
High-throughput semiquantitative analysis of insertional mutations in heterogeneous tumors
- Marco J. Koudijs1,6,9,
- Christiaan Klijn1,9,
- Louise van der Weyden2,
- Jaap Kool3,7,
- Jelle ten Hoeve1,
- Daoud Sie4,8,
- Pramudita R. Prasetyanti1,
- Eva Schut1,
- Sjors Kas1,
- Theodore Whipp2,
- Edwin Cuppen5,
- Lodewyk Wessels1,10,
- David J. Adams2,10 and
- Jos Jonkers1,10
- 1Division of Molecular Biology and Cancer Systems Biology Center, Netherlands Cancer Institute, 1066 CX Amsterdam, The Netherlands;
- 2Experimental Cancer Genetics, The Wellcome Trust Sanger Institute, Hinxton, Cambridge CB10 1SA, United Kingdom;
- 3Division of Molecular Genetics, Netherlands Cancer Institute, 1066 CX Amsterdam, The Netherlands;
- 4Central Microarray Facility, Netherlands Cancer Institute, 1066 CX Amsterdam, The Netherlands;
- 5Hubrecht Institute and University Medical Center Utrecht, Cancer Genomics Center, 3584 CG Utrecht, The Netherlands
-
↵9 These authors contributed equally to this work.
Abstract
Retroviral and transposon-based insertional mutagenesis (IM) screens are widely used for cancer gene discovery in mice. Exploiting the full potential of IM screens requires methods for high-throughput sequencing and mapping of transposon and retroviral insertion sites. Current protocols are based on ligation-mediated PCR amplification of junction fragments from restriction endonuclease-digested genomic DNA, resulting in amplification biases due to uneven genomic distribution of restriction enzyme recognition sites. Consequently, sequence coverage cannot be used to assess the clonality of individual insertions. We have developed a novel method, called shear-splink, for the semiquantitative high-throughput analysis of insertional mutations. Shear-splink employs random fragmentation of genomic DNA, which reduces unwanted amplification biases. Additionally, shear-splink enables us to assess clonality of individual insertions by determining the number of unique ligation points (LPs) between the adapter and genomic DNA. This parameter serves as a semiquantitative measure of the relative clonality of individual insertions within heterogeneous tumors. Mixing experiments with clonal cell lines derived from mouse mammary tumor virus (MMTV)-induced tumors showed that shear-splink enables the semiquantitative assessment of the clonality of MMTV insertions. Further, shear-splink analysis of 16 MMTV- and 127 Sleeping Beauty (SB)–induced tumors showed enrichment for cancer-relevant insertions by exclusion of irrelevant background insertions marked by single LPs, thereby facilitating the discovery of candidate cancer genes. To fully exploit the use of the shear-splink method, we set up the Insertional Mutagenesis Database (iMDB), offering a publicly available web-based application to analyze both retroviral- and transposon-based insertional mutagenesis data.
Transposons and retroviruses are widely used in insertional mutagenesis (IM) screens in mice to discover candidate cancer genes (Collier et al. 2005; Dupuy et al. 2005, 2009; Theodorou et al. 2007; Keng et al. 2009; Starr et al. 2009; Copeland and Jenkins 2010; Rad et al. 2010). IM screens have also been instrumental for the identification of genetic interactions between genes driving tumor evolution (Uren et al. 2008; Kool and Berns 2009; Kool et al. 2010) and for the identification of genes that confer resistance to anticancer drugs (Lauchle et al. 2009) or pathogens (Carette et al. 2009). Finally, retroviruses and transposons are used for germline mutagenesis in a range of experimental organisms (Amsterdam et al. 1999; Golling et al. 2002; Ding et al. 2005; Keng et al. 2005; de Wit et al. 2010).
