
Methylation profiling in individuals with uniparental disomy identifies novel differentially methylated regions on chromosome 15
- Andrew J. Sharp1,9,10,
- Eugenia Migliavacca1,2,
- Yann Dupre1,
- Elisavet Stathaki1,
- Mohammad Reza Sailani1,
- Alessandra Baumer3,
- Albert Schinzel3,
- Deborah J. Mackay4,5,
- David O. Robinson4,5,
- Gilda Cobellis6,
- Luigi Cobellis7,
- Han G. Brunner8,
- Bernhard Steiner3 and
- Stylianos E. Antonarakis1
- 1 Department of Genetic Medicine and Development, University of Geneva, Geneva 1211, Switzerland;
- 2 Swiss Institute of Bioinformatics, Lausanne 1015, Switzerland;
- 3 Institute of Medical Genetics, University of Zurich, Zurich 8603, Switzerland;
- 4 Wessex Regional Genetics Laboratory, Salisbury SP2 8BJ, United Kingdom;
- 5 Human Genetics Division, Southampton University School of Medicine, Southampton SO16 6YD, United Kingdom;
- 6 Department of Pathology, Seconda Universita' di Napoli, Naples 80138, Italy;
- 7 Department of Gynaecology, Obstetrics and Reproductive Medicine, Seconda Universita' di Napoli, Naples 80138, Italy;
- 8 Department of Human Genetics, University Medical Center Nijmegen, Nijmegen 6525GA, The Netherlands
Abstract
The maternal and paternal genomes possess distinct epigenetic marks that distinguish them at imprinted loci. In order to identify imprinted loci, we used a novel method, taking advantage of the fact that uniparental disomy (UPD) provides a system that allows the two parental chromosomes to be studied independently. We profiled the paternal and maternal methylation on chromosome 15 using immunoprecipitation of methylated DNA and hybridization to tiling oligonucleotide arrays. Comparison of six individuals with maternal versus paternal UPD15 revealed 12 differentially methylated regions (DMRs). Putative DMRs were validated by bisulfite sequencing, confirming the presence of parent-of-origin-specific methylation marks. We detected DMRs associated with known imprinted genes within the Prader-Willi/Angelman syndrome region, such as SNRPN and MAGEL2, validating this as a method of detecting imprinted loci. Of the 12 DMRs identified, eight were novel, some of which are associated with genes not previously thought to be imprinted. These include a site within intron 2 of IGF1R at 15q26.3, a gene that plays a fundamental role in growth, and an intergenic site upstream of GABRG3 that lies within a previously defined candidate region conferring an increased maternal risk of psychosis. These data provide a map of parent-of-origin-specific epigenetic modifications on chromosome 15, identifying DNA elements that may play a functional role in the imprinting process. Application of this methodology to other chromosomes for which UPD has been reported will allow the systematic identification of imprinted sites throughout the genome.
Imprinting is a phenomenon in which the expression status of a gene is dependent on the sex of the parent from which it is inherited. Imprinted genes generally exhibit monoallelic expression accompanied by parent-of-origin-specific epigenetic marks such as differential DNA methylation and histone modifications that distinguish the maternal and paternal genomes at these loci (Reik and Walter 2001; Dindot et al. 2009). More than 60 imprinted genes have been identified in humans (http://www.geneimprint.com/), and their clustered nature suggests that many are regulated by regional control mechanisms.
To date, the discovery of imprinted sites in both mouse and human has largely been driven through the use of phenotype-based approaches. The vast majority of loci subject to parent-of-origin effects were first recognized through the observation that maternal and paternal transmission of the same genetic mutation results in different phenotypes (Nicholls et al. 1989). For example, the identification of imprinted gene clusters in 15q11-q13 associated with Prader-Willi/Angelman syndrome, 11p11.5 associated with Beckwith-Wiedemann syndrome, and imprinted loci at 14q32, 6q24, and 20q13.2 were all catalyzed by the initial observation that genetic disease occurred specifically in patients with either uniparental disomy (UPD) or deletions of these regions of specific parental origin. In combination with chromosomal engineering techniques that can systematically generate defined aneuploidies, this notion has been applied to screen the mouse genome for imprinting with great success, resulting in the identification of more than 130 murine imprinted genes (Williamson et al. 2009). However, because this methodology relies on the recognition of overt phenotypic differences between individuals to detect imprinting, it is likely to miss imprinting that may cause subtle phenotype differences or those that manifest in ways that are not easily recognized by typical methods of phenotypic characterization. Further, imprinted genes will also be missed or masked by phenotypes that are lethal.
