
Large-scale methylation domains mark a functional subset of neuronally expressed genes
- 1School of Medicine, Medical Microbiology and Immunology, University of California Davis, Davis, California 95616, USA;
- 2UC Davis Genome Center, University of California Davis, Davis, California 95616, USA;
- 3UC Davis M.I.N.D. Institute, University of California Davis, Sacramento, California 95817, USA
Abstract
DNA methylation is essential for embryonic and neuronal differentiation, but the function of most genomic DNA methylation marks is poorly understood. Generally the human genome is highly methylated (>70%) except for CpG islands and gene promoters. However, it was recently shown that the IMR90 human fetal lung fibroblast cells have large regions of the genome with partially methylated domains (PMDs, <70% average methylation), in contrast to the rest of the genome which is in highly methylated domains (HMDs, >70% average methylation). Using bisulfite conversion followed by high-throughput sequencing (MethylC-seq), we discovered that human SH-SY5Y neuronal cells also contain PMDs. We developed a novel hidden Markov model (HMM) to computationally map the genomic locations of PMDs in both cell types and found that autosomal PMDs can be >9 Mb in length and cover 41% of the IMR90 genome and 19% of the SH-SY5Y genome. Genomic regions marked by cell line specific PMDs contain genes that are expressed in a tissue-specific manner, with PMDs being a mark of repressed transcription. Genes contained within N-HMDs (neuronal HMDs, defined as a PMD in IMR90 but HMD in SH-SY5Y) were significantly enriched for calcium signaling, synaptic transmission, and neuron differentiation functions. Autism candidate genes were enriched within PMDs and the largest PMD observed in SH-SY5Y cells marked a 10 Mb cluster of cadherin genes with strong genetic association to autism. Our results suggest that these large-scale methylation domain maps could be relevant to interpreting and directing future investigations into the elusive etiology of autism.
DNA methylation plays an important role in development, particularly in neurogenesis (Trowbridge and Orkin 2010). DNA methyltransferase 3a (Dnmt3a) null mice, despite having a normal number of neural stem cells, have a reduced number of immature neurons and an increased number of astrocytes and oligodendrocytes, suggesting DNMT3A activity is needed for neural stem cells to proceed along a neuronal cell fate. Surprisingly, although global DNA methylation in Dnmt3a null mice was largely unchanged, reduced methylation of sequence adjacent to gene promoters was actually found to correlate with reduced transcription (Wu et al. 2010). DNA methylation is also important in neuronal maturation, as mice with combined Dnmt1 and Dnmt3a deficiency in post-mitotic neurons have defects in learning and memory (Feng et al. 2010).
In mammals, DNA methylation has traditionally been considered an epigenetic mark on CpG sites that represses gene transcription, especially in promoters and CpG islands. However, recent evolutionary analyses of DNA methylation across species suggests that DNA methylation within gene bodies is an even more ancient mark, predating the divergence of plants and animals. In plants, fish, and insects, gene body methylation is a mark of active transcription, and gene bodies with the highest levels of methylation show moderate expression (Zemach et al. 2010). These new insights into the role of gene body methylation on gene expression have largely come about due to new whole-genome DNA methylation detection technologies.
Although bisulfite sequencing is often considered the gold standard for DNA methylation analyses, until recently the amount of sequencing necessary to apply it genome-wide in humans was prohibitively expensive. Most pre-genomic DNA methylation analyses using bisulfite sequencing have been biased toward promoters, CpG islands, and other regulatory sequences where methylation differences can be seen over short distances and with predictable functional implications. Alternative genome-wide methods were later developed that could assess the methylation of a subset of CpG sites (Irizarry et al. 2008; Ball et al. 2009; Brunner et al. 2009; Harris et al. 2010). Recently, though, one of the most in-depth DNA methylation analyses in humans was published in 2009 by Lister et al. High-coverage, high-throughput bisulfite sequencing (MethylC-seq or Methyl-seq) was used to examine the methylation status of CpG sites genome-wide in both the H1 human embryonic stem cells (hESCs) and IMR90 fetal lung fibroblasts. As expected, low levels of methylation were observed at CpG islands and promoter methylation was inversely correlated with gene expression. In addition, the majority of gene bodies and intergenic sequences in both hESCs and fibroblasts have high levels of methylation (>75%). However, in the IMR90 cells large regions of partial methylation (<70%) were observed, called partially methylated domains (PMDs).
