association between allelic occupancy and expression within 100 bp of transcription start sites (TSSs), and weak association up to 100 kb from TSSs. Sites of differential allelic occupancy were significantly enriched for variants associated with disease, particularly autoimmune disease, suggesting that allelic differences in TF occupancy give functional insights into intergenic variants associated with disease. Our results have the potential to increase the power and interpretability of association studies by targeting functional intergenic variants in addition to protein coding sequences.
Variation in protein coding sequence is interpretable, owing to our knowledge of gene models and the triplet code. Recent studies that utilize exome sequencing take advantage of this knowledge to predict loss-of-function and nonsense mutations (Meyerson et al. 2010; Teer and Mullikin 2010). However, predicting the effects of DNA sequence variation in the large noncoding parts of the genome remains a largely unsolved problem. While transcription factors (TFs) preferentially bind DNA at definable sequence motifs, the motifs are often degenerate and are rarely predictive of binding (Tompa et al. 2005). Recent advances in DNA sequencing technologies allow genome-wide empirical measures of TF occupancy (i.e., chromatin immunoprecipitation followed by sequencing, or ChIP-seq; Johnson et al. 2007; Robertson et al. 2007), revealing that differences in TF occupancy between individuals are common (Kasowski et al. 2010; McDaniell et al. 2010). Furthermore, combining ChIP-seq with personal human genome sequencing has identified instances in which a TF preferentially binds one allele over the other in the same cell type (McDaniell et al. 2010), which we call differential allelic occupancy. Because differential allelic occupancy compares TF binding between alleles in the same nucleus, it is controlled for environmental differences between individuals and cell types and therefore provides a more direct connection between genome sequence and regulatory function than do population-based studies.
To understand the functional consequences of allelic differences in TF occupancy, it is important to measure allelic differences in expression in the same cells. Numerous approaches have been developed to measure differential allelic expression in select genes (e.g., Yan et al. 2002; Gimelbrant et al. 2007; Serre et al. 2008; Main et al. 2009; Zhang and Borevitz 2009; Zhang et al. 2009), with current estimates that 10% of human genes have allele-specific expression (Gimelbrant et al. 2007; Zhang et al. 2009). High-throughput sequencing can identify allelic imbalances in expression when complete genome sequences for both the parents are available, for example in F1 fly hybrids (McManus et al. 2010). When a complete genome sequence is available for a trio of related humans, RNA-seq (Mortazavi et al. 2008) can be used to measure genome-wide allelic imbalances in human gene expression (Degner et al. 2009; Pickrell et al. 2010). However, measurement of differential allelic expression with RNA-seq is limited to genes with heterozygous exonic sequences, which represents less than half of human transcripts.
In this work, we sought to better understand the functional consequences of genomic variation, both on TF occupancy and on gene expression. To do so, we first characterized differential allelic occupancy for 24 TFs and the cofactor EP300, as well as heritability of TF occupancy for a subset of those factors. In addition, we measured differential allelic expression using both RNA-seq as well as ChIP-seq of RNA polymerase II (RNA Pol2). The latter enabled prediction of differential allelic expression of genes with homozygous exons but heterozygous introns (Knight et al. 2003), revealing many additional otherwise undetectable instances of differential allelic expression. Together, the results provide many insights into how genome sequence impacts TF occupancy, and the extent to which that occupancy impacts downstream gene expression. The results may also have the potential to improve our understanding of disease, as we found numerous intergenic variants associated with autoimmune diseases to also be differentially bound by TFs. Ultimately, targeting intergenic regions shown to have functional consequence may improve future microarray- and sequencing-based association studies by increasing coverage with only a modest effect on statistical power.
Results
Transcription factors often cluster together on the same alleles in regions of open chromatin