In order to circumvent this limitation, a variety of genomic techniques have been developed to identify parent-of-origin effects. Several previous studies have attempted to detect imprinting based on the differential expression of parental alleles at imprinted loci. Studies using subtractive cDNA hybridization (Kaneko-Ishino et al. 1995) and high-throughput cDNA sequencing in hybrid mouse strains (Wang et al. 2008) have been used to detect imprinted expression with some success. However, these approaches are limited in that they can only assay the subset of genes expressed in the tissue(s) under investigation, and for some genes, imprinted expression is only observed in specific tissues or at certain developmental stages (Deltour et al. 1995; Rougeulle et al. 1997; Zhou et al. 2006). Furthermore, sequencing-based approaches are only able to assay allelic bias in genes containing transcribed polymorphisms (Daelemans et al. 2010).
Alternative approaches to detect imprinting have used the fact that the maternal and paternal genomes have differential epigenetic marks at most imprinted loci. This approach has the advantage over expression-based methods, in that these differential methylation marks are generally conserved, even in tissues that lack imprinted expression (Dockery et al. 2009). The presence of overlapping euchromatin and heterochromatin marks has been used to highlight imprinted domains in human (Wen et al. 2008), and restriction landmark genome scanning (Hayashizaki et al. 1994) and methylation-sensitive representational difference analysis (Kelsey et al. 1999; Smith et al. 2003) have been applied as methods to detect differentially methylated regions in the mouse genome. However, the reliance of these latter techniques on restriction enzyme digestion means that they can only assay a small subset of CpGs that overlap the enzyme recognition site, and if used in outbred genomes, are liable to artifacts generated by the presence of single nucleotide variants that alter restriction patterns.
Because one of the key features of imprinted genes is the presence of parent-of-origin-specific methylation, we hypothesized that the systematic comparison of DNA methylation patterns in maternal versus paternal chromosomes should represent an optimal method for the detection of imprinted loci. Based on this hypothesis, we have taken advantage of the fact that uniparental disomy provides a unique system that allows the separate study of chromosomes derived from a single parent and combined this with a methodology in which the methylation of entire chromosomes can be analyzed in an unbiased fashion. By analyzing methylation patterns in cases of maternal UPD15 (matUPD15) and paternal UPD15 (patUPD15) using immunoprecipitation of methylated DNA and high-density tiling arrays with complete coverage of human chromosome 15, we generated separate methylation profiles of the maternally and paternally derived alleles. Comparison of the two parental epigenotypes identifies numerous loci on chromosome 15 that show parent-of-origin-specific methylation differences, defining a set of DNA elements that are likely responsible for the establishment and/or maintenance of imprinting on this chromosome. We identify novel imprinted loci both within and outside of the known PWS/AS imprinted domain, suggesting candidate loci that may exert parent-of-origin effects in several human phenotypes.
Results
Systematic comparison of methylation profiles generated from three patients with matUPD15 and three patients with patUPD15 resulted in the identification of a total of 80 5-probe windows, which exceeded our statistical threshold. Prior to further analysis, a 1-kb region was then defined centered on the most significant (central) probe in each DMR. Many of these putative DMRs were composed of multiple overlapping 1-kb windows, which were collapsed into 48 nonredundant loci (Supplementary Table 1). As (1) underlying copy-number variations (CNVs) can sometimes cause false signals in ChIP data that mimic enrichment peaks (Vega et al. 2009; A Sharp, unpubl.), (2) nonunique sequences are highly enriched for CNVs and yield lower quality microarrays data due to cross-hybridization artifacts (Sharp et al. 2005, 2007), and (3) imprinted sites were not expected to occur in nonunique or CNV regions, all sites that overlapped either a high-resolution set of CNVs (Conrad et al. 2010) or segmental duplications (http://www.genome.ucsc.edu/) were removed (n = 7). Prior to incorporating this filtering step, we tested several of these putative DMRs that overlapped known CNVs using bisulfite sequencing, but failed to detect any significant methylation differences between matUPD15 and patUPD15 cases at these loci. On re-examination of the array data, we observed many regions of significant difference that overlapped CNVs and that failed to validate by bisulfite sequencing showed a characteristic pattern that differed from that observed at genuine DMRs (Supplementary Fig. 3). A further filter was applied to the remaining 41 regions to remove those that had a low CpG density (<1 CpG/200 bp, n = 10), as this was considered below the sensitivity of meDIP. This resulted in a filtered set of 31 putative DMRs that showed consistent significant differences between individuals with matUPD15 and patUPD15. Two pairs of these loci were separated by <1 kb and were merged into single regions, resulting in a final set of 29 putative DMRs (Supplementary Table 2).