To date PMDs have not yet been extensively studied. Based on genome-wide DNA methylation analyses there is evidence suggesting that PMDs can be found in fibroblasts (Ball et al. 2009; Lister et al. 2009, 2011; Aran et al. 2011), adipose tissue (Lister et al. 2011), EBV-transformed B-lymphocytes (Ball et al. 2009, Aran et al. 2011), placenta (Popp et al. 2010; Xin et al. 2010; Aran et al. 2011), and cultured breast cancer cells (Shann et al. 2008). However, PMDs are absent from many mature tissues including cerebral cortex (Maunakea et al. 2010; Xin et al. 2010), testes, breast, liver, leukocytes (Shann et al. 2008), lung, kidney (Aran et al. 2011), hESCs (Lister et al. 2009), and induced pluripotent stem cells (iPSCs) derived from fibroblasts (Ball et al. 2009; Lister et al. 2011) and iPSCs from adipose-derived stem cells (Lister et al. 2011). This tissue specificity and the large genomic distances involved explain why their presence has not been noticed until recently.
Very little is known about the function of PMDs. In the IMR90 cells, gene body methylation is positively correlated with gene expression (Lister et al. 2009) and PMDs overlap H3K9me3 and/or H3K27me3 repressive histone marks (Hawkins et al. 2010), suggesting that PMDs are marks of transcriptional repression. In addition, low gene body methylation correlates with late replication timing (Aran et al. 2011). These data suggest that PMDs could mark transcriptionally repressive genomic domains, but the types of genes contained within PMDs or their functional significance has not been previously addressed.
Using MethylC-seq and bioinformatics approaches, we provide the first genomic maps of PMDs in a human neuronal cell line compared to fibroblast and embryonic stem cells. PMDs were observed at a lower frequency in human neuronal cells than fibroblasts and their locations were distinct. We show that these tissue-specific differences in PMDs mark distinct subsets of developmentally regulated genes with particular importance to neuronal synaptogenesis. Autism candidate genes are enriched in N-HMDs, defined as domains marked by the lack of transcriptionally repressive PMDs in neurons compared to fibroblasts. Therefore, these neuronal domain-wide methylation maps are likely to be significant for understanding and interpreting the genetic and epigenetic causes of autism.
Results
MethylC-seq in SH-SY5Y cells reveals PMDs with a distribution distinct from those in IMR90 cells
To determine the methylomic landscape of human SH-SY5Y neuronal cells we sequenced an SH-SY5Y bisulfite-converted MethylC-seq library using two Illumina GAII lanes and aligned the reads to the human genome. Of the 41.9 million reads generated, 26.7 million were of high quality and uniquely mapped to the genome. Bisulfite conversion efficiency (as determined using the percentage of non-CpG cytosines that were unconverted) was 99.40%. Considering all CpG sites on all genome-alignable MethylC-seq reads, SH-SY5Y cells had on average 69.3% methylation, compared to 67.7% for IMR90 cells and 82.7% for H1 cells (Lister et al. 2009).
For an initial comparison of the distribution of genome-wide methylation levels in SH-SY5Y, IMR90, and H1 cells we took 20-kb non-overlapping windows tiled across the genome, omitting tiles with less than 100 covered CpG sites and determined their average methylation level (Fig. 1A). Twenty kilobases was chosen as a wide genomic window that would dampen the methylation effects of CpG islands, which are on average 763 bp in length. For this and all subsequent analyses, the X chromosome was omitted because IMR90 and SH-SY5Y cells came from female donors and X inactivation complicated the detection of PMDs. As previously shown (Lister et al. 2009), H1 embryonic stem cells have high methylation (>80%) at most CpG sites. IMR90 cells, however, have a bimodal distribution with highly methylated domains (HMDs) having a peak at 80%–85% methylation and PMDs having a peak at 50%–60% methylation. SH-SY5Y cells also show evidence of large regions of partial methylation but the distribution of methylation was more heterogeneous in SH-SY5Y cells than in IMR90 cells.
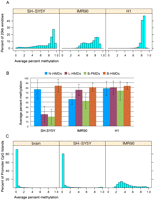
Global DNA methylation levels within the cell lines and methylation domain types. (A) The distribution of average percent methylation across 20-kb windows is shown for each of the three cell lines. (B) Average percent methylation within the four methylation domain types (N-HMD, L-HMD, B-PMD, or B-HMD) defined using the HMM. The H1 cell line is included for comparison. Error bars show standard deviations. (C) Average percent methylation of promoter CpG islands in human cerebral cortex and the SH-SY5Y and IMR90 cell lines. N-HMD, neuronal HMD; L-HMD, lung HMD; B-PMD, PMD in both cell lines; B-HMD, HMD in both cell lines.