In transposon- or retrovirus-induced tumors, efficient isolation of insertion sites is required for the effective identification of candidate cancer genes and genetic interactions between these genes. Insertion sites are typically analyzed by high-throughput sequencing and mapping of PCR-amplified junction fragments from integrated transposons or retroviruses (Largaespada and Collier 2008; Uren et al. 2009). Genomic loci that are found to contain insertional mutations in multiple independent samples are termed common insertion sites (CISs) and are likely to be causally implicated in tumorigenesis. During the multistep process of tumorigenesis, sequential insertions in cancer-relevant loci will result in the formation of cells with increasingly malignant potential (Fig. 1A). Insertional mutations may drive tumorigenesis when they confer a selective advantage to the affected cell such that it expands clonally to give rise to a significant proportion of the tumor mass. To understand how individual insertions contribute to tumorigenesis, clonal, and near-clonal insertion events should be distinguished from background mutations that did not give rise to clonal expansion and are, therefore, present in a single cell, or a few cells, within the tumor. Methods that allow for (semi)quantitative analysis of insertional mutations are, therefore, of critical importance.
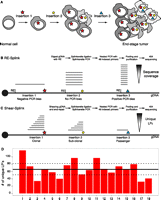
Schematic overview showing the clonality of insertional mutations in tumorigenesis screens, and methods to identify the insertion sites. (A) Clonal expansion of a cell containing an insertion giving a certain growth advantage, which initiates tumorigenesis. In time, additional insertions occur, resulting in a heterogeneous tumor containing a complex collection of insertional mutations. (B) Overview of restriction enzyme based LM-PCR (RE-splink). Amplification of insertion sites using restriction enzymes results in amplicons with a fixed size introducing amplification and sequencing biases, thereby hampering a quantitative identification of insertional mutations within a tumor. (C) Overview of shearing-based LM-PCR (shear-splink), which reduces amplification and sequencing biases and allows the identification of unique ligation points of the splinkerette adapter, each representing a cell within the tumor. (D) Numbers of unique LPs identified for 18 piggyBac insertions in a clonal cell line. The average and 95% confidence interval are indicated by a solid line and dashed line, respectively.
Quantitative assessment of the clonality of insertional mutations is not possible with current methods, which are based on fragmentation of genomic DNA with restriction endonucleases (REs), subsequent splinkerette adapter ligation-mediated PCR (LM-PCR) amplification, and sequencing of the genomic DNA fragments flanking the insertion (Largaespada and Collier 2008; Uren et al. 2009). An important limitation of these methods is the fixed position of RE recognition sites throughout the genome, which results in PCR amplicons of distinct length for each insertion (Fig. 1B). Shorter amplicons will be preferentially amplified over longer amplicons, thereby introducing PCR amplification biases that disturb the linear relationship between the clonality of each insertion and its representation within the pool of sequence reads. Furthermore, many RE-based amplicons are larger than the maximum fragment length that can be analyzed on next-generation sequencing platforms. A direct consequence of these limitations is that RE-based methods are not quantitative. Indeed, the majority of IM studies published to date considered all insertions to have equal importance irrespective of their sequence coverage.
Results
In order to circumvent the aforementioned limitations of RE-based splinkerette adapter LM-PCR (“RE-splink”) methods, we developed a modified method, termed “shear-splink”, which employs random fragmentation of tumor DNA, instead of RE digestion, prior to LM-PCR amplification (Fig. 1C). This method results in a controllable size distribution of DNA fragments for all insertions, thereby limiting amplification and sequencing biases. More importantly, since the splinkerette adapter is ligated to randomly fragmented DNA, we can identify unique ligation points (LPs) which barcode tumor cells with a unique identifier. This feature allows us to use the number of LPs at each insertion site as an estimate of the relative clonality of individual insertions in heterogeneous tumor samples.
Shear-splink analysis of clonal piggyBac insertions
In order to test the utility of the shear-splink method for semiquantitative analysis of insertional mutations, we used shear-splink to identify all piggyBac (PB) insertions in a clonal embryonic stem (ES) cell line (Bouwman et al. 2010), where we expect to identify similar numbers of unique LPs for each PB insertion. In total, we identified 18 piggyBac insertions with an average of 1137 sequence reads per insertion (Supplemental Fig. S1), yielding on average 67 unique LPs per insertion (Supplemental Table S1). To test the significance of the identified number of unique LPs, we performed permutation analysis (1000 permutations) to calculate the expected number of raw reads or unique LPs that one would find if all insertions were completely clonal and not influenced by other biases. When examining unique LPs, we find that 11 out of 18 (61%) PB insertions fall within the 95% confidence interval (Fig. 1D). For the raw read counts, only four out of 18 (22%) insertions fall within the expected interval (Supplemental Fig. S1). These data support our hypothesis that unique LPs are a better representation of clonality than sequence coverage. The observed differences in numbers of unique LPs per insertion are most likely due to amplification and sequencing biases caused by differences in GC content or other features of the genomic DNA sequences flanking the insertions.