Many of these sites were associated with known imprinted genes within the 15q11-q13 region that is commonly deleted in the Prader-Willi/Angelman syndrome, and some had been identified as DMRs in previous studies (Dittrich et al. 1992; Sutcliffe et al. 1994; Buiting et al. 1995; Supplementary Table 2). These included multiple DMRs upstream of or overlapping the SNRPN gene (Supplementary Fig. 4) and others located at the promoter region of MAGEL2, thus showing the ability of our method to detect known imprinted loci. However, many other potential novel DMRs on chromosome 15 were also identified.
In order to validate putative DMRs identified by array analysis, we used bisulfite modification, followed by PCR and sequencing of the candidate loci. Bisulfite sequencing assays were designed for 22 of the 29 putative DMRs and were used to assess methylation at these sites in multiple patients with matUPD15 and patUPD15 and in biparental controls. For 12 loci, bisulfite sequencing confirmed the presence of high levels of methylation in matUPD15 cases, low or absent methylation in patUPD15 cases, and showed intermediate methylation in biparental controls, consistent with the array data indicating mono-allelic methylation of the maternal allele at these loci (Fig. 1; Table 1). These validation studies were performed using both the original six UPD samples used for the microarray screen, in addition to four other UPD cases that had not been tested by array. For each locus, the results of bisulfite sequencing were concordant across all cases of UPD, and in these UPD cases we saw no evidence of polymorphism at any of the loci sequenced. The remaining 10 candidate sites tested were found not to show any methylation difference between maternal and paternal chromosomes, and thus, represent false positives from the microarray resulting from the use of a low-stringency statistical threshold in our DMR analysis.
Twelve differentially methylated regions detected on chromosome 15
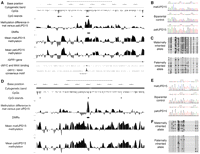
Identification of differentially methylated regions within intron 2 of the IGF1R gene in 15q26.3 and distal to GABRG3 in 15q12. (A,D) Images show the smoothed mean log2 ratios from three individuals with paternal UPD15, three individuals with maternal UPD15, the difference between these two profiles, and putative DMRs identified by statistical analysis uploaded as custom tracks in the UCSC Genome Browser. The DMR within intron 2 of the IGF1R gene contains the sequence RACCACGTGGTY, corresponding to the methylation-sensitive consensus binding motif for MYC/MAX (Solomon et al. 1993), and in vivo binding of both MYC and MAX to this site in K562 cells is confirmed by chromatin immunoprecipitation with massively parallel sequencing (ChIP-seq). Each plot shows a 50-kb window centered on each DMR: (A) chr15:97,203,000–97,253,000; (D) chr15:25,575,000–25,625,000. Also shown are the genomic coordinates (hg18), cytogenetic band, CpG dinucleotides, and CpG islands defined using epigenetic criteria (Bock et al. 2007). (B,E) Sequencing after bisulfite modification of the DNA, which converts unmethylated cytosine residues to uracil, confirms that these two loci both show differential methylation between matUPD15 and patUPD15 samples. A biparental control shows the expected pattern with an approximately equivalent mix of both methylated and unmethylated alleles. (C,F) Individual bisulfite-treated alleles from HapMap individual NA12753 were isolated by cloning and divided by parental origin using heterozygous informative SNPs within each amplicon. In each case, the maternally derived allele is predominantly methylated, while the paternally derived allele is predominantly unmethylated, in agreement with both the array data and bisulfite sequencing of UPD15 cases. Each line represents a separate clone. (●) Methylated CpGs; (○) unmethylated CpGs.
For two DMRs, we further confirmed the presence of maternal-specific methylation (chr15:25,598,371–25,599,471 and chr15:97,226,553–97,227,868) by cloning and sequencing individual alleles amplified from a HapMap control individual heterozygous for a single nucleotide polymorphism (SNP) within each amplicon. At both loci, division of alleles based on their parental origin showed significantly higher levels of methylation on the maternally derived chromosome (Fig. 1).