Using tracks color-coded by percent methylation, PMDs are fairly easy to visually identify (Fig. 2) as was described previously (Lister et al. 2009). In Figure 2, each tick mark in the %methylation tracks represents the average DNA methylation for an individual CpG site (without smoothing). However, we sought an unbiased computational method to divide the genomic sequences of both cell lines into PMDs and HMDs using hidden Markov models (HMMs). HMMs allow for a more sophisticated analysis of PMDs than the wide windowing method used in Figure 1A. Since CpG islands are predominantly hypomethylated, CpG islands were first masked out to prevent improper transitions between PMD/HMD states. Using the model topology in Supplemental Figure 1 and a transition probability p of 1 × 10−30, the emission probabilities for each possible dinucleotide sequence within PMDs and HMDs were estimated separately for each cell line. All further analyses utilize HMM-defined PMDs. Using the HMMs we found that 41% of the IMR90 and 19% of the SH-SY5Y autosomal sequence contained PMDs. When concatenating PMDs together that were separated by CpG islands, we found that PMDs can be >9 Mb in length, disregarding those containing centromeres and telomeres. Since CpG island annotations are computational predictions based on the DNA sequence and actual DNA hypomethylation could occur in unannotated CpG islands, we calculated the average length of PMDs in our data set. We found that the average length of PMDs in both cells lines was >130 kb (median > 31 kb), in contrast to annotated CpG islands which have an average length of 763 bp (median = 560 bp).

Examples of B-PMDs, N-HMDs, and L-HMDs in the three cell lines. Data were loaded as tracks in the UCSC Human Genome Browser. Hidden Markov model PMD annotations for the IMR90 and SH-SY5Y cells are shown as black bars. Percent methylation tracks display MethylC-seq data for individual CpG sites. Red = 80%–100% DNA methylation, green = 60%–80%, blue = 1%–60%, black = 0%, and white = no sequence coverage. PMDs in IMR90 cells appear blue and green. Due to lower sequence coverage, PMDs in SH-SY5Y cells appear black. (A) B-PMDs mark a cluster of 11 olfactory receptor genes and part of an HLA gene cluster. B-PMDs are shaded in gray. (B) N-HMDs mark CNTNAP2, a neurexin and autism candidate gene (Alarcon et al. 2008), and DPP6, a gene highly expressed in brain and critical for membrane excitability (Kim et al. 2008). N-HMDs are shaded in green. (C) L-HMDs mark CFTR, CAV1, and CAV2, all genes with important functions in lung (Mehta 2005; Gosens et al. 2008). L-HMDs are shaded in pink. OR, olfactory receptor; HLA, human leukocyte antigen; CNTNAP2, Contactin-associated protein-like 2; DPP6, dipeptidyl-peptidase 6; CAV1&2, caveolins 1 and 2; CFTR, cystic fibrosis transmembrane conductance regulator.
Since some PMD locations differed between the SH-SY5Y and IMR90 cells (Fig. 2), we divided the human genome into four subdomain types based on the PMD/HMD status in the two cell lines: B-PMDs are PMDs in both cell lines, N-HMDs (neuronal HMDs) have PMDs only in IMR90 cells, L-HMDs (lung HMDs) have PMDs only in SH-SY5Y cells, and B-HMDs have HMDs in both cell lines and comprise the majority of the genome. Thus, N-HMDs are genomic domains that are partially methylated in IMR90 cells but highly methylated in SH-SY5Y cells. In contrast, L-HMDs are genomic domains that are partially methylated in SH-SY5Y cells but highly methylated in IMR90 cells. Figure 1B shows the average percent methylation of these domains in the SH-SY5Y, IMR90, and H1 cell lines. The presence of tissue-specific partial methylation was verified for randomly selected genetic loci in Figure 2 by the independent method of pyrosequencing (Supplemental Fig. 2).