Clonality analysis of MMTV insertions
IM screens for cancer gene discovery in mice yield, in most cases, heterogeneous tumors containing different populations of tumor cells with distinct patterns of insertional mutations. We, therefore, set out to test the utility of shear-splink for clonality analysis of mouse mammary tumor virus (MMTV) insertions by analyzing mixtures of two independent clonal cell lines (BB12 and AE6) derived from one MMTV-induced mammary tumor. Both tumor cell lines contained, in addition to three endogenous MMTV copies and one shared somatic MMTV insertion, a number of unique somatic MMTV insertions (Fig. 2A). We mixed genomic DNA from both cell lines in varying ratios and analyzed the MMTV insertions using shear-splink (absolute read numbers per sample are shown in Fig. 2B). We next determined the number of unique LPs for each MMTV insertion. The BB12 and AE6 cell lines contained five and four somatic MMTV insertions, respectively, that were represented by 15 or more unique LPs. One of the insertions in BB12 was excluded from further analysis because it yielded repeat-rich sequences that could not be uniquely mapped to the mouse reference genome (data not shown). One somatic MMTV insertion site was shared between BB12 and AE6, showing the common origin of these two tumor cell lines. As expected the number of unique LPs for this shared MMTV insertion did not correlate with the ratios at which DNA from both cell lines was mixed (Fig. 2C). In contrast, five out of six cell line-specific MMTV insertions showed a strong correlation (R2 ≥ 0.86) between the number of unique LPs and the DNA mixing ratios (Fig. 2D,E). These data illustrate that shear-splink permits semiquantitative analysis of insertion events within heterogeneous samples, with a sensitivity of ∼10% for biclonal tumors.
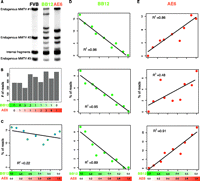
Shear-splink allows semiquantitative analysis of insertional mutations. (A) Southern blot analysis with an MMTV-LTR specific probe shows (in addition to the internal MMTV fragment) three bands representing endogenous MMTV copies in the FVB genome. Clonal MMTV mammary tumor cell lines BB12 and AE6 contain different patterns of somatic MMTV insertions. (B) Overview of total sequence read numbers containing a unique LP for each mixing ratio of BB12 and AE6 DNA. (C) Mixing of BB12 and AE6 DNA does not affect the number of unique LPs for a somatic MMTV insertion that is present in both cell lines. (D,E) Somatic MMTV insertions that are unique for BB12 or AE6 show numbers of unique LPs that correlate with relative clonality of the insertions (R2 > 0.86 for five out of six insertions).
Insertional Mutagenesis Database (iMDB)
In order to fully exploit the increased efficiency of IM screens using shear-splink, we developed a new analysis platform called the Insertional Mutagenesis Database (iMDB). iMDB is a web-based application capable of analyzing mapped retroviral and transposon insertional mutagenesis data. Further, it is designed to handle clonality scores for each insertion by exploiting LP counts. For finding CISs, we implemented the Gaussian Kernel Convolution (GKC) approach, which is able to find statistically significant CISs (de Ridder et al. 2006). Furthermore, we developed tools for calling significantly co-occurring or mutually exclusive insertions and implemented a tool to associate insertions with potential target genes and combine insertion site data with gene expression data (de Ridder et al. 2010). Users may upload their own data, which is kept private, and perform these analyses on the data. All data presented in this paper are publicly accessible from the iMDB. The iMBD can be accessed from http://imdb.nki.nl.