To survey the allelic cis-regulatory landscape, we performed ChIP-seq on 24 sequence-specific human TFs and the transcriptional co-activator EP300 in a lymphoblastoid cell line (LCL), GM12878, generated by EBV immortalization of cells from a female (Supplemental Table 1). Whole genome sequencing has been performed on this cell line and on LCLs derived from both of her parents (The 1000 Genomes Project Consortium 2010), and we aligned sequence reads to both the maternal and paternal versions of the genome (see Methods; Figure 1A). We identified 157,586 high-confidence TF occupied regions, of which 20,013 (13%) overlap at least one heterozygous single nucleotide polymorphism (SNP). We found 1094 (5.5%) of heterozygous sites with a significant difference in occupancy between parental chromosomes for at least one TF (false discovery rate, or FDR, <5%) (Supplemental Table 2). When a single binding site covered multiple variants, we observed a consistent allelic imbalance across all variants in the binding site (Supplemental Fig. 1). Differential allelic occupancy was also reproducible between biological replicates (Supplemental Fig. 2), evenly distributed across autosomes (Supplemental Fig. 3), and not substantially biased in favor of the reference allele (Supplemental Table 3). On the X chromosome, TFs predominantly bound the maternal homolog (Supplemental Fig. 4), consistent with reports of a strong bias toward paternal X inactivation in the GM12878 cell line (McDaniell et al. 2010).
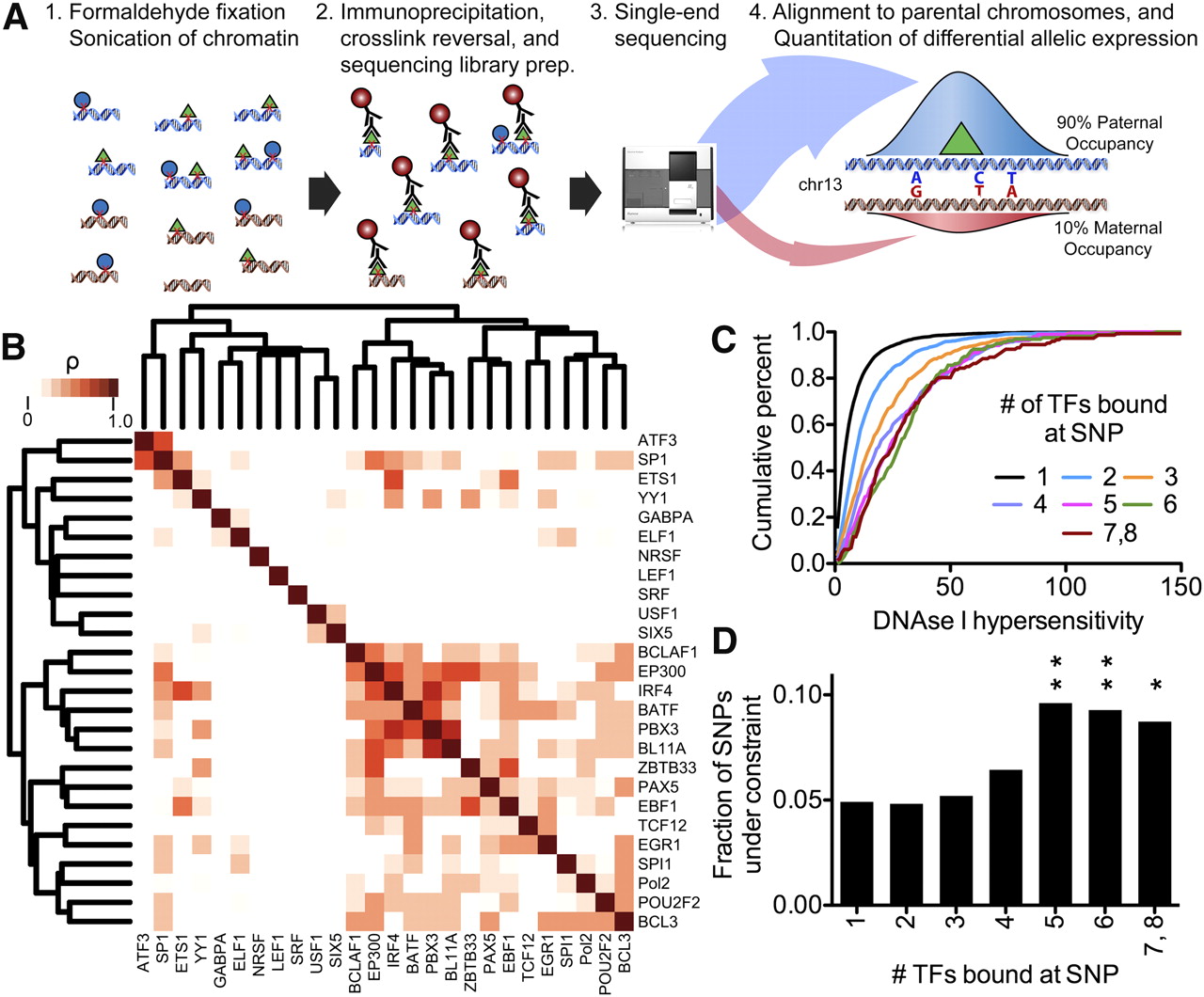
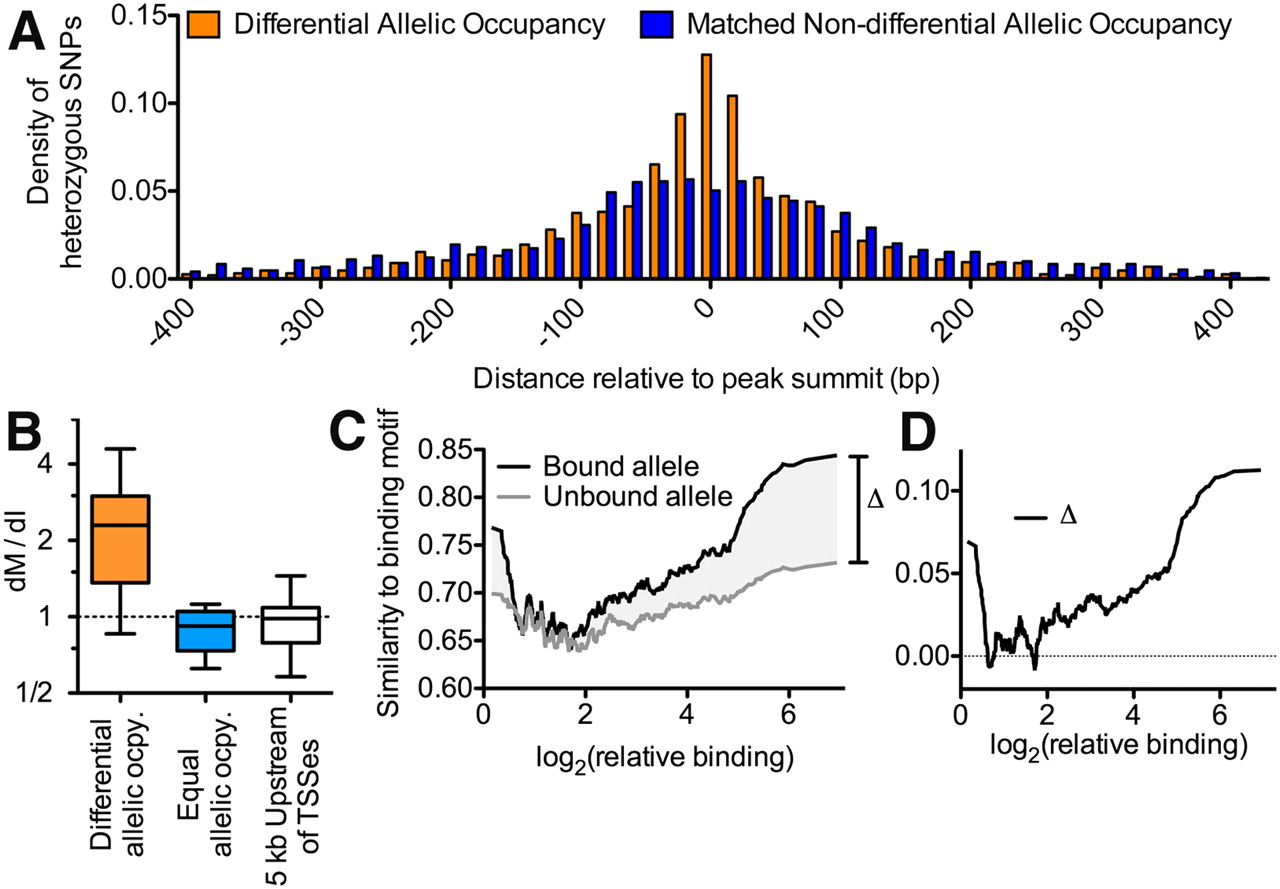
(A) Diagram of method used to measure differential allelic TF occupancy. First, chromatin was formaldehyde-fixed and sonicated. Cross-linked TF-binding complexes were then immunoprecipitated with an antibody specific for the TF of interest. The co-precipitated DNA was recovered and subjected to high-throughput single-end sequencing. Reads were aligned to maternal and paternal versions of the GM12878 genome according to data from the 1000 Genomes Project (The 1000 Genomes Project Consortium 2010). For each binding site, differential allelic occupancy was called when reads aligned to a single allele significantly more often than would be expected by random. (B) Spearman correlation of allelic imbalance at sites of TF co-occupancy throughout the genome. The color of the boxes indicates the correlation coefficient, with white indicating nonsignificant correlation (P > 0.05). The tree shows complete linkage hierarchical clustering. (C) We classified heterozygous variants by the number of TFs binding at that variant. Shown is the cumulative distribution of DNase I hypersensitivity signal at all occupied heterozygous variants in each class, as indicated in the legend. (D) For each class of heterozygous variants (as defined in C), the fractions of variants with phastCons score >0.5. Asterisks ([**] P < 0.01; [*] P < 0.05) indicate statistical significance compared to the uniquely bound variants as described in Methods.
We found evidence that TFs commonly interact with each other on the genome, especially at regions with differential allelic occupancy. Overall, 30% of autosomal TF binding sites with significant differential allelic occupancy overlapped another such site (Supplemental Table 4), and the overlaps appeared to follow a power-law distribution (Supplemental Fig. 5). In comparison, we found on average 15% of binding sites overlapping one another among an equal number of sites for which we did not detect significant differential allelic occupancy. The greater overlap in sites of differential allelic occupancy was unlikely to occur by random (P = 8 × 10−6) according to permutation tests that take into account potential biases resulting from antibody-specific variation in ChIP-seq signal strength and the average size of binding sites between different factors and between binding sites with and without differential allelic occupancy (see Supplemental Methods). When multiple TFs bound the same heterozygous SNP, they frequently resided on the same allele, as indicated by positive correlations between allelic occupancy at co-bound SNPs (Fig. 1B; Supplemental Figs. 6, 7). On the contrary, in no case did we observe pairs of TFs that regularly bound the same position on alternate autosomes. In some cases, the factors may bind together in heteromeric complexes. For example, occupancy of the transcriptional co-activator EP300 correlated with that of many TFs. However, overall, we did not find evidence of known protein–protein interactions supporting our observed correlated occupancy (Persico et al. 2005). Instead, the TF hubs may either include novel TF–TF associations or may be a more general feature of the genomic landscape (MacArthur et al. 2009). Chromatin state may also play a role either in increasing TF occupancy at variants bound by multiple TFs, or in maintaining a state established by pioneer factors. In support of this hypothesis, the DNA near TF hubs had increased sensitivity to DNase I when compared with regions bound by a single factor (Fig. 1C). The result indicates either that these regions of open chromatin were more accessible to TFs before binding, or that the recruitment of many TFs to these regions resulted in more extensive and stable chromatin remodeling. The co-occupied variants may also have particular functional significance, as they were more likely to be evolutionarily conserved than variants bound by a single factor (Fig. 1D). Together, the results reveal the existence of hubs of coordinated differential allelic gene regulation involving multiple TFs throughout the human genome.