In order to verify that the PMDs seen in the SH-SY5Y cells were reproducible at low coverage, a biological replicate was done using a single Illumina GAII sequencing lane resulting in a Spearman correlation of 0.63 between the two data sets (Supplemental Fig. 3). The PMD locations were consistent between the two replicates, showing that very low sequencing coverage (9 million reads) was still sufficient to detect PMDs in the SH-SY5Y cells. In addition, we analyzed the quality of PMD detection at low coverage in IMR90 cells to ensure that the PMD differences in SH-SY5Y cells were actually tissue-specific differences (Supplemental Fig. 4). These results showed that low MethylC-seq coverage is sufficient to detect large (>40%) tissue-specific methylation differences over long genomic domains. It should be noted, however, that low MethylC-seq coverage is not suitable to detect small differences in DNA methylation levels in short sequences of interest (e.g., CpG islands).
We also performed MethylC-seq on human cerebral cortex from a 113-d-old male to compare to the data from the SH-SY5Y cell line. Although the cortex data showed no evidence for large PMDs (data not shown), confirming previously observed high methylation levels in cortex (Xin et al. 2010), we wanted to determine if there was aberrant promoter CpG island methylation in the SH-SY5Y cell line that might be more indicative of a cancerous than a neuronal epigenetic state. Figure 1C shows that, as in normal brain, SH-SY5Y cells have low levels of methylation in promoter CpG islands.
PMDs encompass differentially expressed genes important for developmental tissue function
Lister et al. (2009) showed that PMDs are associated with transcriptionally repressed genes in IMR90 cells. To determine if PMDs associate with repressed genes in SH-SY5Y cells as well, we compared microarray expression data from the NCBI Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo) for genes within L-HMDs, B-PMDs, and N-HMDs (Fig. 3). Genes in B-PMDs have low expression levels in both cell types, while genes in N-HMDs and L-HMDs were expressed in a tissue-specific manner. N-HMD genes were more highly expressed in SH-SY5Y cells compared to IMR90 cells. In contrast, L-HMD genes were more highly expressed in IMR90 cells compared to SH-SY5Y cells, where they were found in PMDs. PMDs therefore appear to be a mark of decreased expression, as has been suggested previously. However, many genes in N-HMDs and L-HMDs have low expression levels in both cell types, suggesting that, although an HMD environment is conducive to expression, other factors are probably also necessary to up-regulate genes in HMDs. In addition, tissue-specific PMDs mark only a specific subset of genes differentially expressed between IMR90 and SH-SY5Y lines. N-HMDs and L-HMDs each account for ∼4% of tissue-specific gene expression (Fig. 3). Microarray expression differences for genes within N-HMDs and N-HMDs were confirmed by qRT-PCR (Supplemental Fig. 5).
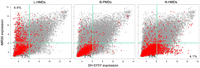
Cell type specific expression of transcripts contained within PMDs. Gray circles are normalized expression values for all microarray probes, averaged over triplicates in both cell lines. Red triangles represent probes to genes within the specified domain type. Light green lines mark the 75th percentile for expression values. Percentages in each quadrant show the percentage of probes in that PMD type for that quadrant.
We found 2590 genes in N-HMDs, 2019 genes in B-PMDs, and 687 genes in L-HMDs (Fig. 4A). In order to determine the types of genes contained within the tissue-specific methylated domains in the two cell lines, we submitted the N-HMD, L-HMD, and B-PMD gene lists to DAVID (Dennis et al. 2003; Huang et al. 2009) for gene ontology (GO) analysis. N-HMDs are significantly enriched for genes involved in homophilic cell adhesion, cell signaling, and synaptic transmission (Fig. 4B). For cellular adhesion, nine cadherins and a large portion of the alpha, beta, and gamma protocadherin clusters are contained within N-HMDs. Cadherins and protocadherins are thought to be important for axon pathfinding, synaptogenesis, and synaptic plasticity (Redies 2000). The N-HMD gene set also included three serotonin receptors, seven gamma aminobutyric acid (GABA) receptors, 17 glutamate receptors, and a neuropeptide Y receptor. Strikingly, 41% of N-HMD genes with GO cellular component annotations were localized to the plasma membrane (26% of all N-HMD genes, P-value = 7.25 × 10−22) and 17% of genes with GO molecular function annotations bound calcium (10% of all N-HMD genes, P-value = 1.06 × 10−38; Supplemental Table 1). In addition, after “neuroactive ligand-receptor interaction,” the “calcium signaling” KEGG pathway was the most significantly represented in the N-HMD gene set (Bonferroni P-value of 6.5 × 10−3; Supplemental Fig. 6). Interestingly, genes in the “long-term depression” KEGG pathway were also overrepresented in N-HMDs (Bonferroni P-value of 4.2 × 10−2; Supplemental Fig. 7). To further determine if N-HMDs mark a functionally important subset of neuronally expressed genes, human cerebral cortex expression microarray data were downloaded from GEO, and 7556 neuronally expressed genes (those with probe signals above the 75th percentile) were collected. Using these neuronally expressed genes as a background gene set for GO analysis, N-HMDs were still significantly enriched for genes involved in cell–cell signaling, synaptic transmission, and neuron differentiation (Supplemental Fig. 8). Overall, these results demonstrate that neuron-specific methylation domains (N-HMDs) mark a specific set of genes important for calcium signaling and synaptic transmission.