Shear-splink analysis of MMTV-induced mammary tumors
To further test the potential of shear-splink and to compare its efficiency with current RE-splink methods, we analyzed 16 MMTV-induced mouse mammary tumors by both RE-splink and shear-splink. Tumor DNA samples were either sheared or digested with BfaI or NlaIII restriction enzymes, which are widely used for RE-splink analysis of insertional mutations (Dupuy et al. 2009; Keng et al. 2009; Starr et al. 2009; Uren et al. 2009). MMTV insertion sites with one sequence read or one unique LP were excluded from further analysis. On average, we obtained 380 mappable sequence reads, corresponding to 27 unique MMTV insertions sites, per tumor (Supplemental Fig. S2; sequencing details per experiment and sample are listed in Supplemental Table S2A,B; all identified insertions are listed in Supplemental Table S3). All identified insertions were plotted on the mouse reference genome (Supplemental Fig. S3) showing a difference in MMTV insertion spectra for the two RE-splink data sets and the shear-splink data set. Despite the fact that the cutoff of >1 LP for shear-splink results in lower numbers of MMTV insertions, there is a significant overlap between the known MMTV common insertion sites identified by shear-splink and both RE-splink methods (Fig. 3A). Of note, the overlapping CISs are strongly enriched for components of the Wnt and Fgf signaling pathways, which are known to cooperate during mammary tumorigenesis in mice (Kwan et al. 1992). This suggests that shear-splink enriches for the most relevant insertions. To further test this, we determined which percentage of reads or unique LPs mapped within 150 kb of published MMTV CISs that are likely to contribute to tumorigenesis (Supplemental Table S4). While the absolute number of MMTV insertions identified in the 16 tumors is higher for the NlaIII and BfaI RE-splink analyses (Fig. 3B), the shear-splink method shows a higher percentage of MMTV insertions near known CISs (Fig. 3C). By increasing the threshold for the number of unique LPs per insertion, a further enrichment for CIS-associated insertions was obtained for the shear-splink method (Fig. 3C). To determine the true-positive rate for the identified insertions, we plotted the observed insertions for shear-splink and the NlaIII and BfaI RE-splink analyses against the list of known CISs in a Receiver Operating Characteristic (ROC) curve (Fig. 3D). For the ROC curve, we ranked the insertions on unique LPs (shear-splink) or on sequence coverage (RE-splink). By moving through these lists, we can see how the true positive and false positive rates evolve by lowering the threshold (i.e., moving from left to right on the curve). In a completely random case, one expects a line of y = x. Enrichment for true positives in the top segment of the ranked lists results in bending of the ROC curve toward the upper left quadrant. As can be seen, the sensitivity and specificity of shear-splink are comparable to both RE-splink analyses, showing that relevant insertions tend to have high unique LPs or sequence coverage. Importantly, this result is obtained with a significantly lower number of informative sequence reads for the shear-splink experiment, indicating a higher signal-to-noise ratio and a reduced fraction of false-positive insertions.
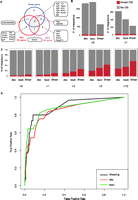
Analysis of insertions in MMTV-induced mammary tumors by shear-splink and RE-splink. (A) Venn diagram showing strong overlap between MMTV insertions at known CISs for the shear-splink and RE-splink methods. The overlapping CISs are strongly enriched for components of the Wnt and Fgf signaling pathways, which are known to cooperate during mammary tumorigenesis. (B) Total number of unique insertions (>0 and >1 sequence coverage and unique LPs) identified in a panel of 16 MMTV-induced tumors using shear-splink or RE-splink with BfaI or NlaIII. A high level of variability is observed in the absolute number of insertions not linked to a CIS, in contrast to a comparable number of insertions mapping to known CISs. (C) Bar diagrams showing percentages of insertions representing known CISs for shear-splink and for RE-splink with BfaI and NlaIII. Increasing the threshold to higher sequence coverage of unique LPs increases the fraction of insertions representing known CISs. For all thresholds tested (>0, >1, >2, >5, >10), the percentage of insertions mapping to known CISs is higher for shear-splink than for RE-splink. (D) Receiver Operating Characteristic (ROC) curves for the RE-splink and shear-splink methods show for shear-splink that enrichment in the identification of relevant insertions does not result in reduced sensitivity. The ROC curves are built upon unique LPs for the shear-splink analysis and sequence coverage for the RE-splink experiments. By moving along the ROC curves from left to right, the ratios between true positives (sensitivity) and false positives (specificity) are visualized.
