
Structural and operational complexity of the Geobacter sulfurreducens genome
- Yu Qiu1,
- Byung-Kwan Cho1,
- Young Seoub Park1,
- Derek Lovley2,
- Bernhard Ø. Palsson2 and
- Karsten Zengler1,3
- 1 Department of Bioengineering, University of California, San Diego, La Jolla, California 92093, USA;
- 2 Department of Microbiology, University of Massachusetts, Amherst, Massachusetts 01003, USA
Abstract
Prokaryotic genomes can be annotated based on their structural, operational, and functional properties. These annotations provide the pivotal scaffold for understanding cellular functions on a genome-scale, such as metabolism and transcriptional regulation. Here, we describe a systems approach to simultaneously determine the structural and operational annotation of the Geobacter sulfurreducens genome. Integration of proteomics, transcriptomics, RNA polymerase, and sigma factor-binding information with deep-sequencing-based analysis of primary 5′-end transcripts allowed for a most precise annotation. The structural annotation is comprised of numerous previously undetected genes, noncoding RNAs, prevalent leaderless mRNA transcripts, and antisense transcripts. When compared with other prokaryotes, we found that the number of antisense transcripts reversely correlated with genome size. The operational annotation consists of 1453 operons, 22% of which have multiple transcription start sites that use different RNA polymerase holoenzymes. Several operons with multiple transcription start sites encoded genes with essential functions, giving insight into the regulatory complexity of the genome. The experimentally determined structural and operational annotations can be combined with functional annotation, yielding a new three-level annotation that greatly expands our understanding of prokaryotic genomes.
Genomes can be characterized at three different organizational levels, resulting in structural, operational, and functional annotations (Fig. 1). Structural genome annotation provides the foundation for further operational and functional annotation and consists of coding (open reading frames [ORFs]) and noncoding genes, as well as intergenic regions. Elucidating the precise structural genome annotation subsequently allows decoding the operational genome annotation, which consists of operons and transcriptional units. As a higher level of genome organization, the operon structure is a key to decipher the flow of information encoded in the genome. A functional genome annotation assigns the function of a gene and can be considered as a last step in the flow of information from genotype to phenotype, as it describes the biochemical properties of the gene products.
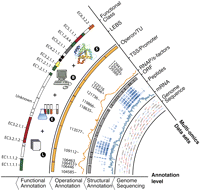
Overview of systems approach to determine structural, operational, and functional genome annotation. Data sets include genome sequence, transcription profiles, peptide reads, RNA polymerase (RNAP), sigma factor binding profiles, transcription start site (TSS) reads, as well as literature data (L), experimental data (E), bioinformatic data (B), and structural information (S).
Precise annotation at the structural, operational, and functional level solely by bioinformatics tools is not possible at present (Kyrpides 2009). We thus developed a systems approach using a combination of genome-wide omics methods to determine the structural and operational genome organization of prokaryotic genomes and applied them to Geobacter sulfurreducens. Since its isolation over 15 yr ago, G. sulfurreducens has been studied intensively, in part because of its impact on the natural environment and its capability of harvesting electricity from waste organic matter (Caccavo et al. 1994; Lovley et al. 2004). Validation and elucidation of its structural and operational annotation by experimental methods, however, is still missing.
Results
Structural annotation
To elucidate the structural genome annotation of the G. sulfurreducens genome, we first determined coding regions by combining a proteogenomics (Jaffe et al. 2004) with a transcriptomics-based approach. We applied liquid chromatography coupled to Fourier transform ion cyclotron resonance mass spectrometry (LC-FTICR-MS) and accurate mass and time tag (AMT tag) (Zimmer et al. 2006) to validate predicted genes and determine translated genes on a genome scale. A total of 28,701 unique peptides were obtained from 12 different growth conditions. Mapping these peptides to the genome sequence using a G. sulfurreducens' genome translation stop-to-stop database (Cho et al. 2009), a total of 2963 potential open reading frames (pORFs) were determined (Supplemental Table S1). A total of 2371 of these pORFs were present in the current annotation, accounting for 69% of all annotated ORFs (3446 total). To verify transcription of pORFs, we applied a transcriptomics-based approach using strand-specific high-density tilling microarrays to identify all transcribed regions of the genome and unambiguously determine antisense transcripts. To reduce cultivation-dependent effects, transcription data were obtained from five different growth conditions; this resulted in a cumulative coverage of transcripts of >96% of the entire genome (Supplemental Table S2). The transcriptomic profiles were subsequently integrated with proteomics-derived data to verify potential ORFs. A total of 537 out of 592 pORFs not previously annotated were removed due to low peptide coverage and weak support from transcription data, resulting in a total of 55 new ORFs that were missed by the current annotation of the G. sulfurreducens genome (Methé et al. 2003). These new ORFs were consequently added to the revised structural genome annotation. A total of 36 out of the 55 ORFs were found in intergenic regions, whereas 19 ORFs were annotated in a different frame or on the opposite strand (Fig. 2A). Additionally, we confirmed 241 ORFs that had previously been predicted as hypothetical proteins. Compared with the current annotation, the proteogenomics approach resulted in ∼9% of newly discovered and validated ORFs (Table 1).