Most differential allelic occupancy results from variation outside the DNA binding motif
To better understand the mechanisms underlying differential allelic occupancy, we investigated the genetic contributions to allelic occupancy. Kasowski and colleagues previously found that variation of NFKB binding between different individuals significantly associated with disruption of the NFKB binding motif (Kasowski et al. 2010), and others have suggested a similar relationship may be found for differential allelic occupancy (McDaniell et al. 2010). We therefore sought to determine generally across many TFs how often differences in the primary TF binding site correspond to differential allelic occupancy. We first evaluated the location of heterozygous SNPs in autosomal TF binding sites. We found that, after controlling for biases in read coverage and variant density, differentially occupied sites were strongly enriched for heterozygous SNPs within 50 bp of the position of maximal ChIP signal (Fig. 2A), indicating that they may be the most functionally important nucleotides. We then compared the rate at which heterozygous SNPs occurred at motif versus non-motif intergenic positions (Supplemental Table 5), a ratio we designate dM/dI. Generally across all factors and limited to autosomes, we found that heterozygous variants in motifs were nearly three times more likely to occur in differentially bound sites (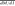 = 2.47) than in equally bound sites (
= 2.47) than in equally bound sites (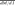 = 0.80) (Fig. 2B). Compared with an estimated background rate calculated from randomly chosen 5-kb promoter regions (
= 0.80) (Fig. 2B). Compared with an estimated background rate calculated from randomly chosen 5-kb promoter regions (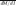 = 0.98), we found motif-disrupting mutations were significantly enriched in differentially bound regions and significantly depleted in equally bound regions (P < 1 × 10−100 for both cases; see Methods). As expected and consistent with reports of inter-individual variation of NFKB binding (Kasowski et al. 2010), the bound alleles were overall more similar to the consensus motif than the unbound alleles (Supplemental Fig. 8). Differential allelic occupancy ranged from subtle to absolute. Binding sites with the greatest allelic difference in occupancy corresponded to the presence of a canonical binding motif and to mutation of that motif (Fig. 2C,D). However, variants in known binding motifs explained only ∼12% of instances of differential allelic occupancy. While the exact percentage is dependent on many factors, it appears that the minority of differential allelic occupancy can be attributed to mutation of a canonical TF binding motif. Instead, our results suggest that there are different regimes of variation in TF binding. At the minority of differentially occupied binding sites, mutation of a canonical binding motif drives strong allelic differences in TF occupancy. Meanwhile, at the majority of differentially occupied sites, TFs bind DNA at weak or noncanonical binding motifs. In such cases, smaller differences in occupancy occur, perhaps via genetic disruption of a cofactor binding site or differences in chromatin structure (McDaniell et al. 2010; Gertz et al. 2011)
= 0.98), we found motif-disrupting mutations were significantly enriched in differentially bound regions and significantly depleted in equally bound regions (P < 1 × 10−100 for both cases; see Methods). As expected and consistent with reports of inter-individual variation of NFKB binding (Kasowski et al. 2010), the bound alleles were overall more similar to the consensus motif than the unbound alleles (Supplemental Fig. 8). Differential allelic occupancy ranged from subtle to absolute. Binding sites with the greatest allelic difference in occupancy corresponded to the presence of a canonical binding motif and to mutation of that motif (Fig. 2C,D). However, variants in known binding motifs explained only ∼12% of instances of differential allelic occupancy. While the exact percentage is dependent on many factors, it appears that the minority of differential allelic occupancy can be attributed to mutation of a canonical TF binding motif. Instead, our results suggest that there are different regimes of variation in TF binding. At the minority of differentially occupied binding sites, mutation of a canonical binding motif drives strong allelic differences in TF occupancy. Meanwhile, at the majority of differentially occupied sites, TFs bind DNA at weak or noncanonical binding motifs. In such cases, smaller differences in occupancy occur, perhaps via genetic disruption of a cofactor binding site or differences in chromatin structure (McDaniell et al. 2010; Gertz et al. 2011)
(A) Histogram of the distance of heterozygous SNPs from the location of maximal ChIP-seq signal for sites with (orange) and without (blue) differential allelic TF occupancy. To control for potential observation biases resulting from high read coverage at variants near the center of binding sites, the sites of equal allelic occupancy were chosen to match the differential allelic occupancy in two ways. First, for each site of differential allelic occupancy, we required the total number of aligned reads covering heterozygous variants in the matched site to be equivalent. Second, we required that the total number of variants in each binding site was also equivalent. If a suitably matched site did not exist, the site was excluded from the sites of differential allelic occupancy for this analysis. Using this strategy, the distribution of aligned reads at heterozygous variants was not significantly different between the sites of differential allelic occupancy and the matched set of equal allelic occupancy (P = 0.15, two-sided Wilcoxon rank-sum test). (B) The ratio of the rate of motif-disrupting to non-motif-disrupting intergenic mutations (dM/dI) across all sited of differential allelic TF occupancy (orange), and at TF binding sites that lack significant differential allelic occupancy (blue). To allow comparison with cis-regulatory DNA, the distribution of dM/dI is also shown for regions 5 kbp upstream of 10,000 randomly chosen TSSs (white). Whiskers show 95% confidence intervals. We excluded TFs for which we only observed a single motif-disrupting variant across all binding sites. (C) For the bound (black) or unbound (gray) allele at all sites of differential allelic occupancy, the similarity to TF binding motif (as a fraction of the optimal match) at sites of heterozygosity (y-axis) plotted against relative binding (the ratio of reads aligning to the bound vs. unbound allele; x-axis). Data were smoothed over a 32-data-point sliding window. The shaded region labeled Δ indicates the amount of difference in motif similarity between bound and unbound alleles, and is plotted in panel D.
RNA Pol2 occupancy predicts differential allelic expression of genes with homozygous exons
To evaluate the effects of differential allelic occupancy on expression, we used ultrahigh-throughput mRNA sequencing (RNA-seq) (Mortazavi et al. 2008) to measure differential allelic gene expression across the human genome (Pant et al. 2006; Gimelbrant et al. 2007; Zhang et al. 2009). To avoid biases from mapping to the reference genome (Degner et al. 2009; Pickrell et al. 2010), we assembled complete paternal and maternal GM12878-specific versions of all RefSeq transcripts. We then sequenced the transcriptome and aligned the reads to the parental transcripts (Fig. 3A; Supplemental Table 6). We identified significant (FDR < 5%) differential allelic expression for 381 (9%) of the 4194 expressed RefSeq transcripts with heterozygous variants in exonic regions (Fig. 3B). The results were reproducible between biological replicates (r2 = 0.88, P < 2 × 10−27) (Supplemental Fig. 9), and validation with Sanger sequencing reproduced results from six of six tested genes (Supplemental Fig. 10; Gertz et al. 2011). Differences in allelic expression were often subtle: 166 (52%) of the 322 autosomal genes identified had less than a twofold difference in expression between alleles. Known imprinted genes (Morison et al. 2005; Pollard et al. 2008) and X-linked genes were the exception, nearly all of which had a greater than twofold allelic expression difference. Most X-chromosomal genes were transcribed from the maternal copy (Supplemental Figs. 11, 12), as expected, given the paternal X inactivation bias in GM12878 cells (McDaniell et al. 2010; Kucera et al. 2011). We also identified differential allelic expression of seven long noncoding RNAs (Supplemental Fig. 13). Monoallelic expression of XIST (Brown et al. 1991) and KCNQ1OT1 (Weksberg et al. 2003; Nagano and Fraser 2009) is necessary for silencing gene expression on the opposite alleles, and it remains to be seen if any of the additional five that we identified have a similar function (Mohammad et al. 2009; Malecova and Morris 2010).
(A) Diagram of our method for using RNA-seq to measure differential allelic expression. First, poly(A)+ RNA was isolated using magnetic beads conjugated to oligo(dT) nucleotides. After RNA fragmentation, dsDNA was synthesized and subjected to paired-end sequencing on an Illumina Genome Analyzer. Reads were then aligned to GM12878-specific maternal and paternal versions of all RefSeq transcripts. Differential allelic expression was called when significantly more reads aligned to a single allele than would be expected by random. (B) Distribution of the fraction of maternal expression for all heterozygous genes (black), autosomal genes with differential allelic expression (orange), and X-chromosomal genes with differential allelic expression (white). (C) Prediction of differential allelic expression (y-axis) along the X chromosome (x-axis) using allelic occupancy of RNA Pol2. (Black lines) Significant differential allelic RNA Pol2 occupancy; (gray lines) nonsignificant binding. The shaded region on the left indicates the pseudoautosomal region that is not inactivated. All significant differential allelic occupancy predicted expression as expected. Genes that do not achieve statistical significance in the inactivated region of the X were a mix of genes that are known to escape inactivation as well as false negatives.