Shear-splink analysis of Sleeping Beauty–induced lymphomas
Although retroviral IM screens have been widely used for identification of leukemia and mammary tumor genes (Mikkers and Berns 2003; Kool and Berns 2009), the potential of in vivo IM screens has expanded considerably with the advent of genetically engineered IM models based on (tissue-specific) mobilization of Sleeping Beauty (SB) or piggyBac transposons in mice (Copeland and Jenkins 2010). To test the utility of our method for semiquantitative analysis of transposon-based IM screens, we compared the shear-splink method to NlaIII and BfaI-based RE-splink in its ability to enrich for cancer-relevant transposon insertions in a panel of 127 SB-induced lymphomas. For the shear-splink analysis, we identified, in total, 3292 insertions with more than one LP, and 7318 and 6124 insertions with more than 1 sequence read for NlaIII and BfaI, respectively (sequencing details per experiment and sample are listed in Supplemental Table 2A,C), all identified insertions are listed in Supplemental Table S5). Using our iMDB web-based application, we identified 32 and 35 CISs for the BfaI and NlaIII based RE-splink experiments, respectively, and 48 CISs for the shear-splink experiment (Supplemental Table S6). We manually assigned target genes for these CISs and determined the overlap between unique target genes recovered for each experiment (Fig. 4A). Shear-splink identified 13 unique target genes, whereas the RE-splink method yielded only four and three unique target genes for BfaI and NlaIII, respectively. We next determined for each of the three experiments how many CISs were identified per 1000 insertions. Shear-splink identified 14.6 CISs for each 1000 insertions, whereas RE-splink yielded 5.2 and 4.8 CISs per 1000 insertions for the BfaI and NlaIII experiments, respectively (Fig. 4B). Interestingly, combining both RE-splink datasets before CIS calling did not increase the number of CISs per 1000 insertions. The efficiency of shear-splink is further illustrated by the fact that, of all insertions identified by shear-spink, 14.0% contributes to a CIS, compared to 5.6% for the individual RE-splink experiments and 7.7% for the combined RE-splink data (Fig. 4C). These results confirm that the shear-splink method outperforms RE-splink in its efficiency to identify CISs.
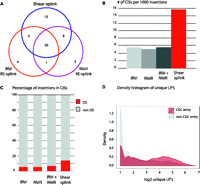
Analysis of insertions in Sleeping Beauty–induced lymphomas by shear-splink and RE-splink. (A) Venn diagram showing the overlap of CISs in 127 Sleeping Beauty–induced lymphomas, as identified by shear-splink and RE-splink using BfaI or NlaIII . In total, 53 CISs are identified. Shear-splink detected more CISs (45) than BfaI- and NlaIII-based RE-splink (31 and 33 CISs, respectively). (B) Shear-splink enriches for insertions contributing to CISs, as shown for the number of CISs identified per 1000 insertions. Combining both RE-splink data sets does not increase the efficiency, since the number of CISs per 1000 insertions is similar to the individual RE-splink analysis. (C) The percentage of insertions contributing to a CIS is higher for shear-splink than for the individual or combined RE-splink datasets, confirming that shear-splink enriches for relevant insertions. (D) Insertions near cancer-related genes are represented by higher numbers of unique LPs. Density plot showing the distribution of unique LPs for SB insertions neighboring a Cancer Genome Census (CGC) gene (red) vs. those not flanking a CGC gene (gray).
We next asked whether SB insertions with a high number of unique LPs are more frequently linked to cancer-relevant genes, as described in the Cancer Gene Census (CGC) database (http://www.sanger.ac.uk/genetics/CGP/Census/). For this, we separated SB insertions that mapped within 150-kb distance of CGC genes from insertions in nonCGC regions and plotted a smooth histogram of the unique LPs for CGC- and nonCGC-related SB insertions (Fig. 4D), showing that insertions near known cancer related genes are represented by a higher number of unique LPs. In addition, we plotted the density of insertions as a function of unique LP number (Supplemental Fig. S4). We observed a significant increase in unique LPs for SB insertions within all tested distances (25–250 kb) from cancer-relevant genes.