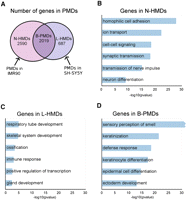
The characteristics of genes in PMDs. (A) Venn diagram showing overlap of PMDs between SH-SY5Y and IMR90 cells. The numbers of genes in each domain type are shown. (B–D) GO biological process classifications for genes in each domain type using DAVID. Bonferroni correction was applied to the P-values. Redundant GO terms containing similar lists of genes were excluded and a full list can be found in Supplemental Table 1. Because of the large number of olfactory receptor genes in B-PMDs, the “sensory perception of smell” category had a Bonferroni P-value of 2.9 × 10−277.
L-HMDs are most significantly enriched for genes involved in respiratory tube development, but are also enriched for genes involved in skeletal system, immune response, and gland development (Fig. 4C). This may be due to the fact that some of these tissues share a common developmental lineage (Liu et al. 2009; Ogawa et al. 2010). B-PMDs contain genes that are expressed in neither lung fibroblasts nor neurons and could be locations of tissue-specific HMDs in other cell types. Strikingly, B-PMDs contain 337 out of 431 olfactory receptor genes, suggesting that PMDs might mark epigenetic domains that regulate olfactory receptor expression. Other statistically significant biological processes for genes in B-PMDs included keratinization and epithelial cell differentiation (Fig. 4D).
PMDs mark a subset of autism candidate genes
Since multiple autism candidate genes were observed in our N-HMD gene list, we explored whether autism candidate genes were overrepresented in PMDs. Autism candidate gene lists were taken from two sources: the Simons Foundation Autism Research Initiative (SFARI) (Basu et al. 2009; Banerjee-Basu and Packer 2010) and Pinto et al. (2010). A χ2 test was performed using the genome-wide distribution of genes in these domains as the null distribution (Table 1). The number of autism candidate genes in PMDs from both gene sets was significantly different than the null distribution, with more autism candidate genes in N-HMDs and L-HMDs than expected. For the N-HMDs in particular, there were over twice as many autism candidate genes than expected by chance, and half of these had some function in the axon and 15 of the 59 had neuronal receptor activity (Supplemental Table 2).
χ2 test for overrepresentation of autism candidate genes in PMDs
Surprisingly, we also found that the largest PMD in the SH-SY5Y cells that did not include a centromere or telomere (9.5 Mb) was on 5p13.3–5p14.3 (Fig. 5). This domain includes four cadherin genes including CDH10 and CDH9. Between CDH10 and CDH9 lie a number of SNPs such as rs4307059 that were found to be strongly associated with autism spectrum disorders but perplexingly located megabases away from either gene (Wang et al. 2009). Most of the domain is actually a B-PMD, which might not be surprising since CDH10 is highly expressed in a specific subset of brain regions including the cerebral cortex (Wang et al. 2009), while SH-SY5Y cells are thought to originate from the sympathetic nervous system (Xie et al. 2010). Thus, neuronal genes whose expression is associated with HMDs in neuronal lineages other than sympathetic adrenergic neurons may appear in B-PMDs when SH-SY5Y cells are used to define N-HMDs. In addition, another recent autism genome-wide study showed association with a SNP in the intron of MACROD2 (Anney et al. 2010), which was found in a N-HMD in our analysis (Supplemental Table 3). These results suggest that domain-wide methylation analyses may aid in the functional interpretation of human genetic analyses by providing maps of epigenetically defined genomic regions.

B-PMD at the 5p14.1 locus implicated in autism. Diagram of the largest PMD in SH-SY5Y cells that does not include centromeric or telomeric sequence (9.7 Mb). The domain includes four cadherin (CDH) genes. Both the rs4307059 and rs10038113 SNPs have been implicated in autism spectrum disorder (Wang et al. 2009). Notably, rs7727656 was associated with hippocampal atrophy (Potkin et al. 2009).