Experimentally derived structural and operational annotation of the Geobacter sulfurreducens genome
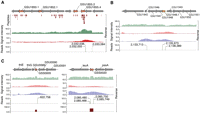
Experimental elucidation of the structural genome annotation. (A) Determination of new open reading frames, ORFs, (orange arrows) by proteogenomics compared with current annotation (gray arrows). Peptide reads (brown bars) were mapped onto the genome sequence. Strand-specific transcription data (green), binding of RNA polymerase (RNAP) (red), and single-base pair resolution TSS data were used for confirmation. (B) New ORFs (orange arrow) determined by transcriptomic data (green), RNAP binding (red), sigma D binding (blue), and TSS reads. (C) Examples of sRNAs (orange arrows) determined by transcription profiling (green), RNAP (red) and sigma D binding profiles (blue), and TSS reads. Secondary structures confirmed sRNA models (TPP and T-Box) predicted by computational methods (brown).
Next, we used the transcriptomic approach to identify new genes that were not covered by proteomics. Typically, contiguous transcriptomic data do not allow for identifying individual ORFs directly and rely on computational methods to infer transcription boundaries (Venkatraman and Olshen 2007). However, mechanisms such as RNA degradation and RNA polymerase (RNAP) pausing can lead to differential expression levels even within a single ORF (Selinger et al. 2003; Bernstein et al. 2004; Kireeva and Kashlev 2009). At the same time, deep-sequencing of transcripts with processed 5′ ends and cross-mapping of transcripts can also affect data analysis, resulting in overestimation of ORFs (Jäger et al. 2009; de Hoon et al. 2010). We therefore integrated contiguous transcription profiles with promoter profiles derived from RNAP binding regions (using rifampicin treatment to generate a static binding map) (Cho et al. 2009) as well as RpoD (sigma70, sigma factor D) and RpoN (sigma54, sigma factor N) binding regions obtained by chromatin immunoprecipitation with microarray hybridization (ChIP-chip). The reasoning is that RNAP holoenzyme initiates transcription at the promoter region, and determination of the RNAP holoenzyme components (RNAP and the two sigma factors) therefore allows segregating contiguous transcripts into transcription segments. In addition, we experimentally determined the transcription start sites (TSSs) of primary mRNAs genome-wide to support the promoter profiling approach. This TSS determination with single-base-pair resolution was accomplished by applying a recently described 5′-RACE method (Cho et al. 2009) that had been modified so that only mRNAs with triphosphate 5′ end were considered.
The integration of high-resolution strand-specific transcriptomic data and genome-wide promoter profiles with TSS data resulted in RNAP-guided transcription segments (Cho et al. 2009). A total of 753 and 700 RNAP-guided transcription segments (RTSs) were determined on the forward and reverse strand, respectively (Fig. 2B; Supplemental Table S2). These RTSs had an average length of 2518 base pairs and contained 2.2 genes on average. Beside evidence for transcription, 96% of these transcription segments contained additional information of either RNAP binding, sigma factor binding, or TSS, and over 85% of RTS contained at least two of these additional experimental evidences. Analysis of RNAP-guided transcription segments (RTS) resulted in 111 new experimentally verified transcripts (∼8% of all RTS) that were not present in the current annotation (Fig. 2B; Supplemental Table S2). The average length of these new transcription segments was 580 bp. The majority of them (∼70%) represent antisense transcripts. A subset of those was validated by Northern blot. Furthermore, we corrected 70 ORFs in the current annotation that had predicted translation starts upstream of experimentally validated TSSs (Table 1; Supplemental Fig. S1).