In summary, the shear-splink method allows us to apply an intelligent filter on insertional mutagenesis data by discarding background insertions marked by single LPs. Consequently, two separate RE-splink analyses are outranked by a single shear-splink experiment, which requires less sequence read and as such reduces the costs and labor involved in large scale IM screens.
Discussion
The implementation of next-generation sequencing has greatly increased the throughput of IM screens, resulting in the identification of hundreds of insertions per tumor. Current methods, however, consider each unique insertion site as equally relevant, independent of their prevalence within the tumor cell population. Subsequent determination of CISs is, consequently, hampered by a low signal-to-noise ratio, resulting in long lists of CISs that are likely to contain a substantial fraction of false positives. We show that the shear-splink method permits semiquantitative analysis of insertions in genetically heterogeneous tumor samples, resulting in efficient recovery of relevant insertion sites with less sequencing effort and improved signal-to-noise ratio. One limitation of the 454 Life Sciences (Roche) sequence data is the relatively low number of reads containing the LP, because ∼30% of all 454 reads lack the splinkerette sequence at the 3′ end. We attempted to increase this efficiency by optimizing the shearing conditions to obtain 200–300 bp fragments, which are ideally suited for the 454 GS FLX system. Unfortunately, this did not result in increased numbers of informative reads, probably due to the amplification bias of short, unmappable fragments and/or the automatic clipping of sequence data with low quality scores by the Roche 454 software. However, this limitation can be overcome by generating paired-end sequencing data on the Illumina or Applied Biosystems (Life Technologies) SOLiD next-generation sequencing platforms.
The most important implication of semiquantitative identification of insertional mutations is the ability to apply experimental filters to IM data. Insertion sites from individual tumors can be excluded or rated by the number of unique LPs prior to CIS analysis (de Ridder et al. 2006). This is of particular interest for SB-based IM screens, where RE-splink analysis of tumor samples yields, on average, between 100 and 150 SB insertions, of which 50%–80% are represented by only one sequence read (Dupuy et al. 2009). Without intelligent filtering, the very large numbers of SB insertions may show random clustering on the genome, resulting in the identification of false-positive CISs. Shear-splink enables exclusion of random insertions represented by single LPs and thus enriches for biologically relevant insertions present in a substantial fraction of the tumor mass, as was demonstrated by our comparative analysis of shear-splink and RE-splink data from 127 SB-induced lymphomas.
One major challenge in the field of insertional mutagenesis screens is to distinguish early versus late insertions in the multistep process of tumorigenesis. This analysis is hampered by the fact that RE-splink and shear-splink methods cannot discriminate between heterogeneity resulting from clonal evolution of a monoclonal tumor vs. the existence of multiple distinct cell clones within a bi- or oligoclonal tumor. Nevertheless, the unique features of shear-splink, in combination with iMDB, our publicly available database and analysis pipeline, will significantly improve the efficiency of in vivo IM screens and facilitate the analysis of IM-based in vitro fitness screens, in which abundance of individual cell clones in a polyclonal population is measured under selective vs. nonselective conditions. In in vivo IM screens, shear-plink allows us to study co-occurring or mutually exclusive mutations implicated in tumor formation and in acquired traits, such as metastasis or drug resistance, with a higher reliability. In conclusion, shear-splink effectively enriches for the most relevant insertions by providing a rationale to exclude irrelevant insertions with single or few unique ligation points.
Methods
Generation of tumors
MMTV-induced mammary tumors were derived from MMTV-C3H-infected FVB mice according to Theodorou et al. (2007). SB-induced lymphoma samples (spleen or thymus) were isolated from bitransgenic mice (mixed C57Bl6/129S5SvEvBrd background), carrying the SB11 Transposase (Dupuy et al. 2005) and a transgenic array of T2Onc transposable elements on chromosome 1 (Collier et al. 2005).