Discussion
This study provides the first MethylC-seq analysis of a neuronal cell line and the first computationally derived maps of cell line-specific PMDs. Our study also included a functional investigation of PMDs, showing that genes contained within tissue-specific methylation domains have tissue-specific functions and expression. We also show that relatively low sequence coverage (1-2 Illumina GAII lanes) is sufficient to detect PMDs by our HMMs, making our method cost-efficient for PMD detection. Finally, we demonstrate neuronal cells have a functionally distinct methylation landscape compared to fibroblasts, reflecting HMDs marking genes involved in synapse development and autism risk.
The question remains why PMDs were observed in the SH-SY5Y cells but not cerebral cortex tissue. Some have argued that aberrant DNA methylation could be an artifact of prolonged tissue culture or cancer progression (Shann et al. 2008). Studies have shown that prolonged tissue culture propagation leads to increases in methylation at high CpG promoters (Meissner et al. 2008), but we did not observe large methylation differences between SH-SY5Y cells and brain tissue at promoter CpG islands. Aran et al. (2011) also showed that DNA methylation changes occur with increasing cell passage, but they found that transcriptionally inactive regions lose DNA methylation whereas active, highly methylated genes remain highly methylated. They concluded, however, that this was not solely due to tissue culture conditions because proliferative tissues such as placenta and fibroblast also have gene body hypomethylation whereas nonproliferative tissues such as brain, lung, and kidney do not (Aran et al. 2011). In addition, not all cells grown in culture show hypomethylation. H1 cells are propagated in tissue culture yet maintained high genomic methylation (Lister et al. 2009) and, although fibroblasts have PMDs, iPS cells derived from fibroblasts lose their PMDs (Ball et al. 2009; Lister et al. 2011). These combined results provide strong evidence that tissue culture is not the chief cause of hypomethylation.
Cancer cells are also known to have aberrant DNA methylation, especially hypermethylation in promoter regions and CpG islands and hypomethylation in intergenic regions and repetitive elements (Ehrlich 2009). For example, a restriction digest-based methylomic study showed breast cancer cell lines have PMDs whereas normal breast tissue does not (Shann et al. 2008). This study found breast cancer PMDs covering genes also found in our B-PMDs such as olfactory receptor, neuronal, and immune-specific gene clusters. However, it is telling that many of the neuronal genes found in PMDs in breast cancer cells, such as CNTNAP2, DPP6, and PTPRN2, were actually N-HMDs in our data set. From our examination of PMDs in different cell lines, we identify a subset of tissue-specific genes differentially expressed in those cell types. The functions of most of the genes in the methylation domains in the two cell lines are not cancer-related. Shann et al. (2008) argue that PMDs in cancer are associated with known chromosomal rearrangement breakpoint regions and therefore might contribute to genome instability in cancer cells. However, our evidence of very tissue-specific partial methylation patterns in SH-SY5Y cells suggests that, even if PMDs do not exist in mature neuronal tissue, the PMDs we observe are marking regions that are developmentally important and differentially regulated in healthy neurons. We therefore suggest that hypomethylation may be observed in human tumors because cancer cells are stalled in or regress to a more immature state than their mature tissue counterparts.
We hypothesize that PMDs are present in a transient subset of normal cells that are undergoing commitment to a particular cellular lineage but have not yet fully differentiated or ceased replicating. Thus, a PMD landscape may be an intermediate epigenetic mark to repress transcription that is replaced by other marks such as histone modifications in the fully differentiated cells. Alternatively, PMDs may not actively repress transcription but instead be a consequence of a late-replicating, heterochromatic state that may not achieve full methylation in proliferating cells. Either way, PMDs mark an important subset of tissue-specific genes that are potentially epigenetically regulated via a common mechanism. One interesting implication from our results is that in order to find genes regulated by this mechanism in a particular cell type, its PMD maps must be compared to those of other cell types to identify HMDs specific to the cell type of interest. Therefore it will be useful in future studies to identify more tissues with PMDs to fully characterize the full list of tissue-specific genes regulated this way.