Noncoding genes
Bacterial genomes contain large numbers of noncoding genes such as rRNA, tRNA, and small RNA genes (sRNAs). Computational methods allow for annotation of highly conserved rRNAs and tRNAs; sRNAs, however, have traditionally been difficult to annotate precisely because of their size (50–300 nt) and are therefore often underestimated in annotations (Zhang et al. 2004). Numerous new sRNAs have recently been predicted computationally using genome and metagenome sequences (Livny et al. 2008; Shi et al. 2009), and experimental methods to determine their functions have just been reported (Hobbs et al. 2010). Here, we applied a computational prediction (Nawrocki et al. 2009) to predict noncoding RNAs (E-value ≤ 0.001), including putative sRNAs (psRNAs) in the G. sulfurreducens genome (Supplemental Table S3). These predictions were consequently mapped to our RNAP-guided transcription segments. By doing so, we experimentally validated all eight rRNAs, 49 tRNAs, as well as tmRNA and RNase P in G. sulfurreducens. Moreover, we identified 34 sRNAs out of 271 computationally predicted psRNAs that had previously not been annotated and were transcribed under our experimental conditions. Most of these sRNAs (33) were identified in intergenic regions; only one represented an antisense sRNA (Fig. 2C). A large fraction (16 of 34) of sRNAs contained a GEMM motif (genes for the environment, for membranes, and for motility), widespread in members of the delta-proteobacteria such as G. sulfurreducens (Weinberg et al. 2007). None of these sRNAs were part of the 111 new RNAP-guided transcription segments identified. All six sRNAs that were randomly chosen for further validation were confirmed by Northern blot (Supplemental Fig. S2), suggesting that the large majority presents bona fide sRNAs. Most sRNAs were expressed under a variety of growth conditions, while others showed differential expression, e.g., expression of GSS0019 was down-regulated 3.5-fold under molecular nitrogen fixing conditions (Supplemental Table S4).
Overall, our experimental approach resulted in an improved structural genome annotation that contained 270 new genes, 34 of them sRNAs and 70 that have been corrected, representing around 8% of the genome (Table 1; Supplemental Table S5). Furthermore, 361 hypothetical proteins were confirmed by either peptide or transcription evidence, overall an increase of more than 18% over current knowledge. However, this percentage is likely to increase further if technological challenges will be overcome in the future, given the fact that the coverage of proteomics data was only 69% and correction of the ORF start codon position could only be accomplished for the first gene within a transcript with an experimentally determined TSS. No information was obtained for 254 out of 3446 genes in the current annotation by the combined proteogenomics and transcriptomic approach.
Structural complexity
The large number of antisense transcripts in the G. sulfurreducens genome with one antisense gene per every 18 genes (5.6%) was unexpected. This number is substantially higher than for Escherichia coli (2.4%) (Cho et al. 2009), Bacillus subtilis (3.7%) (Rasmussen et al. 2009), and Vibrio cholera (4.5%) (Liu et al. 2009), but significantly smaller than what has recently been reported for the Archaea Sulfolobus solfatarius (6.8%) (Wurtzel et al. 2010), Halobacterium salinarum (8.1%) (Koide et al. 2009), and the genome-reduced bacterium Mycoplasma pneumoniae (12.1%) (Fig. 3; Guell et al. 2009). Why the number of antisense transcripts varies between different prokaryotic genomes has so far not been addressed conclusively. We found that the percent of experimentally verified antisense transcripts in these bacteria and archaea reversely correlates with the genome size and number of genes (Fig. 3). One can hypothesize that the reduction in genome size leads to an increase in antisense transcripts, thus countering to a certain degree the loss of genome complexity. If this correlation is universal for prokaryotes it might have implications for generating organisms with reduced genomes (Fig. 3; Posfai et al. 2006) and for the design of synthetic microorganisms.
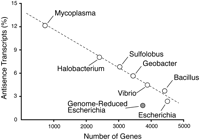
Percentage of antisense transcripts per genome. Experimentally determined number of antisense transcripts in Archaea (Koide et al. 2009; Wurtzel et al. 2010) and Bacteria (Cho et al. 2009; Guell et al. 2009; Liu et al. 2009; Rasmussen et al. 2009; this study) correlated with genome size (open circles). Percentage of antisense transcripts in a genome-reduced E. coli strain (Posfai et al. 2006) is shown as a gray circle. Number of antisense transcripts per genes detected (i.e., experimental coverage) was extrapolated to whole genomes.