Amplification and sequencing of MMTV insertions
Identification of MMTV insertion sites using restriction enzymes was performed as described previously (Uren et al. 2009). Two μg of genomic DNA in 100 μl H2O was sheared to 100 bp–1 kb fragments using a Covaris S2 sonicator for 45 s (6×16 AFA fiber Tube, duty cycle: 5%, intensity: 1, cycles/burst: 200, frequency sweeping). Sheared DNA was precipitated and dissolved in 20 μl MQ and blunt-ended using the End-it kit (Epicentre Biotechnologies) by adding 2.94 μl dATP, 2.94 μl dNTPs, 2.94 μl buffer, and 0.57μl enzyme mix and incubated for 45 min at RT. End-repaired DNA was purified using the Qiagen PCR purification kit, and concentrations were normalized to 20 ng/μl. Splinkerette adapters were generated by denaturing 400 pmol of oligonucleotides at 95°C, re-annealing by gradually decreasing temperature to 20°C, and diluted to a concentration of 40 pmol. Blunt-ended splinkerette adapters were ligated in 100-fold excess to 200 ng sheared DNA fragments using 4 U T4 DNA ligase (Roche) at 16°C for 16 h. To prevent amplification of internal MMTV fragments, samples were digested with DraI (New England Biolabs) for 3 h and inactivated at 70°C for 20 min. Primary amplification was performed using ThermoStart Taq (Thermo Scientific) polymerase with a primer specifically amplifying the exogenous MMTV-LTR (primer MMTV4 contains mismatch with the endogenous sequence at the most 3′ end) and a splinkerette-specific primer (P7) with the following cycle conditions: 94°C for 30 s, 68°C for 30 s, and 72°C for 1 min for 15 cycles, followed by 10 rounds of amplification with annealing temperature of 66°C. Secondary PCR was performed using 454B-T7 primers for the splinkerette adapter and oligonucleotides containing 48 different 10-bp indexes and a 9- bp spacer in combination with the 454-A adapter sequence on the viral end. Cycle conditions of the secondary PCR were similar to primary amplification, with the exception that annealing was performed at 55°C. All oligonucleotides were purchased from Integrated DNA Technologies. Primer, adapter, and barcode sequences are listed in Supplemental Table S7. Secondary PCR samples were pooled (generally 48×), purified using the Qiagen PCR purification kit, and sequenced on one quarter of a picotiter plate on the 454 GS FLX platform, according to the manufacturer's protocol (Roche).
Amplification and sequencing of PB and SB transposon insertions
PB insertions were identified as described previously (Bouwman et al. 2010). SB insertions were identified using the method described for MMTV insertions, except that XhoI was used to remove internal SB fragments. SB samples are sequenced on the 454-Titanium platform according to the manufacturer's protocol. Primer sequences are listed in Supplemental Table S7.
Mapping and processing of sequence reads
A general overview of the complete analysis pipeline of MMTV- and SB-induced tumors is presented in Supplemental Figure S5. All raw and processed sequencing data and barcode information are submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE22657. Sequences from each sample were sorted on barcodes, filtered for the presence of the 5′ end of the viral or transposon genome (not present in the oligonucleotides sequence) and the splinkerette sequence using Exonerate, mapped to the genome using BLAT (95% similarity) and aggregated by genomic coordinate of the transition of viral or transposon sequence to genomic DNA. A reference genome was generated using all unique BLAT hits in order to include short sequence reads that originally could not be mapped in a second round of mapping against this reference sequence. Unique LPs were determined by determining unique BLAT coordinates of reads per insertion site. All reads were processed using Perl, and all information was stored in an SQLite database.
Read filtering and insertion determination
All subsequent filtering and processing steps were performed in R. To remove background and nontumor-related insertions, we excluded all unique genomic coordinates that were supported by one read (for the restriction enzyme experiments) or one unique ligation point (for the shear-splink experiment). To determine insertion sites and to compensate for small (1–10 bp) shifts in genomic coordinate at the viral or transposon end, we used hierarchical clustering to aggregate reads into insertions. Clustering was performed with absolute genomic distance in bp and single linkage. The resulting dendrogram was cut at a distance of 10 bp. This cutoff resulted in a mean within-cluster distance that was seven orders of magnitude lower than the mean nearest between-cluster distance, indicating a very tight clustering. The resulting clusters represent the unique insertions that we recovered in the analyses.