Intriguingly, N-HMDs are enriched in genes involved in the calcium signaling and long-term depression (LTD) pathways. In neurons, calcium signaling is important for differentiation, migration, and synaptogenesis (Cohen and Greenberg 2008; Greer and Greenberg 2008) and has been implicated in autism (Krey and Dolmetsch 2007). N-HMDs contain all three ryanodine receptors, which release calcium from internal stores in the endoplasmic reticulum (Berridge 1998). N-HMDs also contain all the subunits necessary for complete NMDA and AMPA receptors, which are also important for calcium signaling and LTD (Collingridge et al. 2010). LTD, along with long-term potentiation (LTP), are important for synaptic plasticity and long-term learning and memory (Massey and Bashir 2007). This suggests that, in SH-SY5Y cells, N-HMDs mark genes important for synaptogenesis and synaptic plasticity.
Although autism has a strong genetic component and 10%–20% of autism cases have a known genetic cause (Bailey et al. 1995; Abrahams and Geschwind 2008), the genetics of autism is complex and likely also involves epigenetic and environmental influences. While prior epigenetic studies in autism have focused on the promoters of a few candidate genes and found subtle differences (Nagarajan et al. 2006, 2008; Gregory et al. 2009; Nguyen et al. 2010), future investigations of the epigenetics of autism would benefit by looking at the broader methylomic patterns near autism candidate genes. Our data show that autism candidate genes are overrepresented in N-HMDs and most of the autism genes in this subset have functions at the neuronal synapse including neuronal adhesion, neurotransmitter receptors, axon guidance, and scaffold structure. Although the majority of autism candidate genes lie outside PMDs in both cell lines, PMDs could give important insights into the epigenetic regulation during synaptic maturation of an important subset of genes implicated in the pathogenesis of autism. While we do not currently know how PMD/HMD boundaries are formed, it is tempting to speculate that disruption of boundary elements by copy number variation or other genetic polymorphism could cause epigenetic dysregulation of genes within N-HMDs and contribute to some cases of autism. For example, the polymorphism in the 3 Mb intergenic region between CDH9 and CDH10 is strongly associated with autism, but the functional reason for this genetic association to gene expression is unknown (Wang et al. 2009). Remarkably, the genome wide association peak is at the center of a very large PMD containing four cadherin genes and thus this genetic difference might have relevance to the disruption of the PMD during brain development. We therefore expect that the maps of PMD/HMD boundaries discovered in the current study will help guide future genetic and epigenetic etiologic investigations in autism and other neurodevelopmental disorders.
An interesting caveat of this investigation is that MethylC-seq using bisulfite conversion cannot distinguish between 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) (Huang et al. 2010). Some methods, such as MeDIP, are specific to 5mC (Jin et al. 2010), while many of those that rely on common methyl-sensitive restriction enzymes are not (Nestor et al. 2010). Care should be taken when interpreting results using these different methods since many tissues are known to have 5hmC, including brain and hESCs (Kriaucionis and Heintz 2009; Szwagierczak et al. 2010). However, since bisulfite sequencing recognizes both of these cytosine modifications, we know that PMDs are deficient in both forms of cytosine modification. It would be interesting, however, to look at the relative contributions of 5hmC and 5mC to HMDs in these cell lines, especially in N-HMDs.
DNA methylation is an important epigenomic mark in cell differentiation, neuronal development and function, and tumorigenesis. The fairly recent discoveries of 5hmC and non-CpG methylation in human cells (Lister et al. 2009) suggest there is still much to learn about the human epigenomic landscape and its effects on gene expression. Our analysis of PMDs in IMR90 and SH-SY5Y cells suggests that large-scale genomic regions could be regulated epigenetically, contributing to the tissue-specific identity and differentiation of a cell.
Methods
MethylC-seq
SH-SY5Y cells were grown in MEM media supplemented with 10% FBS and 1× Pen-Strep-Glut (Gibco). Human cerebral cortex sample was obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD. DNA from the SH-SY5Y cells and frozen human cerebral cortex was purified using Qiagen's Puregene kit and fragmented to ∼300 bp using Diagenode's Bioruptor. To create the sequencing libraries, 5 μg of DNA was end-repaired using 1× T4 DNA ligase buffer, 400 μM dNTPs, 15 U T4 DNA polymerase (NEB), and 50 U PNK (NEB) for 30 min at 20°C. After PCR purifying (Qiagen), adenine bases were appended to the ends using 1× NEB 2 buffer, 200 μM dATP, and 15 U Klenow Fragment (3′ to 5′ exo-, NEB) for 30 min at 37°C. After another DNA purification using the PCR MinElute kit (Qiagen), 3 μL of Illumina's methylated sequencing adapters were ligated on using 1× ligase buffer and 5 μL Quick T4 DNA Ligase (NEB) for 30 min at room temperature. After a final PCR purification, 500 ng of library was bisulfite converted using Zymo's EZ DNA Methylation-Direct kit according to the manufacturer's instructions. The library was then amplified using 2.5 U PfuTurbo Cx Hotstart DNA Polymerase (Stratagene) for 12 cycles using Illumina's standard amplification protocol. The library's quality was assessed on a Bioanalyzer (Agilent) and sequenced (76 bp, single-ended) on an Illumina GAII. The SH-SY5Y library was sequenced on two sequencing lanes and the SH-SY5Y biological replicate and cerebral cortex libraries were sequenced on one lane.