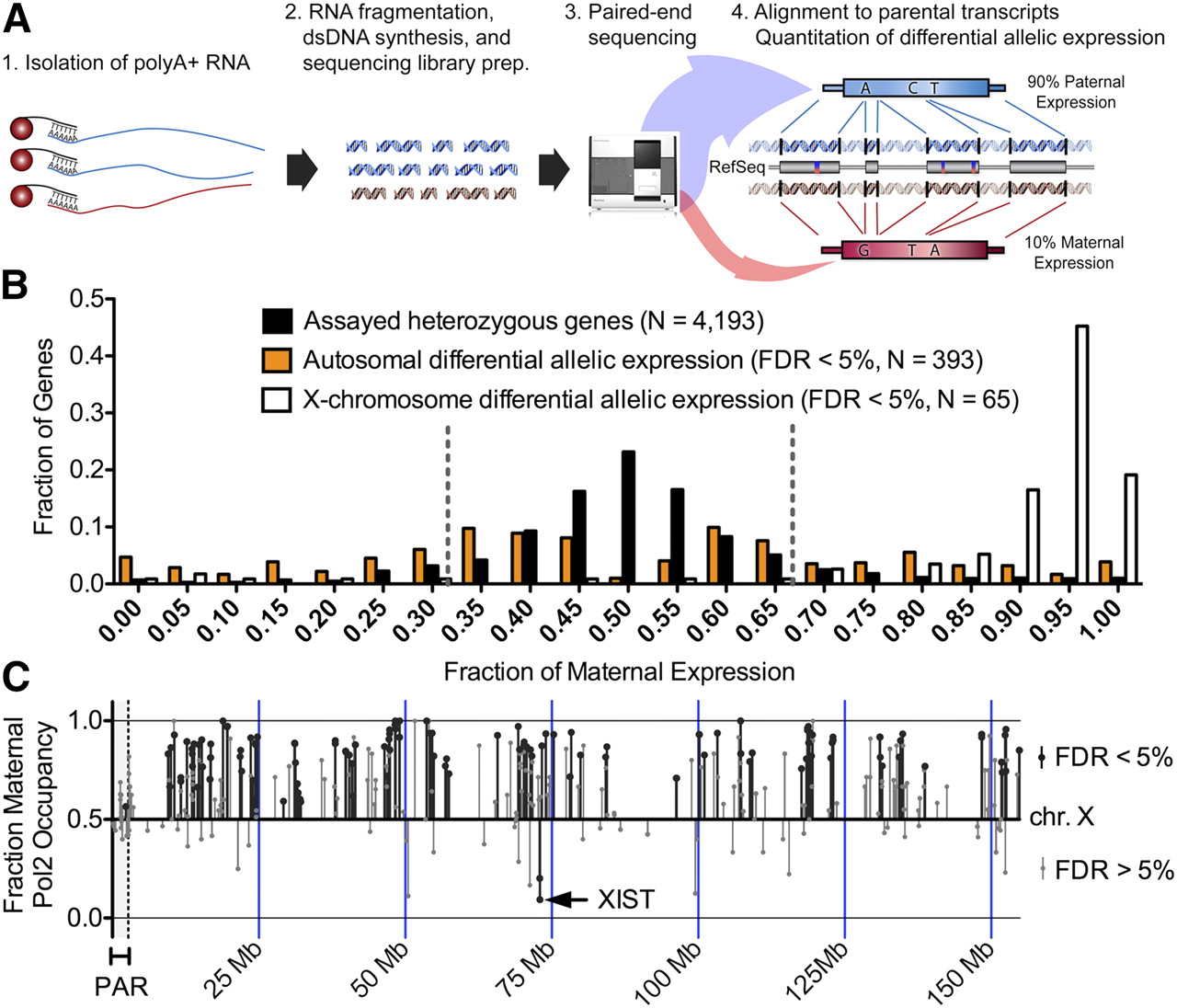
Allelically imbalanced gene regulation likely results from regulatory sequences that are not in exons, and therefore both heterozygous and homozygous genes may have differential allelic expression. However, measurement of differential allelic expression with RNA-seq is limited to genes with heterozygous exonic sequences, which represents only 39% of the transcripts in GM12878. Chromatin immunoprecipitation of RNA Pol2 isolates DNA from both exons and introns, enabling genome-wide prediction of differential allelic expression of genes with homozygous exons but heterozygous introns (Knight et al. 2003). Aggregating allelic RNA Pol2 ChIP-seq signals across gene bodies, we predicted differential allelic expression for 654 (6.3%) of the 10,353 genes with sufficient coverage of RNA Pol2 at heterozygous variants. The genes included 456 autosomal that lacked exonic heterozygous variants and could not be evaluated with RNA-seq. When we found significant differential allelic expression of X-linked genes, we predicted expression from the expected allele giving us perfect specificity (Fig. 3C). However, not all X-linked genes reached our significance threshold, some of which may escape inactivation. Comparing to a chromosome-wide study of genes subject to or escaping from X inactivation (Carrel and Willard 2005), we estimated that our analysis of RNA Pol2 occupancy achieves 66% sensitivity in predicting X inactivation or escape. Given the perfect specificity, relaxing our significance criteria combined with deeper sequencing may improve the sensitivity. However, for the purposes of this study, we were more concerned with ensuring a high true positive rate. As a further positive control, we measured differential allelic expression and RNA Pol2 occupancy in complementary clonal isolates of GM12878 with paternal or maternal X chromosomes inactivated. For both RNA-seq and RNA Pol2 occupancy, we predicted that >80% of genes with differential allelic expression were transcribed from the expected X chromosome in these clonal cell populations (Supplemental Figs. 14–16). On the autosomes, however, we see strong concordance in allelic expression among clonal isolates as well as with the original cell population (Supplemental Fig. 17). Searching for evidence of random monoallelic expression that could explain the observed differential allelic expression (Gimelbrant et al. 2007), we found that 13.5% of genes with differential allelic expression in one clone were either bi-allelic or expressed from the homologous chromosome in a different clone (Supplemental Table 7). While only a limited number of clones were studied, the result suggests that the minority of differential allelic expression results from random monoallelic expression. Across the autosomes, allelic differences in RNA Pol2 across the gene body positively predicted allelic differences in expression for 135 (92%) of the 146 genes that were also detected in RNA-seq (P = 1 × 10−27, Fisher's exact test). That variation in differential allelic RNA Pol2 occupancy significantly but imperfectly explains variation in gene expression (r2 = 0.48, P < 1 × 10−16) (Supplemental Fig. 18) may result both from technical noise in genome-wide measurements of allelic RNA Pol2 occupancy as well as from biological sources such as differential rates of transcriptional initiation or elongation, or by allelic differences in RNA stability. Combining evidence of differential allelic expression from RNA-seq and from RNA Pol2 ChIP-seq, we thus identified 910 genes with differential allelic expression in GM12878. The list of all genes with differential allelic expression is provided in Supplemental Materials.
Transcription factor occupancy is more directly inherited than gene expression
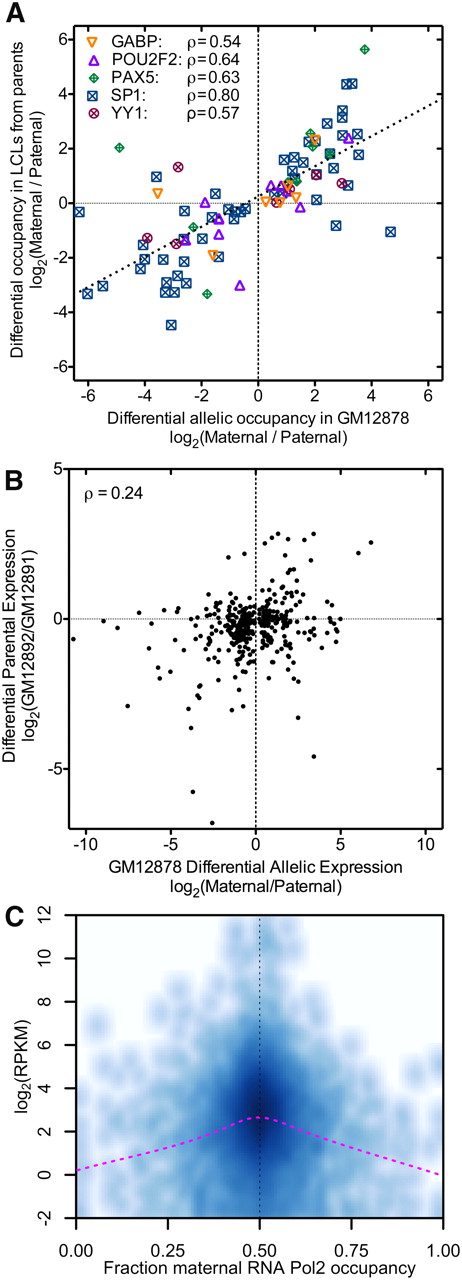
While differential allelic occupancy and expression are prevalent in an individual, understanding the extent to which these traits are inherited is critical to understanding how they contribute to heritable disease risk. To investigate, we measured genome-wide both the occupancy of five TFs (GABPA, POU2F2 a.k.a. OCT2, PAX5, SPI.1 a.k.a. PU.1, and YY1) and also gene expression in LCLs derived from both the mother and the father of the GM12878 donor. When a TF had differential allelic occupancy at a heterozygous autosomal variant in GM12878, and each parent was homozygous for one of the alleles, the allele with stronger binding in GM12878 had greater ChIP-seq signal in the corresponding parent in 81% of cases, significantly more often than previously reported for CTCF (McDaniell et al. 2010) (P = 1.5 × 10−5, binomial test). We also found that the extent of differential allelic occupancy in GM12878 strongly correlated with differential occupancy between the parental LCLs (Spearman's ρ = 0.75) (Fig. 4A). On the contrary, differential allelic expression of autosomal genes was less directly heritable than differential allelic occupancy (ρ = 0.24, P = 2.1 × 10−6) (Fig. 4B), with the more highly expressed allele in GM12878 having greater expression in the corresponding parental cell line for 60% of genes (P = 3 × 10−4; Fisher's exact test). The reduced heritability of expression likely reflects the integration of a complex mixture of regulatory contributions from both parents, acting both in cis and in trans, as well as epigenetic contributions. In comparison, individual TF binding sites appear to be more strongly determined by local sequence signals and less affected by the surrounding genomic milieu.
(A) Inheritance of allelic TF occupancy. The log-ratio of occupancy of the indicated TFs in the maternally versus paternally derived LCLs (y-axis) is plotted against the allelic occupancy of the same factors in GM12878 (x-axis). For each site plotted (N = 85), we required that both parents were homozygous for alternate alleles. Combining all points together, the overall correlation is ρ = 0.75, and for 88% of sites, the more bound allele in GM12878 was also more bound in the corresponding parent. (B) Similar to A, the log-ratio of expression from the parental LCLs plotted as a function of the allelic expression in GM12878. (C) Genes with differential allelic expression have overall lower expression in GM12878. For each gene with expression >0.25 RPKM, the gene expression (y-axis) is shown as a function of differential allelic RNA Pol2 occupancy (x-axis). (Darker shading) Greater density of values; (magenta line) less smoothing over the data.