Operational annotation
A validated operational genome annotation of G. sulfurreducens is currently unavailable. Here, we experimentally determined promoter regions, TSSs, ORFs, regulatory noncoding regions, and untranslated regions (UTRs) (Supplemental Table S2). A total of 1374 TSSs were determined (Fig. 4A; Supplemental Table S2) and mapped to the overall 1453 RNAP-guided transcription segments (i.e., operons). Over 73% (1063) of all operons had a TSS assigned to them. Most operons had a single TSS associated, whereas 237 operons (22%) contained multiple TSSs (Fig. 4A; Supplemental Table S2), thus resulting in an increase in transcriptome complexity by usage of alternative transcripts (Cho et al. 2009). A large fraction of operons with multiple TSSs encoded genes with essential functions, e.g., genes involved in amino acid biosynthesis, central metabolism, gluconeogenesis, and electron transport (Supplemental Tables S2, S5). Several of these genes, such as NADH dehydrogenase, helicase, and genes involved in amino acid biosynthesis and central metabolism had both RpoD- and RpoN-dependent promoters associated (Supplemental Table S2). The use of different holoenzymes (Eσ70 and Eσ54) for these essential genes might guarantee constant expression levels under different conditions through regulatory mechanisms. This hypothesis is fortified by expression data that show steady expression levels for these genes under all conditions.
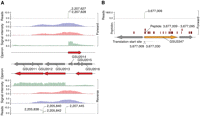
Experimental elucidation of the operational genome annotation. (A) Multiple data sets were mapped strand specifically onto experimentally determined structural annotation (gray arrows). Data included peptide reads (data not shown), transcription profiles (green), RNA polymerase binding (red), sigma D binding (blue), and transcription start site reads. Integration of these data sets resulted in genome-wide operon structure (red arrows) with single base pair resolution start site information. (B) Identification of leaderless mRNAs. Position of the TSS matched the start codon (SC). Peptide reads (brown bars) between the TSS and the next possible SC in frame confirmed leaderless mRNA.
Leaderless mRNA transcripts
In addition, we investigated the 5′ UTR length of ORFs in G. sulfurreducens. The median length of the 5′-UTR region was 37 bp, with no preferences to functional categories (Supplemental Fig. S3), similar to what had recently been described for E. coli (Cho et al. 2009) but opposite to reports for yeast (David et al. 2006), hinting at a nondistinctive regulatory function of 5′ UTRs in bacteria. A total of 52 operons were identified that had no 5′ UTR (UTR length ≤ 5 bp), suggesting the formation of leaderless mRNAs for these operons (Fig. 4B; Supplemental Table S6; Moll et al. 2002). Two of the 52 potential leaderless mRNAs were confirmed by matching peptide data upstream of the next possible translation start codon (Fig. 4B). The potential leaderless mRNAs were encoding proteins of various functions (Supplemental Table S6). Whereas translation initiation using leaderless mRNA seems a more common feature in Archaea, e.g., in Halobacterium salinarum and Sulfolobus solfatarius (Hering et al. 2009; Wurtzel et al. 2010), it is still considered a rare exception in bacteria (Laursen et al. 2005). The large number of potential leaderless mRNAs in a Gram-negative bacterium is unprecedented and precedes the number of leaderless mRNAs in bacteria known so far (∼40) (Laursen et al. 2005). Very recently, a total of 34 leaderless mRNAs were reported in the human pathogen Helicobacter pylori (Sharma et al. 2010), suggesting that leaderless mRNA are much more widespread in bacteria than previously thought.
Operational complexity
G. sulfurreducens not only contains more genes per operon (2.2) than E. coli (1.4), it also has less operons with multiple TSSs (22% compared with 35% in E. coli) (Cho et al. 2009), suggesting a simpler transcriptional regulatory network (TRN) and reduced operational genome complexity. Recently, genome-wide elucidation of TSSs in the methanogenic archaeon Methanosarcina mazei revealed that only a small fraction of operons (∼6%) (Jäger et al. 2009; RA Schmitz, D Jäger, pers. comm.) contained multiple TSSs. One can hypothesize that the genome complexity at the operational level reflects on the organism's lifestyle. A more generalist species (E. coli) has to adjust to a variety of environmental cues requiring a complex TRN. Specialists such as G. sulfurreducens, and even more so, M. mazei, can be successful with a less complex TRN by thriving in a narrower range of environments. The operational genome complexity may therefore be modulated to adjust to complex lifestyles by increasing the number of operons with multiple TSSs.