Mapping MethylC-seq reads to the genome
Reads were mapped to the hg18 version of the human genome using BS Seeker (Chen et al. 2010), allowing for two mismatches (not including bisulfite conversion mismatches). At any given genomic position, only one clonal read (starting at the same genomic position, potentially due to PCR amplification) was kept. CpG site methylation data were combined from both DNA strands.
Hidden Markov model
Using ∼19 Mb of human chromosome 7 that had been visually classified as HMD or PMD for IMR90 and SH-SY5Y cells, we designed and trained a simple two-state HMM (Supplemental Fig. 1). Because of the large differences in sequencing coverage between the MethylC-seq data from SH-SY5Y cells and published data from IMR90 cells (Lister et al. 2009), models were trained individually for each cell line. Unlike most HMMs that emit nucleotide or protein sequences, this HMM emits a six-symbol alphabet: N, 1, 2, 3, 4, 5. The letter N represents any nucleotide without methylation data from MethylC-seq. The numbers 1–5 indicate the methylation state of each CpG with MethylC-seq coverage: 1 = 80%–100% methylation, 2 = 60%–80% methylation, 3 = 25%–60% methylation, 4 = 0%–25% methylation, and 5 = 0% methylation. The emission probabilities were second order Markov models, meaning that the probability of the emission depends on the previous two emissions. The transition probabilities between the states were set at 10−30. This very low probability was chosen to minimize frequent state changes during decoding and not to reflect the underlying biology. The HMM was implemented in the StochHMM software (P Lott, unpubl.).
Testing the HMM on a subset of chromosome 1 revealed that CpG islands force the model to switch in and out of the PMD state. Rather than redesign the model, we masked the CpG islands (Karolchik et al. 2004) using the annotation provided by the UCSC Genome Browser for hg18 (http://genome.ucsc.edu/). Since StochHMM is not able to process entire chromosomes, we analyzed 2 Mb of sequence every 250 kb. This results in eight predictions for every nucleotide. Positions were identified as HMDs or PMDs if five or more of the predictions resulted in the same decoding.
Mapping genes to PMDs
N-HMD, L-HMD, and B-PMD domains in the genome were defined by looking at the overlap between PMDs in the IMR90 and SH-SY5Y cell lines. Using these three domain lists, genes were assigned to one of the three domain types based on which covered the greatest length of the gene. If none covered >20% of the length of the gene (including introns), it was assigned to a B-HMD.
Microarray data analysis
Affymetrix expression data for IMR90 cells (GSM470491, GSM470492, and GSM470493) (B Stab and R Helm, unpubl.), SH-SY5Y cells (GSM102825, GSM102842, and GSM102870) (Peddada et al. 2006), and human cerebral cortex (GSM379859, GSM379862, and GSM379864) (Pollard et al. 2009) were downloaded from NCBI's GEO (Edgar et al. 2002). Probe intensities across the experiments were RMA normalized using Bioconductor packages in R. To collect the subset of genes highly expressed in cerebral cortex, all genes with expression above the 75th percentile in cortex were selected.
Data access
Sequence data for MethylC-seq have been submitted to GEO under accession number GSE25930.
Acknowledgments
We thank members of the LaSalle and Korf labs for helpful comments and suggestions and Weston Powell and Mike Gonzales for critical review of the manuscript. This work was funded by NIH 2R01HD041462 and NIH 1R01HG00064.
Footnotes
-
↵4 Corresponding author.
E-mail jmlasalle{at}ucdavis.edu.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.119131.110.
- Received December 9, 2010.
- Accepted July 5, 2011.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press