Bisulfite sequencing of the DMR distal to GABRG3 (chr15:25,598,371–25,599,471) revealed apparent polymorphic imprinting at this site. While the majority of cases examined showed results consistent with mono-allelic methylation, three of 26 individuals with normal biparental inheritance of chr15 showed complete absence of methylation, suggesting that this site shows loss of imprinting in a minority of individuals.
Allele-specific expression studies of IGF1R using RNA extracted from peripheral blood and placenta of a normal control, and of OCA2 using a cultured melanocyte line, did not show any allelic bias in their expression in the samples analyzed.
We investigated two genomic features that have been suggested to be associated with imprinted loci: (1) periodicity of CpG dinucleotides (Jia et al. 2007), and (2) DNA secondary structure (Dindot et al. 2009). Periodicity analysis within the 12 confirmed DMRs showed no consistent pattern with the distribution of CpG dinucleotides, either within any individual region or in a composite dataset (Supplementary Figs. 5, 6). Analysis of predicted sites of guanine-quadruplex DNA within 15q11-q13 showed that the strongest predicted region of G-quadruplex within the entire 6-Mb region coincides with a DMR overlapping a CpG island within intron 1 of the SNRPN gene (chr15:22,643,652–22,645,255, Supplementary Fig. 7). The predicted G-quadruplex structure maps to a (CGGGGG) n tandem repeat that lies within the putative AS imprinting centre (Buiting et al. 1995).
Discussion
Methylation profiling in uniparental tissues allows the systematic identification of imprinted loci, and we describe 12 regions on chromosome 15 that show differential methylation between paternal and maternal alleles (Table 1). Of note, we identify a novel DMR within intron 2 of the IGF1R gene at 15q26.3, a region not previously thought to be imprinted (Fig. 1). The 11 other confirmed regions of differential methylation all occurred within 15q11-q13, a region that is known to contain multiple imprinted transcripts implicated in the phenotypes of Prader-Willi and Angelman syndromes. Six DMRs occur overlapping or upstream of SNRPN, two in the promoter region of MAGEL2, while three are located intergenically more than 150 kb from the nearest annotated gene (Fig. 2). We hypothesize that some or all of these DMRs represent DNA elements that play a role in establishing or maintaining the imprinted expression patterns of genes in this region. As such, we suggest that they represent excellent candidate sites for cryptic mutation in cases of PWS/AS with no known genetic defect.

Differentially methylated regions in 15q11-q13 identified by comparative methylation profiling in patients with UPD15. Eleven of the 12 DMRs we identified lie within a 5-Mb region of proximal chromosome 15q (black bars). This includes multiple DMRs at the known imprinted genes SNRPN and MAGEL2 in addition to several novel intergenic DMRs. The DMR distal to GABRG3 lies within a 687-kb region of 15q12-q13.1 (chr15:25,059,076–25,747,771) previously defined as likely to contain an imprinted locus which, when excess maternal copies are present, confers an increased risk of psychosis in PWS patients (Webb et al. 2008). Genes are colored according to their expression status, as reported by previous studies. A dense cluster of small nucleolar RNAs and several other apparently noncoding transcripts located distal to SNRPN are not shown for clarity. The region shown is chr15:21,000,000–26,000,000.
In each case, differentially methylated regions were relatively GC-rich regions, extending over hundreds of base pairs and including multiple CpG residues. Strikingly, all 12 confirmed DMRs were methylated on the maternal chromosome and unmethylated on the paternal chromosome, consistent with the known excess of maternally methylated imprinted regions identified to date (Kobayashi et al. 2006). While many occur outside of CpG islands defined by classical criteria, aside from a small number specifically at the promoters of imprinted genes, all overlap a more recent map of CpG islands identified using epigenetic data (Bock et al. 2007). These observations indicate that these more sophisticated definitions of CpG islands are much better predictors of genomic loci subject to epigenetic modification compared with those defined solely by sequence characteristics using arbitrary criteria (Gardiner-Garden and Frommer 1987).
Two DMRs were identified ∼180 kb upstream of NDN in 15q11.2, which overlap sites of RNA polymerase II and CTCF binding that are enriched for histone H3K4 mono-, di-, and trimethylation. This epigenetic signature suggests that these may represent either sites of unidentified imprinted transcripts or alternatively regulatory or insulator elements for genes in cis (Supplementary Fig. 8).