Genes with differential allelic expression are expressed at lower levels in many human cell lines
To investigate the comparatively weak inheritance of gene expression, we first looked for evidence of mechanisms that compensate for allelic differences in the expression of autosomal genes. To do so, we used RNA Pol2 occupancy to identified genes with and without evidence of differential allelic expression, and used RNA-seq to compare expression between the two sets of genes. To control for potential biases due to sample size and RNA Pol2 coverage, for each gene with differential allelic expression we selected a matched gene with a similar amount of RNA Pol2 coverage at heterozygous positions (see Supplemental Methods). If allelic imbalances in autosomal gene expression were compensated, we would expect an overall similar level of expression between the two sets of genes. Contrary to this hypothesis, we found that genes with differential allelic expression have substantially and significantly lower expression than genes expressed equally from both alleles (Fig. 4C). The result is independent of the read coverage threshold, as we have reproduced the result at the RNA Pol2 ChIP-seq coverage threshold ranging from 25× to 120× (Supplemental Table 8). To see if the increased allelic variability of lowly expressed genes was specific to GM12878 cells, we measured gene expression of eight additional cell lines and found that the same genes were significantly less expressed in those cell lines as well (Supplemental Fig. 19). Therefore, it appears that genes with differential allelic occupancy generally have lower expression, perhaps due to fundamental differences in the cis-regulatory landscape surrounding these genes. With the exception of immunoglobulin genes and the protocadherin-gamma cluster, both known to exhibit monoallelic expression patterns (Kaneko et al. 2006), we did not find evidence that genes with differential allelic expression were enriched for particular classes or functions of proteins.
Transcription factor occupancy explains expression up to 100 kb from transcription start sites
One of the major advantages of studying differential allelic occupancy and expression is the potential to link intergenic variants implicated in diseases with functional changes in TF occupancy and gene expression. It is therefore important to know the extent to which allelic TF occupancy correlates with allelic gene expression, especially considering our finding that gene expression was weakly heritable. Overall, we found more TF and cofactor occupancy at variants associated with regulation of gene expression (Montgomery et al. 2010) than would be expected by random (see Supplemental Methods), strongly suggesting that the occupancy we measured does indeed impact gene expression. To investigate further, we evaluated the local cis-regulatory landscape of autosomal genes to determine if differential allelic TF occupancy occurred near genes with differential allelic expression. We found that differential allelic occupancy was significantly closer to genes with differential allelic expression than without (P = 5.0 × 10−15, Wilcoxon test comparing the distance to the nearest TSS of a gene with differential vs. equal allelic expression) (Fig. 5A). In contrast, binding sites with equal allelic occupancy were on average no closer to genes with imbalanced or balanced allelic expression (P = 0.21, two-sided Wilcoxon test) (Fig. 5B). The fact that differential allelic occupancy occurred closer to genes with differential allelic expression did not result from differences in the total number of observed binding sites, but instead from a greater fraction of the TF binding sites around genes with differential allelic expression having differential allelic occupancy. Specifically, 6.8% of sites within 100 kb of a TSS with differential allelic expression had differential allelic occupancy, compared to 3.9% of sites within 100 kb of a TSS without differential allelic expression (P < 1 × 10−20, Fisher's exact test). Finally, we did not observe a significant difference in the total number of binding sites in the same regions. The association between differential allelic occupancy and expression suggests we may be able to observe a functional relationship between the two.
(A) Cumulative distribution of the distance from the TSS (x-axis) to the nearest site of differential allelic occupancy for all autosomal genes with differential allelic (orange) or equal allelic (blue) expression. (Left) All genes with differential allelic expression, where the difference between the two distributions is highly significant. (Right) Genes with equal allelic expression, and there is no significant difference between the two distributions. (B) Spearman's correlation (y-axis) of allelic occupancy with allelic expression within the distance from autosomal TSSs indicated on the x-axis. For each point, we aggregated all allelic occupancy (both for sites with and without a significant allelic imbalance) at the indicated distance around all genes with significant differential allelic expression. Then, for every gene with at least a single site with a significant differential allelic occupancy, we calculate Spearman's correlation coefficient and plot. Detailed scatter plots are included in Supplemental Figure 20. (C) Differential allelic occupancy of multiple factors at variants either directly or through perfect linkage disequilibrium (R2 = 1; red dash) with celiac disease. Nearby, RMI2 (also known as C16orf75) is predominantly expressed from the maternal allele, and the regulatory interaction is supported by expression quantitative trait loci (eQTL) mapping. (D) Similar to C, allelic occupancy of EBF1 at a variant associated (via linkage disequilibrium) with psoriasis corresponds with differential allelic expression of COG6. Again, the regulatory interaction is supported by eQTL analysis.
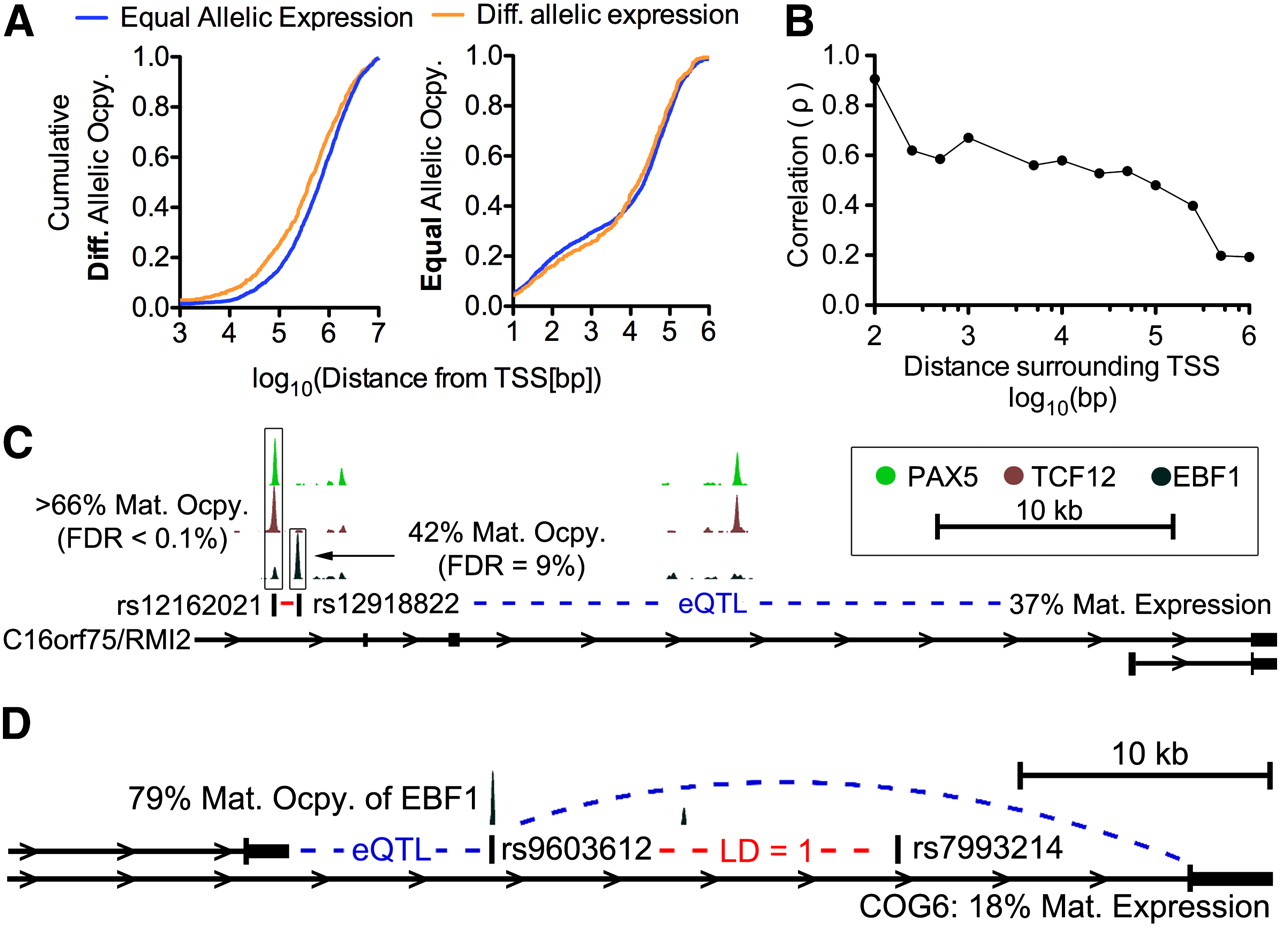
Limited to autosomal cases in which we found allelic imbalance both in occupancy and in expression, the ability of allelic occupancy to explain allelic expression depended on the proximity of binding to the transcription start site (TSS). In the few cases where we observed allelic occupancy within 100 bp of the TSS, we found strong positive correlation between allelic occupancy and expression from the same allele (ρ = 0.91, N = 13). Meanwhile, allelic occupancy at intervals between 1 and 100 kb from the TSS weakly explained expression (ρ = 0.45, N = 290). More than 100 kb from the TSS, differential allelic occupancy did not significantly explain expression (ρ = 0.06, N = 760) (Fig. 5B). The results show that differential allelic occupancy does indeed correspond to differential allelic expression, and may therefore give functional hypotheses to intergenic disease-associated variants. Notably, while the analysis included binding from all TFs and did not attempt to distinguish activating from repressive binding sites or factors, we observed an overall positive correlation. The result suggests either that the TFs chosen in the study are more commonly activating than repressing, or alternatively that activating sites are more amenable to detection by ChIP-seq.