Discussion
An accurate genome annotation on all three organizational levels, structural, operational, and functional, is paramount for studies in the post-genome era. It provides the framework for a wide variety of genome-wide applications such as metabolic engineering, as well as metabolic and transcription regulatory network reconstructions (Feist and Palsson 2008). The majority of prokaryotic genomes, however, are annotated using error-prone computational methods (Kyrpides 2009). Only recently, studies exploring transcriptome complexity by next-generation sequencing technologies or high-density tiling array techniques revealed a much more complex picture than what was previously expected based on in silico tools (Sorek and Cossart 2010). However, a comprehensive elucidation of multiple genome annotations at three organizational levels has not been archived by experimental methods. It is expected that by making experimentally validated annotations available for multiple organisms, we will be able to gain insights into the governing constraints of complexity at these different levels.
To address this need, we first used a proteogenomic approach to validate ORF predictions, discover previously unannotated genes, and correct annotation errors. Using proteomics data from 12 different growth conditions we identified 55 new ORFs and verified the translation of 241 hypothetical proteins (Table 1). Since the proteogenomic analysis directly verifies and corrects annotations at the translation level, it is critical to define protein-coding ORFs. Although the predicted proteome is fairly accurate, it is known that N terminus prediction can be quite erroneous due to the prediction bias of the algorithms toward large ORFs (Armengaud 2009). Even though we achieved relatively high proteome coverage (69% of the theoretical proteome), relatively low sequence coverage for most proteins makes it difficult to accurately determine the N terminus by proteomics data alone. In the future, proteins of low abundance might be specifically targeted to improve the proteome coverage. Also, N-terminal-oriented proteomic approaches (Armengaud 2009) could be applied to accurately determine translation start positions. This would help to address several biological questions, such as N-terminal modification, usage of uncommon start codons, and 5′ UTR determination. Even without an N-terminal-oriented proteomics approach, the combination of proteomics and transcriptomics results presented here enabled the correction of a large number of translation start sites. Furthermore, this combination allowed identification of several key features of the structural annotation, such as sRNAs and novel transcripts in the genome (Table 1).
To elucidate the operational annotation, we integrated data from three experimental approaches: ChIP-chip-based binding profiles of RNAP or sigma factors, tiling-array-based expression profiles, and deep-sequencing-based TSS determination. Although these three approaches can be used independently to determine levels of the transcriptional architecture (Mooney et al. 2009; Sorek and Cossart 2010), certain limits exist. For instance, the binding profiles of RNAP and sigma factors can be used to determine promoter regions (Mooney et al. 2009), but this method has relatively low resolution (hundreds of base pairs) and might fail to detect weak promoters. The tiling-array-based method is applied to determine transcript boundaries (i.e., operons or transcription units) by detecting “change points” in the transcription abundance map (Bonneau et al. 2007). However, the detected transcription level inside an operon may not be uniform; thus, results in false positives and some “change points” might as well be the result of RNA functional decay (Bernstein et al. 2004). Furthermore, complex transcription architectures like internal promoters may not be determined using datasets from a limited number of conditions. Our integrated approach used data from all three platforms together, and therefore generated an accurate operational annotation of G. sulfurreducens by cross-validating findings. Moreover, instead of inferring complex operon structures from large numbers of experiments, this approach allowed us to elucidate operon structures with data from only a few growth conditions. This integrated approach covered >90% of predicted genes using data from only five different growth conditions. Growth conditions were chosen (e.g., nitrogen fixation and growth on electrode) to represent a most diverse set of life styles, allowing transcription of a large percentage of the genome (96%). Iterative integration of these five data sets showed that the coverage starts to flatten after already three rounds of iteration (Supplemental Fig. S4). Data from additional growth conditions might further increase coverage, but this increase will be subtle. Dozens and even hundreds of conditions may be surveyed before complete coverage can be achieved. Applying next-generation sequencing technology could slightly increase the resolution of the transcription data, but a similar number of conditions will be needed for full coverage.