Analysis of PB insertions in ES cells
We determined PB insertions by selecting unique genomic coordinates for which we could determine at least 20 unique LPs. Expected unique LPs and reads were calculated by sampling from a uniform distribution of 18 PB insertions. The number of samplings was equal to the total unique LPs or sequence reads. Samplings were repeated 1000 times to determine a 95% confidence interval per insertion.
Analysis of MMTV insertions in tumor cell lines
We determined MMTV insertions in tumor cell lines by selecting unique genomic coordinates for which we could determine at least 15 unique LPs. We determined the number of unique LPs for the selected MMTV insertions for all dilutions. Correlation coefficients (R2) were determined using the stats package in the R software.
Analysis of MMTV and SB insertions in mouse tumors
Endogenous MMTV sequences and potential cross-contaminations were eliminated by removing all MMTV and SB reads that reported a unique, identical genomic coordinate over more than two tumors. In the case where two tumors contained reads reporting the same unique genomic coordinate, the reads were only kept if they had a fivefold higher read-depth in one tumor compared to the other. In all other cases, the reads were discarded. SB insertions on chromosome 1 were excluded because local transposition on the chromosome containing the multicopy array of T2Onc transposon elements is a known feature of SB that complicates the identification of common insertion sites (Collier et al. 2005).
Statistical analyses
The Fisher-Exact test as implemented in the R software was used to compare the significance of the enrichment for known CIS genes between the restriction enzyme experiments and the shearing experiments. ROC curves were calculated and visualized using the ROCR package for R. The ranking required by the ROC analysis was based on the total number of reads per insertion for the restriction enzyme experiments and the total number of unique ligation points for the shearing experiments.
Generation of clonal cell lines from MMTV-induced tumors
Cryopreserved MMTV-induced tumor fragments were grown in DMEM medium, supplemented with 10% FCS, 50 U/mL penicillin, 50 μg/mL streptomycin, 10 μg/mL bovine insulin, and 60 μg/mL EGF. Cells were immortalized by overexpression of SV40 large T antigen as described previously (Jat and Sharp 1986). Epithelial cells were enriched using partial trypsinization, and single-cell dilutions were seeded in 96-well plates (1 and 0.3 cells per well) and selected for single colonies. Cell lines were expanded and passaged for 4 wk, harvested, and analyzed for MMTV insertions according to the aforementioned protocol.
Southern blot analysis of clonal cell lines
Southern blot analysis was performed using an MMTV-LTR probe generated by PCR using the LTR-specific primers 5′-GTTGTTTCCCACCAAGGAC-3′ and 5′-TTCTAGGCCTGTGGTCAATAG-3′. Genomic DNA was digested using PstI, which enables detection of the 3′ LTR plus the flanking genomic region as well as an internal MMTV fragment, which is identical for all MMTV insertions. Hybridization was performed according to standard protocols. A schematic overview of the probe design is shown in Supplemental Figure S6.
Data access
The sequence data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE22657.
Acknowledgments
We thank Maarten van Lohuizen, Joost Moes, and Wendy Lagcher (Netherlands Cancer Institute, Division of Molecular Genetics, Amsterdam, The Netherlands) for providing tumor samples, and Ewart de Bruijn and Michal Mokry (Hubrecht Institute, Utrecht, The Netherlands) for technical assistance. We thank Anthony Uren and Waseem Akhtar for critically reading the manuscript. This research was funded by AICR grant 07-0585, NWO Horizon Breakthrough grant 40-41009-98-9109, and the Cancer Systems Biology Center (CSBC). D.J.A. is supported by Cancer Research-UK and the Wellcome Trust.
Footnotes
-
↵10 Corresponding authors.
E-mail j.jonkers{at}nki.nl.
E-mail l.wessels{at}nki.nl.
E-mail da1{at}sanger.ac.uk.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.112763.110.
- Received July 14, 2010.
- Accepted August 11, 2011.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press