Our detection of a DMR within IGF1R (insulin-like growth factor 1 receptor) is arguably not surprising as the related genes IGF2 and IGF2R are both known to be imprinted in human and/or mouse (Barlow et al. 1991; Giannoukakis et al. 1993). Monoallelic maternal expression of IGF1R was observed in one case of Beckwith-Wiedemann syndrome, but previous studies of IGF1R in several normal human tissues have failed to detect any consistent allelic expression bias (Howard et al. 1993; Ogawa et al. 1993), and our analyses in blood and placenta also revealed no apparent bias in IGF1R expression. However, these results do not exclude that IGF1R shows imprinted expression that is limited to specific tissues or developmental time points, phenomena that are observed for many imprinted genes (Deltour et al. 1995; Rougeulle et al. 1997). For some genes, imprinting can manifest in subtle ways such as alternative splicing and polyadenylation events (Kosaki et al. 2000; Wood et al. 2008), or as seems likely in the case of IGF1R, differential transcription-factor binding (Kim et al. 2003). The IGF1R DMR contains the 12-bp MYC/MAX consensus-binding motif RACCACGTGGTY (Solomon et al. 1993), and immunoprecipitation experiments confirm that this region is bound by both MYC and MAX in vivo (Fig. 1). As previous studies in mouse have shown that binding of Myc/Max is dependent on the methylation state of its binding site (Prendergast et al. 1991), the paternal and maternal copies of IGF1R are likely differentially regulated by MYC/MAX binding as a result of maternal-specific methylation. IGF1R plays a fundamental role in growth regulation and the insulin-signaling pathway, has links with survival to old age (Suh et al. 2008), and is important in a variety of cancers (Klinakis et al. 2009; Neuhausen et al. 2009; Sachdev et al. 2009). We suggest that IGF1R and its DMR, therefore, represent interesting candidates for future studies of parent-of-origin effects and epigenetic changes in these conditions. However, we also note that no consistent parent-of-origin effect on growth has been observed in patients carrying mutations or copy-number changes of IGF1R (Abu-Amero et al. 1997; Walenkamp et al. 2006; Tatton-Brown et al. 2009).
Previous mapping of chromosome 15 rearrangements has defined a minimal 687-kb region of 15q12-q13.1 (chr15:25,059,076–25,747,771) as likely to contain an imprinted locus which, when excess maternal copies are present, confers an increased risk of psychosis in PWS patients (Webb et al. 2008). This critical region lies adjacent to a cluster of three gamma-aminobutyric acid receptor genes that act as receptors for the major inhibitory neurotransmitter of the brain. Several lines of evidence suggest these genes as candidate loci for neuropsychiatric disorders, including mouse knockout and association studies linking them with autistic phenotypes (Ma et al. 2005) and the observation that maternal, but not paternal, duplications of this region are seen in both autism and schizophrenia (Cook et al. 1997; Webb et al. 2008). While previous studies have yielded evidence suggesting that the GABAA gene cluster in 15q12 is imprinted in humans (Meguro et al. 1997; Bittel et al. 2005), there is also conflicting evidence suggesting that they are not imprinted (Gabriel et al. 1998; Hogart et al. 2007). We demonstrate that this 687-kb critical region contains a novel DMR ∼150 kb distal to the 3′ end of GABRG3, which we suggest, therefore, likely represents the imprinted element responsible for the increased risk of psychosis in PWS patients with matUPD15 or maternal duplications of 15q12. We hypothesize that this DMR represents an excellent candidate locus, genetic or epigenetic variation of which might represent a more general risk factor for psychosis. Given that maternal duplications of 15q11-q13 are also one of the most frequent chromosomal abnormalities found in autism (Schroer et al. 1998), and alterations of GABAA gene expression is frequent in autistic brains (Hogart et al. 2007), it is tempting to speculate that this imprinted locus may also have links with autism and other psychiatric phenotypes. Of note, we observed that this DMR distal to GABRG3 showed complete loss of methylation in ∼10% of apparently normal individuals, suggesting that imprinting at this site is polymorphic. Similar polymorphic imprinting has previously been reported at some other imprinted genes, such as IGF2R (Monk et al. 2006). We note that of all of the DMRs that we detected on chr15, this region showed the weakest parental difference in methylation (Fig. 1). While most of the other DMRs we examined showed complete methylation of maternal alleles and complete demethylation of paternal alleles, the difference observed at GABRG3 was only partial, suggesting that imprinting at this site is relatively weakly regulated. We suggest that future work to investigate methylation levels at this site in neuropsychiatric disorders might be fruitful.