The structural genome organization of prokaryotes seems to be tailored to compensate for genome reduction by increasing the number of antisense transcripts. What kinds of genes are transcribed in antisense and how these antisense transcripts influence transcription and potential translation efficiency is currently under investigation. However, these possible constraints resulting from structural genome complexity can be offset by an increased operational genome complexity, i.e., multiple TSSs per operon. These different TSSs can be utilized by different holoenzymes. On one hand, this can increase the regulatory flexibility; on the other hand, it can lead to a more robust TRN, allowing for constant gene expression of essential genes under a variety of conditions, as demonstrated here. We also found several antisense TSSs similar to what recently has been described for Helciobacter pylori (Sharma et al. 2010). However, the vast majority of our antisense TSSs were not supported by any other additional evidence, such as RNAP or sigma factor binding as well as expression data, and were therefore removed from our data set. In this manner, structural and operational genome annotations can help to decipher genome complexity on levels beyond sequence information in prokaryotic genomes. When experimentally derived functional genome annotation is added, a new three-level annotation for prokaryotic genomes emerges. Such multiscale annotation will greatly increase our understanding of genome function of the target organisms and is likely to lay the foundation for a new era in comparative genomics that in turn will help elucidate fundamental constraints and features of genome design.
Methods
Bacterial strains, medium, and growth conditions
G. sulfurreducens (ATCC 51573) was grown under strictly anoxic conditions at 30°C in mineral salt medium as previously described (Lovley et al. 1993; Shelobolina et al. 2008), with acetate as electron donor and fumarate or ferric citrate as electron acceptor. For growth in the absence of fixed inorganic nitrogen, ammonium chloride was omitted from the medium and N2 was the only N source. Cells in microbial fuel cells were grown as described previously (Nevin et al. 2008).
Transcriptome analysis
Cells were harvested in mid-log phase, and total RNA was extracted with TRIzol reagent (Invitrogen). Removal of residual DNA was performed with the RNeasy Mini kit (Qiagen). Although the RNeasy Mini column has less binding affinity for RNAs smaller than 200 bp, it does not completely remove sRNAs (Y Qiu, unpubl.). A total of 10 μg of purified total RNA sample was reverse transcribed to cDNA with amino-allyl dUTP. The amino-allyl-labeled cDNA samples were then coupled with Cy3 monoreactive dyes (Amersham). Cy3-labeled cDNAs were fragmented to a 50∼300-bp range with DNase I (Epicentre). High-density oligonucleotide tiling arrays consisting of 381,174 50-mer probes spaced 20-bp apart across the whole G. sulfurreducens genome were used (Roche NimbleGen). Hybridization, wash, and scan were performed according to the manufacturer's instructions. Three biological replicates were utilized for each growth condition. Probe level data were normalized with RMA (robust multi-array analysis) algorithm (Irizarry et al. 2003) without background correction, as implemented in NimbleScan 2.4 software.
ChIP-chip
A ChIP-chip protocol previously described (Cho et al. 2008a,b) was adapted for G. sulfurreducens. Genome-wide binding sites for RNA polymerase (RNAP), RpoD, and RpoN were determined for cells grown to mid-log phase in triplicates under various conditions. Prior to microarray hybridization, real-time quantitative PCR targeting previously known binding regions were carried out to verify enrichment of IP DNA fragments. qPCR and amplification of DNA was performed as previously described (Cho et al. 2008b). Microarray hybridization, wash, and scan were performed in accordance with manufacturer's instruction (Roche NimbleGen).
Transcription start site determination
Total RNA samples were isolated as described above. RNA with a 5′-monophosphate end was removed with Terminator 5′-phosphate-dependent exonuclease (Epicentre). The 5′-triphosphate end of primary RNA was then converted to a 5′-monophosphate end with RNA 5′ polyphosphatase (Epicentre). 5′-RNA adapter (5′-GUUCAGAGAGUUCUACAGUCCGACGAUC) was ligated to the 5′ end of mRNA. cDNAs were then synthesized from the adapter-ligated mRNA using 3′-adapter (5′-CAAGCAGAAGACGGCATACGANNNNNNNNN). A fraction of the cDNA between 100 and 300 bp was then gel purified. The cDNA samples were amplified with primer mix (5′-CAAGCAGAAGACGGCATACGA and 5′-AATGATACGGCGACCACCGACAGGTTCAGAGTTCTACAGTCCGA). The final amplified DNA libraries were sequenced on an Illumina Genome Analyzer. The data were then aligned onto the G. sulfurreducens PCA genome (NC_002939) using Mosaik Aligner (http://bioinformatics.bc.edu/marthlab/Mosaik). Only reads that aligned to only one genomic location and had at least three counts were retained. The genomic coordinate of the 5′ end of these uniquely aligned reads were defined as potential TSSs.