Allelic variation in TF occupancy in GM12878 provides insights into autoimmune disease
The majority of genomic variants associated with disease using genome-wide association studies (GWAS) are intergenic and have unclear regulatory consequences. TF binding sites may give functional insights into the variants identified. Using our observations of TF binding and differential allelic occupancy, we investigated a compilation of disease-associated variants (Hindorff et al. 2009) for potential overlaps that suggest function. Overlap with differential allelic occupancy is particularly interesting because the variant may also explain the difference in TF occupancy between the two alleles. We found 155 unique autosomal variants that were either directly associated with disease, or that were in perfect linkage disequilibrium (R2 = 1) with a disease-associated variant, that also occurred in a heterozygous TF binding site. The overlap was unlikely to occur by random when compared to a set of variants matched on distance relative to a TSS and on minor allele frequency (Supplemental Table 9). Of those variants, we found 21 instances of disease-associated variants that occurred in a site of differential allelic occupancy. More than 75% of the disease-associated variants are associated with autoimmune diseases, including variants associated with multiple sclerosis, celiac disease, Type 1 diabetes, systemic lupus erythematosus, and psoriasis (Supplemental Table 10). The result is especially compelling considering that the functional differences are identified in a cell type relevant for immune modulation (B-cells), and is in agreement with recent findings of a study evaluating genome-wide chromatin states in the same cells (Ernst et al. 2011). As an example, we found a cluster of TFs including EBF1 and PAX5—two key factors in B-cell development—binding with a more than twofold preference to the maternal (protective) allele at variants in complete linkage disequilibrium with the celiac disease-associated variant rs12928822 (Dubois et al. 2010). The variants are found near isoforms of RMI2, a gene important for genomic stability. In our study, RMI2 also shows differential allelic expression, but from the opposite homolog. Furthermore, evidence from expression quantitative trait loci (eQTL) mapping (Dubois et al. 2010) substantiates the presence of a regulatory interaction between the variant and the RMI2 (Fig. 5C). In another example, we found differential allelic occupancy of EBF1 at the psoriasis-associated variant rs9603612 and expression of the nearby gene COG6, a gene involved in the structure of the Golgi apparatus, again from the opposite homolog (Fig. 5D; Liu et al. 2008). Again, eQTL linkage between the variant and COG6 supports the presence of a regulatory interaction (Zeller et al. 2010).
Discussion
Understanding the impact of genetic variation on gene regulation remains a major challenge in deciphering the human transcriptional regulatory code. To uncover functional noncoding variants we used ultra-high throughput sequencing to measure genome-wide gene expression and occupancy of RNA Pol2, of the transcriptional co-activator EP300, and of 24 sequence-specific TFs in the female LCL GM12878. By aligning sequence reads to versions of the reference human genome modified to include homozygous and heterozygous variants identified by the 1000 Genomes Project (The 1000 Genomes Project Consortium 2010), we measured allelic differences both in gene expression and in TF occupancy. In doing so, we have produced an extensive and detailed map of transcripts that show allelic bias in expression and alleles that impact TF binding.
Comparing genomic occupancy between multiple TFs, we found that hubs of TF occupancy occur frequently in the human genome: ∼15% of the TF binding sites in our study overlapping a binding site for another factor. An abundance of TF-binding hubs have also been found in fly (MacArthur et al. 2009) and may be a common feature of the cis-regulatory landscape in complex genomes. The hubs often exhibited a coordinated reaction to functional variants. In such cases, the co-occupying factors bound similarly to the same allele, suggesting a cooperative behavior at such sites. The overabundance of allelically imbalanced hubs also suggests that TF hubs are particularly sensitive to genetic variation, and that genetic polymorphism can destabilize occupancy across the entire hub as opposed to that of a single factor. We also found that the DNA in the most populated hubs had greater evolutionary conservation, suggesting they may play an important role as enhancers of distal gene regulation.
To link allelic TF occupancy to gene expression outcomes, we also characterized differential allelic gene expression across the genome. We used a combination of techniques to measure allelic gene expression. While RNA sequencing gives a direct measurement of allelic gene expression, we found that the majority of protein-coding genes have no heterozygous variants in their exons. Leveraging the ability of ChIP-seq to detect elongating RNA Pol2 at heterozygous variants in introns and to serve as a proxy for gene expression, we developed a complementary approach to measure genome-wide allelic expression of exonically homozygous genes. Our findings suggest that differential allelic expression is as common in genes with genetically identical transcripts as in genes with genetically different transcripts, and that the majority of differential allelic expression is therefore not detectable by comparing mRNA abundance. Comprehensively characterizing such cases of cryptic differential allelic expression may be important in better understanding haploinsufficiency-based disease by revealing many more instances of monoallelic gene expression than are currently known.
Looking across all genes with differential allelic expression, we found that such genes are more likely to be lowly expressed, even in unrelated cell lines. The finding may indicate a closer link between gene expression and evolutionary conservation than has previously been shown. The protein-coding sequences of highly expressed genes are in general more conserved than that of lowly expressed genes (Pal et al. 2001; Subramanian and Kumar 2004; Wall et al. 2005), and our findings suggest that the transcriptional regulation of highly expressed genes is also more conserved. Similarly, it has also been shown that genes with expression limited to specific tissues have less constrained protein coding sequence (Duret and Mouchiroud 2000), and we found evidence that genes with differential allelic expression are expressed in fewer tissues (Supplemental Fig. 21). It may be that the evolutionary pressures or other mechanisms of constraint introduced by increased and organism-wide expression act more broadly than protein coding sequence and also limit allelic variation in the regulation of the same genes.
With a more complete characterization of differential allelic expression, we were able to link allelic TF occupancy to these genes, showing that differential allelic occupancy is more prevalent near differential allelic expression. Ultimately, we found allelic occupancy within 100 bp of the TSS to be highly predictive of expression. However, while we detected significant associations between occupancy and expression up to 100 kb away from a TSS, the associations were comparatively very weak. The finding highlights the ongoing challenge of understanding the extent to which distal cis-regulatory elements contribute to expression, and may underlie the weak penetrance that genetic variation at many intergenic variants has in genome-wide association studies. It is also important to note that, while many factors are known to act both as an activator and a repressor, we did not observe any systematic inverse relationships between allelic TF occupancy and expression. The result may be explained by studies in inducible systems that have found the repressive activity of TFs to be predominantly associated with occupancy distal to the TSS (e.g., Cheng et al. 2009; Reddy et al. 2009).
Targeted exon sequencing is becoming a common tool for identifying rare coding variants that may be associated with disease. From genome wide association studies it is clear that many regulatory variants are also associated with disease, but due to their predominantly intronic or intergenic location (Hindorff et al. 2009) as well as the complex nature of cis-regulation, such variants are more difficult to functionally interpret. The compendium of functional noncoding variants we have identified provide a resource for identifying noncoding polymorphisms that are likely to have an effect on genomic function, suggesting a compromise between GWAS and exon sequencing. By using a capture approach that includes functional intergenic regions in addition to exons, targeted sequencing can explore a greater fraction of the potentially functional genome while limiting the number of hypotheses being tested. By expanding exon sequencing to include targeted regulatory regions, it may therefore be possible to identify rare intergenic variants that are significantly associated with disease. Meanwhile, the prior knowledge of particular TFs bound in each region provides a mechanistic hypothesis to investigate in more detail, overcoming another of the major challenges in existing association studies (Freedman et al. 2011). That many of the functional variants identified in this study overlap with previously identified disease associated SNPs provides hope that augmenting disease studies with targeted sequencing of functional regulatory variation will ultimately be a successful strategy.
Methods
Cell growth
Biological replicates of GM12878, GM12891, and GM12892 cells were grown in RPMI 1640 media with 2 mM L-glutamine, 15% fetal bovine serum, and 1% penicillin-streptomycin at 37°C under 5% carbon dioxide.