We did not observe differential methylation at some sites identified in previous studies. This included the promoter regions of NDN (Lau et al. 2004) and MKRN3, although the latter was reported to show only very slight differential methylation in most somatic tissues (Jong et al. 1999). We also did not observe any DMRs around RASGRF1, a gene that is known to be imprinted and differentially methylated in mouse (Yoon et al. 2002). Based on the sequence properties of a training set of imprinted and nonimprinted genes, bioinformatic analyses has also predicted the presence of up to 13 novel imprinted transcripts on chromosome 15 (Luedi et al. 2007). However, we did not observe DMRs at or neighboring any of these 13 predicted imprinted genes.
It should be noted that the use of meDIP and array hybridization has some limitations. Firstly, its resolution is limited, being only able to detect methylation changes at clusters of multiple CpGs and potentially missing differences comprised of only a few CpGs. More specifically, for the study of imprinting, some DMRs associated with imprinted genes are only differentially methylated in specific tissues, and other imprinted genes have been identified that apparently lack nearby DMRs. Thus, the use of a single tissue type for detecting imprinting will never be comprehensive. Furthermore, while this technique is able to identify DNA elements that carry parent-of-origin-specific epigenetic modifications, it does not necessarily allow the definition of which genes have imprinted expression, as in some cases these may lie hundreds of kilobases away from the closest DMR. Despite these caveats, we have identified many novel sites that show parent-of-origin epigenetic modifications that have been missed by other techniques, indicating that analysis of uniparental tissues is a powerful method for detecting imprinting.
As the arrays we used also had coverage of chromosomes 12, 13, 14, and 16, we also investigated potential methylation differences at other loci outside of chromosome 15. Five loci on chromosomes 12 and 14 that showed possible methylation differences between individuals in our cohort were selected, but in each case, bisulfite sequencing showed no significant methylation difference at these loci. Therefore, while our testing was limited, within the sensitivity limits of our technique we did not find any evidence to support the notion that uniparental disomy might cause methylation differences in trans.
In order to identify specific sequences that might play a role in specifying DNA elements that are subject to imprinting, we performed a preliminary investigation of two sequence-dependent features of the DMRs that we identified: CpG periodicity and guanine-quadruplex structure. DNMT3A is the enzyme responsible for establishing methylation at imprinted sites in the maternal germ line (Okano et al. 1999). Based on its crystal structure, which comprises a dimer with two active sites capable of methylating cytosine residues spaced ∼40 Å apart, it has been suggested that CpGs at maternally imprinted loci show a periodicity of 8–9 bp (Ferguson-Smith and Greally 2007; Jia et al. 2007). We tested whether the spacing of CpGs within the maternally methylated DMRs we identified on chromosome 15 exhibited similar periodicity, but did not observe any discernable pattern at any of these 12 DMRs (Supplementary Figs. 5, 6). Thus, our data do not support the hypothesis that the regular spacing of CpGs, such as an 8–9-bp periodicity suggested as the ideal substrate for de novo methylation by DNMT3A, is a factor in determining imprinted loci.
Investigation of predicted secondary DNA structure resulting from guanine-quadruplexes within 15q11-q13 showed that the highest peak of potential G4 DNA occurs at a (CGGGGG) n tandem repeat motif that is differentially methylated between maternal and paternal alleles. This tandem repeat lies within the 1.15-kb minimal region found in Angelman syndrome patients with imprinting center mutations, deletion of which disrupts imprinting within the 15q11-q13 region (Buiting et al. 1995; Ohta et al. 1999). Tandem repeat motifs have also been identified within DMRs at several other imprinted loci (Dindot et al. 2009), and in mouse, transgene experiments have shown that tandem repeat sequences located within the Igf2r DMR are sufficient to establish imprinting (Reinhart et al. 2002, 2006). Our observations raise the possibility that the secondary structure of tandem repeats at DMRs is an important factor for establishing the correct pattern of imprinting during gametogenesis.