Predicting potential ORFs (pORFs) with proteomics data
Proteomics data using cells grown under various conditions by using LC-FTICR mass spectrometry were obtained, and pORF predictions were performed as described previously (Lipton et al. 2002; Cho et al. 2009).
Identification of RNAP and sigma factor binding regions
Binding regions of RNAP, RpoD, and RpoN were determined as described before (Cho et al. 2009). All RNAP, RpoD, and RpoN binding regions were then combined together to define potential binding regions of RNAP.
Determination of RNAP-guided transcript segments
We used “Transcription Detector” algorithm (TD) (Halasz et al. 2006) to determine probes expressed above background as described before (Cho et al. 2009). Genome-wide summary of piecewise constant expression segments (i.e., RNAP-guided transcript segments [RTSs]) were obtained by assembling the expressed probes between two RNAP binding regions and then assigning genomic coordinates of first/last expressed probes to start/end genomic coordinates of each assembled region, respectively. Potential TSSs determined previously were then mapped onto the 5′ end of RTSs. Multiple TSSs were determined if a TSS had no less than 60% counts compared with the TSS with the highest count of the same RTS. At least two experimental evidences (RNAP binding, sigma factor binding, TSS, or transcription change point, which was determined by the cbs package in R) (Venkatraman and Olshen 2007) were required to break a continuously transcribed region to smaller RTSs.
5′ UTR calculation and start codon adjustment
The 5′ UTR was calculated from each TSS to the start codon of the first gene in the RTS. If a TSS is downstream from the annotated start codon of the first gene results in negative 5′ UTR, the gene was shortened to a new start codon (in frame) that is the most upstream one after the TSS (Supplemental Fig. S1).
Identification of potential sRNA
Potential small RNAs (psRNAs) in G. sulfurreducens were predicted with Infernal (http://infernal.janelia.org) (Nawrocki et al. 2009). Rfam 9.1 was used as model for the prediction. Hits with E-value < 0.001 were mapped to RTSs previously identified, and hits located inside RTSs were considered psRNAs.
Northern blot
RNA samples (10 μg) were denatured for 5 min at 70°C in Novex TBE-urea sample buffer (Invitrogen), resolved by 6% TBE-urea gel (Invitrogen), and transferred to positively charged nylon membranes by electroblotting. The membranes were hybridized with 5′ biotin-modified oligonucleotides in ULTRAhyb buffer (Ambion). The target RNAs were visualized using the BrightStar BioDetect Kit for biotinylated nucleic acid detection system (Ambion) according to the procedure specified by the manufacturer. As an RNA size marker in denaturing gel electrophoresis, RNA Century-Plus marker (Ambion) was introduced and labeled with biotin by using the BrightStar Psoralen-Biotin Kit (Ambion). Primer sequences used in this study are available on request.
Data visualization and availability
Experimental data as well as annotated features were formatted into gff or wig file format (http://www.genome.ucsc.edu/FAQ/FAQformat.html) and visualized in either SignalMap (Roche NimbleGen) or The Integrated Genome Browser (IGB, http://www.bioviz.org). Raw microarray data sets have been submitted to the NCBI Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE17838 and GSE22512. Processed experimental data and annotation features identified in this study are available at http://www.gcrg.ucsd.edu. We also provided these data as Supplemental Data set 1, which can be used for visualization in IGB (see Supplemental Method).
Acknowledgments
We thank Samuel Purvine, Kim Hixton, Mary Lipton, Richard Smith, and other members of the EMSL user facility, a national scientific facility sponsored by the Department of Energy's Office of Biological and Environmental Research and located at the Pacific Northwest National Laboratory for providing the proteomics data. We also thank Kelly Nevin at UMASS for providing cells of G. sulfurreducens grown on electrodes and as biofilms, as well as Katy Juárez (UNAM, Mexico) for providing the RpoN antibody. This work was supported by the Office of Science (BER), U.S. Department of Energy, grant DE-FC02-02ER63446.
Footnotes
-
↵3 Corresponding author.
E-mail kzengler{at}ucsd.edu.
-
[Supplemental material is available online at http://www.genome.org. The microarray data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession nos. GSE17838 and GSE22512.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.107540.110.
- Received March 10, 2010.
- Accepted June 16, 2010.
- Copyright © 2010 by Cold Spring Harbor Laboratory Press