ChIP-seq
We performed ChIP experiments and prepared the immunoprecipitated DNA for sequencing on an Illumina Genome Analyzer as described (Johnson et al. 2007). We selected factors to include both ubiquitous TFs and cofactors (e.g., SP1 and EP300), and factors specific to the development of B-cells (e.g., POU2F2, SPI1, PBX3, BCL3, and EBF1). Antibodies used are listed in Supplemental Table 1. For each factor, we produced ≥12 million 36 nucleotide reads per biological replicate. We aligned reads to the GM12878-specific reference genome using Bowtie (Langmead et al. 2009) with options “-n 2 -l 36 -k 1–best”, and removed alignments mismatching at any heterozygous SNP. To avoid potential biases resulting from amplification artifacts, we collapse all sequences identified multiple times to a single instance. To define binding regions, we used QuEST (Valouev et al. 2008) with “stringent peak calling parameters”. For each binding region, we estimated the fraction of maternal (paternal) occupancy as the fraction of mini-contig alignments that mapped to the maternal (paternal) chromosome.
For RNA Pol2, we produced 64 million additional paired-end 100-bp reads by using a similar protocol and the Illumina HiSeq 2000 sequencer. We aligned each end independently against the GM12878-specific reference genome using Bowtie (Langmead et al. 2009) with options “–best–strata -n 2 -m 10 -k 1”, and excluded alignments that mismatched at any heterozygous SNP. We predicted the fraction of maternal expression as the fraction of mini-contig alignments across each RefSeq gene that mapped to the maternal allele. To ensure stringency, we only considered genes with reads aligning to at least three heterozygous SNPs.
RNA-seq
Paired-end RNA-seq experiments were performed in biological replicate as described previously (Trapnell et al. 2010). Replicate one and two were sequenced to a depth of 44 and 25 million paired-end 75-bp reads, respectively. We aligned reads to the reference transcriptome using Bowtie (Langmead et al. 2009) with parameters “-a–best–strata” and default paired-end settings. The parameters were chosen to allow alignment to multiple isoforms. We then removed any alignments that resulted in mismatches at heterozygous SNPs. Finally, we aligned RNA-seq reads to the reference transcriptome, and estimated the fraction of expression from the maternal (paternal) chromosome as the fraction of reads mapping to a heterozygous SNP that contain the maternal (paternal) allele.
Sequence alignment and determination of differential allelic occupancy and expression
To measure differential allelic occupancy, we constructed a GM12878-specific reference genome that allowed concurrent alignment to both the maternal and paternal genome as suggested by Degner et al. (2009). Maternal and paternal genome sequences were determined using variants in the March 2010 data release by the 1000 Genomes Project (The 1000 Genomes Project Consortium 2010). To construct the maternal and paternal genomes, we first altered homozygous SNPs in the hg18 reference genome to match the GM12878 genotype. Then, for each heterozygous SNP with discernable parent-of-origin, we replaced the SNP and the flanking 35 bp (for a 36-bp read length) with a paternal and a maternal version of the sequence. We then combined overlapping sequences such that any read aligning to a parental sequence will overlap a heterozygous SNP and vice versa. For RNA Pol2, we used RefSeq genes instead of peak calls, and only considered genes with reads aligning to at least three heterozygous SNPs.
To measure differential allelic expression, we aligned RNA-seq reads to a GM12878-specific reference transcriptome that included both maternal and paternal versions of all transcripts with a heterozygous variant in an exon. To do so, we first assembled sequences for all RefSeq transcripts from the hg18 reference human genome. We then corrected all homozygous SNPs to match the sequence of GM12878. Then, we created a paternal and maternal version of each transcript with a heterozygous exon by changing heterozygous nucleotides to match the parental chromosome, if known.
We performed a number of additional filtering steps to remove false positives. First, to remove artifacts due to incorrect genome sequence and copy number variation, we removed from analysis variants with a substantial allelic bias in sequencing of input control DNA (i.e., DNA from chromatin that was cross-linked and sonicated, but not immunoprecipitated). We also removed variant calls that were discordant with sequencing of the GM12878 genome as performed by Complete Genomics (Drmanac et al. 2010). Next, we filtered reads that aligned to positions in the genome for which either the maternal or paternal sequence were not unique and could have therefore arisen from a different location, as sequences aligning to such positions are inherently biased to a single allele (Degner et al. 2009). To do so, we simulated every possible 36-bp read that would overlap a heterozygous variant. We then aligned all such reads to the maternal and paternal genomes, and noted every genomic position that did not have a unique 36-bp alignment for either the maternal or paternal version (i.e., reads for which the maternal or paternal variant could also align elsewhere in the genome, or could originate from elsewhere in the genome). The additional screening step reduced the number of sites of differential allelic occupancy by 1.5%. Lastly, we removed 10 (<0.05% of total) SNPs that overlapped regions of aneuploidy as measured by microarray experiments (Supplemental Table 11).
To determine statistical significance of differential allelic expression or occupancy, we used a binomial test against the null hypothesis that an equal number of reads maps to each chromosome. For all statistical testing, we require a 7× coverage threshold because it is the minimum number of reads required to achieve significance with a binomial test. We corrected for multiple hypotheses using the method of Benjamini and Hochberg (Benjamini and Hochberg 1995) implemented in the R statistical package.
Identification of differential allelic occupancy at disease-associated variants
Disease-associated variants were obtained from the National Human Genome Research Institute's Catalog of Published Genome-Wide Association Studies on April 19, 2011. We then expanded the list to include all variants known to be in perfect linkage disequilibrium (R2 = 1) in individuals of central European ancestry according to the HapMap project. Comparing the list with resequencing of the GM12878 genome, we identified all disease-associated variants that are heterozygous in GM12878. Finally, we identified all such variants that also had significant differential allelic occupancy by one or more TFs at the same SNP.
To determine if the overlap with TF occupancy was greater than expected by random, we used a permutation approach. To do so, we randomly assigned disease association among the phased (i.e., where the inheritance of each allele is unambiguous) heterozygous variants in GM12878, controlling for observation biases in GWAS studies in three ways: (i) maintaining a matched distribution of minor allele frequencies (with 5% absolute value difference), (ii) maintaining a matched distance to the TSS of the nearest RefSeq gene (with 1 kb), and (iii) maintaining both similar minor allele frequency (within 10% absolute value difference) and similar distance to the nearest RefSeq TSSs (within 2 kb). For the third group, we used relaxed stringency in order to assure that we could find enough matched sets. For (i) and (ii), we performed 1000 random sets and for (iii) we used 150 random sets. We then count the number of unique variants that overlap TF binding from our study, and describe the resulting distribution in Supplemental Table 9.
Data access
All ChIP-seq and RNA-seq data are publicly available from the ENCODE repository on the UCSC Genome Browser. Details of accession numbers can be found in Supplemental Tables 12 and 13. In addition, processed data specific to our study including allele-specific alignments, aggregation over variants, binding site calls, and aggregation of allelic alignments over those called binding sites are available online at http://hudsonalpha.org/sites/default/files/DataSets/Myerslab/Differential_allelic_occupancy_and_expression.
Acknowledgments
We thank Chris Gunter, Greg Cooper, and the members of the Myers lab for contributions and suggestions. This work was funded by NHGRI ENCODE Grant 5U54HG004576 to R.M.M. and B.W. Support for T.E.R. was from NIH/NIAMS fellowship 5T32AR007450.
Authors' contributions: T.E.R., J.G., K.E.V., H.F.W., and R.M.M. conceived and designed the study, T.E.R. performed and interpreted the analysis, and wrote the manuscript. J.G. carried out the cloning-based validation of the RNA-seq experiments. F.P. and K.M.N. carried out the ChIP-seq experiments and RNA-seq experiments for the clonal isolates of GM12878. L.S. and G.E.C. performed and contributed to the interpretation of the DNase I hypersensitivity experiments. K.S.K. and H.F.W. designed and created the clonal GM12878 isolates, including determining the X inactivation state. G.K.M., A.M., B.A.W., and B.W. designed and performed the RNA-seq experiments. All authors contributed to the editing of the manuscript.
References
- ↵The 1000 Genomes Project Consortium. 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073.
- ↵Y Benjamini, Y Hochberg. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300.
- ↵CJ Brown, A Ballabio, JL Rupert, RG Lafreniere, M Grompe, R Tonlorenzi, HF Willard. 1991. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 349: 38–44.
- ↵L Carrel, HF Willard. 2005. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434: 400–404.
- ↵Y Cheng, W Wu, SA Kumar, D Yu, W Deng, T Tripic, DC King, KB Chen, Y Zhang, D Drautz, . 2009. Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression. Genome Res 19: 2172–2184.
- ↵JF Degner, JC Marioni, AA Pai, JK Pickrell, E Nkadori, Y Gilad, JK Pritchard. 2009. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 25: 3207–3212.
- ↵R Drmanac, AB Sparks, MJ Callow, AL Halpern, NL Burns, BG Kermani, P Carnevali, I Nazarenko, GB Nilsen, G Yeung, . 2010. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327: 78–81.
- ↵PC Dubois, G Trynka, L Franke, KA Hunt, J Romanos, A Curtotti, A Zhernakova, GA Heap, R Adany, A Aromaa, . 2010. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet 42: 295–302.
- ↵L Duret, D Mouchiroud. 2000. Determinants of substitution rates in mammalian genes: expression pattern affects selection intensity but not mutation rate. Mol Biol Evol 17: 68–74.