Our analysis of DNA methylation patterns in patients with UPD15 provides the first comprehensive map of imprinted loci on chromosome 15. Unlike previous studies that have utilized candidate gene or phenotype-driven approaches, we show that methylation profiling in uniparental tissues represents an unbiased method to prospectively identify the specific DNA elements that are epigenetically modified depending on their parental origin. We identify DMRs close to genes such as IGF1R and GABRG3, both of which are known to be involved in several common human diseases, suggesting that imprinting is probably more common than is currently appreciated (Cheverud et al. 2008). Given that uniparental disomy has been reported for almost every human chromosome (Kotzot and Utermann 2005), we predict that further application of this technique will allow the comprehensive assessment of parent-of-origin effects throughout most of the human genome. Furthermore, the use of such imprinting maps to incorporate parent-of-origin effects into disease association studies may identify previously unrecognized epigenetic influences in many human phenotypes (Kong et al. 2009).
Methods
Genomic DNA was extracted from peripheral blood samples from (1) six unrelated patients with complete matUPD15 and a clinical diagnosis of Prader-Willi syndrome, and (2) four unrelated patients with complete patUPD15 associated with Angelman syndrome. In each case, analysis of multiple microsatellite markers distributed along chromosome 15 in the proband, and their parents, suggested complete UPD for chromosome 15. Two of the patients with patUPD15 were isodisomic, while all of the other cases used were heterodisomic.
Three cases with matUPD15 and three cases with patUPD15 were methylation profiled by microarray hybridization, as follows. Methylated DNA was immunoprecipitated using monoclonal antibodies that recognize methylated cytosine (Weber et al. 2005). Briefly, 15 μg of DNA was sonicated to generate fragments 200–800 bp in size (Branson 450D Sonifier), incubated with 10 μg of anti 5-methyl cytidine (Diagenode), immunoprecipitated using protein A sepharose beads (Life Technologies), and purified by phenol:chloroform extraction. Immunoprecipitated and input DNA from each case were labeled by random priming using Cy5 and Cy3-conjugated random nonamers (TriLink BioTechnologies) and hybridized to tiling oligonucleotide arrays.
We used arrays composed of 2.1 million 50–75-mer oligonucleotides covering chromosomes 12, 13, 14, 15, and 16 at a median probe density of 1 per 100 bp (Roche Nimblegen). Of these, 612,834 probes cover the 82.1-Mb sequenced portion of chromosome 15. DNA labeling, array hybridizations, and washes were performed according to manufacturer's recommendations, and slides were scanned using a G2565 scanner at 5 μm resolution (Agilent Technologies). Array images were analyzed using NimbleScan v2.5 software (Roche NimbleGen) with default parameters incorporating spatial correction, and the resulting files of probe log2 ratios were used for subsequent analysis. All experiments were performed in duplicate.
We developed a custom analysis pipeline to detect regions of differential methylation between individuals with matUPD15 and patUPD15. To avoid systematic bias when comparing different arrays, for example, resulting from differences in the antibody enrichment or labeling efficiency between different DNA samples, we applied quantile normalization (Bolstad et al. 2003; Supplementary Fig. 1). Subsequently, a sliding window analysis was implemented to identify low-confidence data points (outliers) based on the deviation in the log2 ratio of a probe from its immediate neighbors. This approach utilizes the fact as the mean probe spacing (∼100 bp) is considerably less than the mean size of DNA fragments hybridized to the array (∼500 bp), closely spaced probes are expected to show correlated values. Outlier probes were identified using a sliding window that identified all clusters of five consecutive probes, which spanned a physical distance of 1 kb or less. For each group of probes, if the difference in log2 values between the central probe and the median value of the probe cluster was larger than the interquartile range of log2 values on chr 15 (0.97), it was considered an outlier, and its value was replaced with the median log2 ratio of the remaining four probes in the group. This procedure resulted in the replacement of an average of 3.2% of probes per array. Overall, these normalization and filtering steps resulted in significant noise reduction and improvements to data quality, with the mean correlation between log2 ratios in replicate hybridizations for the six individuals tested increasing from 0.83 in the raw data to 0.93 after quantile normalization and outlier replacement. We then applied a linear smoothing function (Pelizzola et al. 2008) with a window size of 1 kb, although this was only used for visualization purposes, not for differential methylation analysis. An example of the results of outlier replacement and weighted smoothing are shown in Supplementary Figure 2.