- ↵J Ernst, P Kheradpour, TS Mikkelsen, N Shoresh, LD Ward, CB Epstein, X Zhang, L Wang, R Issner, M Coyne, . 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49.
- ↵ML Freedman, AN Monteiro, SA Gayther, GA Coetzee, A Risch, C Plass, G Casey, M De Biasi, C Carlson, D Duggan, . 2011. Principles for the post-GWAS functional characterization of cancer risk loci. Nat Genet 43: 513–518.
- ↵J Gertz, KE Varley, TE Reddy, KM Bowling, F Pauli, SL Parker, KS Kucera, HF Willard, RM Myers. 2011. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet 7: e1002228. doi: 10.1371/journal.pgen.1002228.
- ↵A Gimelbrant, JN Hutchinson, BR Thompson, A Chess. 2007. Widespread monoallelic expression on human autosomes. Science 318: 1136–1140.
- ↵LA Hindorff, P Sethupathy, HA Junkins, EM Ramos, JP Mehta, FS Collins, TA Manolio. 2009. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci 106: 9362–9367.
- ↵DS Johnson, A Mortazavi, RM Myers, B Wold. 2007. Genome-wide mapping of in vivo protein-DNA interactions. Science 316: 1497–1502.
- ↵R Kaneko, H Kato, Y Kawamura, S Esumi, T Hirayama, T Hirabayashi, T Yagi. 2006. Allelic gene regulation of Pcdh-α and Pcdh-γ clusters involving both monoallelic and biallelic expression in single Purkinje cells. J Biol Chem 281: 30551–30560.
- ↵M Kasowski, F Grubert, C Heffelfinger, M Hariharan, A Asabere, SM Waszak, L Habegger, J Rozowsky, M Shi, AE Urban, . 2010. Variation in transcription factor binding among humans. Science 328: 232–235.
- ↵JC Knight, BJ Keating, KA Rockett, DP Kwiatkowski. 2003. In vivo characterization of regulatory polymorphisms by allele-specific quantification of RNA polymerase loading. Nat Genet 33: 469–475.
- ↵KS Kucera, TE Reddy, F Pauli, J Gertz, JE Logan, RM Myers, HF Willard. 2011. Allele-specific distribution of RNA polymerase II on female X chromosomes. Hum Mol Genet 20: 3964–3973.
- ↵B Langmead, C Trapnell, M Pop, SL Salzberg. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. doi: 10.1186/gb-2009-10-3-r25.
- ↵Y Liu, C Helms, W Liao, LC Zaba, S Duan, J Gardner, C Wise, A Miner, MJ Malloy, CR Pullinger, . 2008. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet 4: e1000041. doi: 10.1371/journal.pgen.1000041.
- ↵S MacArthur, XY Li, J Li, JB Brown, HC Chu, L Zeng, BP Grondona, A Hechmer, L Simirenko, SV Keranen, . 2009. Developmental roles of 21 Drosophila transcription factors are determined by quantitative differences in binding to an overlapping set of thousands of genomic regions. Genome Biol 10: R80. doi: 10.1186/gb-2009-10-7-r80.
- ↵BJ Main, RD Bickel, LM McIntyre, RM Graze, PP Calabrese, SV Nuzhdin. 2009. Allele-specific expression assays using Solexa. BMC Genomics 10: 422. doi: 10.1186/1471-2164-10-422.
- ↵B Malecova, KV Morris. 2010. Transcriptional gene silencing through epigenetic changes mediated by non-coding RNAs. Curr Opin Mol Ther 12: 214–222.
- ↵R McDaniell, BK Lee, L Song, Z Liu, AP Boyle, MR Erdos, LJ Scott, MA Morken, KS Kucera, A Battenhouse, . 2010. Heritable individual-specific and allele-specific chromatin signatures in humans. Science 328: 235–239.
- ↵CJ McManus, JD Coolon, MO Duff, J Eipper-Mains, BR Graveley, PJ Wittkopp. 2010. Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Res 20: 816–825.
- ↵M Meyerson, S Gabriel, G Getz. 2010. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet 11: 685–696.
- ↵F Mohammad, T Mondal, C Kanduri. 2009. Epigenetics of imprinted long noncoding RNAs. Epigenetics 4: 277–286.
- ↵SB Montgomery, M Sammeth, M Gutierrez-Arcelus, RP Lach, C Ingle, J Nisbett, R Guigo, ET Dermitzakis. 2010. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature 464: 773–777.
- ↵IM Morison, JP Ramsay, HG Spencer. 2005. A census of mammalian imprinting. Trends Genet 21: 457–465.
- ↵A Mortazavi, BA Williams, K McCue, L Schaeffer, B Wold. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628.
- ↵T Nagano, P Fraser. 2009. Emerging similarities in epigenetic gene silencing by long noncoding RNAs. Mamm Genome 20: 557–562.
- ↵C Pal, B Papp, LD Hurst. 2001. Highly expressed genes in yeast evolve slowly. Genetics 158: 927–931.
- ↵PV Pant, H Tao, EJ Beilharz, DG Ballinger, DR Cox, KA Frazer. 2006. Analysis of allelic differential expression in human white blood cells. Genome Res 16: 331–339.
- ↵M Persico, A Ceol, C Gavrila, R Hoffmann, A Florio, G Cesareni. 2005. HomoMINT: an inferred human network based on orthology mapping of protein interactions discovered in model organisms. BMC Bioinformatics (Suppl 4) 6: S21. doi: 10.1186/1471-2105-6-S4-S21.
- ↵JK Pickrell, JC Marioni, AA Pai, JF Degner, BE Engelhardt, E Nkadori, JB Veyrieras, M Stephens, Y Gilad, JK Pritchard. 2010. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464: 768–772.
- ↵KS Pollard, D Serre, X Wang, H Tao, E Grundberg, TJ Hudson, AG Clark, K Frazer. 2008. A genome-wide approach to identifying novel-imprinted genes. Hum Genet 122: 625–634.
- ↵TE Reddy, F Pauli, RO Sprouse, NF Neff, KM Newberry, MJ Garabedian, RM Myers. 2009. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res 19: 2163–2171.
- ↵G Robertson, M Hirst, M Bainbridge, M Bilenky, Y Zhao, T Zeng, G Euskirchen, B Bernier, R Varhol, A Delaney, . 2007. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods 4: 651–657.
- ↵D Serre, S Gurd, B Ge, R Sladek, D Sinnett, E Harmsen, M Bibikova, E Chudin, DL Barker, T Dickinson, . 2008. Differential allelic expression in the human genome: a robust approach to identify genetic and epigenetic cis-acting mechanisms regulating gene expression. PLoS Genet 4: e1000006. doi: 10.1371/journal.pgen.1000006.
- ↵S Subramanian, S Kumar. 2004. Gene expression intensity shapes evolutionary rates of the proteins encoded by the vertebrate genome. Genetics 168: 373–381.
- ↵JK Teer, JC Mullikin. 2010. Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet 19: R145–R151.
- ↵M Tompa, N Li, TL Bailey, GM Church, B De Moor, E Eskin, AV Favorov, MC Frith, Y Fu, WJ Kent, . 2005. Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol 23: 137–144.
- ↵C Trapnell, BA Williams, G Pertea, A Mortazavi, G Kwan, MJ van Baren, SL Salzberg, BJ Wold, L Pachter. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515.
- ↵A Valouev, DS Johnson, A Sundquist, C Medina, E Anton, S Batzoglou, RM Myers, A Sidow. 2008. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat Methods 5: 829–834.
- ↵DP Wall, AE Hirsh, HB Fraser, J Kumm, G Giaever, MB Eisen, MW Feldman. 2005. Functional genomic analysis of the rates of protein evolution. Proc Natl Acad Sci 102: 5483–5488.
- ↵R Weksberg, AC Smith, J Squire, P Sadowski. 2003. Beckwith-Wiedemann syndrome demonstrates a role for epigenetic control of normal development. Hum Mol Genet 12: R61–R68.
- ↵H Yan, W Yuan, VE Velculescu, B Vogelstein, KW Kinzler. 2002. Allelic variation in human gene expression. Science 297: 1143.
- ↵T Zeller, P Wild, S Szymczak, M Rotival, A Schillert, R Castagne, S Maouche, M Germain, K Lackner, H Rossmann, . 2010. Genetics and beyond—the transcriptome of human monocytes and disease susceptibility. PLoS ONE 5: e10693. doi: 10.1371/journal.pone.0010693.
- ↵X Zhang, JO Borevitz. 2009. Global analysis of allele-specific expression in Arabidopsis thaliana. Genetics 182: 943–954.
- ↵K Zhang, JB Li, Y Gao, D Egli, B Xie, J Deng, Z Li, JH Lee, J Aach, EM Leproust, . 2009. Digital RNA allelotyping reveals tissue-specific and allele-specific gene expression in human. Nat Methods 6: 613–618.
Notes
[1] [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.131201.111.