Prior to further analyses, probes were prefiltered for variance, and those with a standard deviation <0.2 were removed (n = 76,485, or 12.5% of the probes on chr 15). Finally, a modified t-statistic as implemented in the LIMMA software package (Smyth 2004) was calculated to determine the significance value of differential methylation for each remaining probe between matUPD15 and patUPD15 samples. Since we were analyzing both biological and technical replicates, a mixed model analysis was used (Smyth et al. 2005). False discovery rate (FDR) (Benjamini and Hochberg 1995) multiple testing correction was applied. Putative regions of differential methylation were identified by analyzing all clusters of five consecutive probes with a span of <1 kb. Putative regions of differential methylation were identified based on the presence of clusters of multiple independent probes that exceeded a relatively low-stringency statistical threshold, suited for the discovery of novel DMRs. Clusters were scored as differentially methylated if the central probe had an FDR-adjusted P < 0.1 and at least two of the four neighboring probes had an unadjusted P < 0.05. Overlapping intervals identified by this approach were merged to form a nonredundant set. All statistical analyses were performed using software from the Bioconductor project (Gentleman et al. 2004).
For validation of putative DMRs, we designed primers to amplify bisulfite converted DNA using Methyl Primer Express v1.0 (Life Technologies). Primers were designed for 22 of the 29 sites identified by array analysis, while for the remaining seven regions it was not possible to design successful assays, due either to the presence of common repeat elements and/or the difficulties of designing specific primers for reduced-complexity bisulfite-treated DNA (Supplementary Table 1). A total of 2 μg of genomic DNA from patients with matUPD15, patUPD15, and control individuals was bisulfite converted and purified using Epitect Bisulfite Kits (Qiagen). Bisulfite-treated DNA was amplified using JumpStart REDTaq DNA Polymerase (Sigma), unincorporated primers and nucleotides were removed by incubation with Exonuclease I and Shrimp Alkaline Phosphatase (New England BioLabs), and the products were subjected to Sanger sequencing. In addition to bisulfite sequencing of 10 cases of UPD15, we tested normal control DNAs derived from peripheral blood. A minimum of four controls were tested at each locus.
To allow separate bisulfite sequencing of the maternal and paternal alleles in a normal control sample with biparental inheritance of chromosome 15, a HapMap individual (NA12753) heterozygous for informative polymorphisms within each PCR amplicon was identified from publicly available SNP genotype data (http://hapmap.ncbi.nlm.nih.gov/). After amplification of bisulfite converted DNA, PCR products were cloned by TOPO TA cloning (Life Technologies) and transformants grown on agar plates supplemented with X-Gal. Individual colonies containing an insert were reamplified and sequenced, maternal and paternal alleles separated based on their SNP genotype, and the methylation state of each CpG dinucleotide visualized using CpGViewer (http://dna.leeds.ac.uk/cpgviewer/).
Allele-specific expression studies were performed by Sanger sequencing of both DNA and cDNA in individuals carrying transcribed polymorphisms of IGF1R and OCA2. For IGF1R, we used RNA extracted from placenta and cord blood from newborns, and for OCA2, RNA was obtained from cultured melanocytes. We were unable to examine GABRG3 due to its tissue-limited expression pattern.
Putative sites of guanine-quadruplex DNA were identified using QRGS Mapper (http://bioinformatics.ramapo.edu/QGRS/analyze.php), which identifies and assigns a score to DNA motifs based on their sequence. To assess the relative potential of genomic regions to adopt G-quadruplex structures on a local level, QRGS Mapper was run using default parameters, and the total summed scores of all overlapping sites for each 100-bp window within 15q11-q13 were calculated.
We assessed the periodicity of CpG sites within validated DMRs on chromosome 15. CpG periodicity was measured for each DMR that overlapped an updated set of CpG islands (Bock et al. 2007) and the results assessed both individually and as a single combined group to give improved power using a larger dataset. In each case, periodicity was analyzed by compiling an all-by-all matrix that measured the distance of each CpG from every other CpG within that CpG island, and the frequency with which each inter-CpG distance occurred was plotted graphically.
Acknowledgments
The research leading to these results has received funding from the Fondation Jerome LeJeune, the European Commission Seventh Framework Program under grant agreement 219250 to A.J.S., and the European Commission Sixth Framework Program under grant agreement LSH-2005-1.1.5-1 (anEUploidy). E.M. thanks Mauro Delorenzi for useful discussions.
Footnotes
-
↵10 Corresponding author.
E-mail andrew.sharp{at}mssm.edu; fax (646)537-8527.
-
[Supplemental material is available online at http://www.genome.org. The microarray data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE22188.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.108597.110.
- Received March 31, 2010.
- Accepted June 16, 2010.
- Copyright © 2010 by Cold Spring Harbor Laboratory Press








