
On the origin of prokaryotic species
Abstract
The notion that all prokaryotes belong to genomically and phenomically cohesive clusters that we might legitimately call “species” is a contentious one. At issue are (1) whether such clusters actually exist; (2) what species definition might most reliably identify them, if they do; and (3) what species concept—by which is meant a genetic and ecological theory of speciation—might best explain species existence and rationalize a species definition, if we could agree on one. We review existing theories and some relevant data. We conclude that microbiologists now understand in some detail the various genetic, population, and ecological processes that effect the evolution of prokaryotes. There will be on occasion circumstances under which these, working together, will form groups of related organisms sufficiently like each other that we might all agree to call them “species,” but there is no reason that this must always be so. Thus, there is no principled way in which questions about prokaryotic species, such as how many there are, how large their populations are, or how globally they are distributed, can be answered. These questions can, however, be reformulated so that metagenomic methods and thinking will meaningfully address the biological patterns and processes whose understanding is our ultimate target.
“ … in the end, I think the debate about species reality boils down, sadly, to different interpretations of the word ‘real’.”
Our quotation is from a review of Coyne and Orr's recent authoritative monograph, Speciation (Coyne and Orr 2004). The book deals overwhelmingly with the problems and practices of systematists who work with nonmicrobes (mostly animals) and the arguments of philosophers and historians who have taken an interest in what these systematists do. But Mallet's conclusion applies equally to debates among microbiologists. We too remain deeply divided, in our case about whether or not prokaryotes (i.e., Bacteria and Archaea; pace Pace 2006) have real species and if so how we might recognize, enumerate, and integrate them into existing theoretical frameworks in ecology, population genetics, and evolutionary biology. To the philosophically inclined, this should be more interesting than sad, however. At the end of this essay we will conclude that prokaryotic genomics shows us that there is no reasonable interpretation of the word “real” that can be applied to microbial species generally, but that thinking about species has been highly productive—and learning to do without them will be even more so.
The current status of species, for eukaryotes
Microbiologists often write as if the ontology of the species category has now been largely agreed upon in zoology and botany, and that mere practical problems (microbes' small size and gross morphological sameness, their difficulty of cultivation, and lack of regular sex) keep us from adapting this ontology in microbial and particularly prokaryotic systematics. But this is not so: nonmicrobes still have a major species problem, and a whole scholarly industry devoted to perpetuating arguments about it.
There is some common ground, of course, and for the last half-century and more, Ernst Mayr's Biological Species Concept (BSC)—according to which species are maximally inclusive groupings whose members can produce fertile offspring through mating—has enjoyed a certain hegemony as an algorithm for deciding which eukaryotes belong in which species. Species are a uniquely real taxonomic rank, according to the BSC, defined by real biological interactions between their members (mating) and not by the arguable phenotypic similarities and differences used to delimit higher taxonomic ranks, in what is inevitably an arbitrary way.
The BSC's one-size-fits-all approach has always had problems of applicability, however. In an essay entitled “What is a species, and what is not?” Mayr (1996) himself admitted that “the BSC is based on the recognition of properties of populations. It depends on the fact of non-interbreeding with other populations. For this reason the concept is not applicable to organisms which do not form sexual populations.” Some vertebrates, many invertebrates, and many fungi and plants can never make up species recognized by the BSC, for all that they may form tight and nonoverlapping clusters of populations at either phenotypic or genotypic levels. And then there are all those nested clusters of populations of phenotypically and ecologically similar organisms whose reproductive compatibilities are simply unknown or not conveniently tested. These too beg to be recognized and named.
In consequence, there are more than a dozen alternate eukaryote-specific “species concepts” now on the go, embracing different genetic/ecological processes and principles for recognition (de Queiroz 2007). Faced with such a wealth of ways of thinking about species, systematists are obliged to make commitments to either monism or pluralism. Species monists hold the view that one concept is nevertheless right for all organisms—we just have yet to find it. Species pluralists either accept different concepts as appropriate for different organisms (Mishler and Brandon 1987) or allow that multiple incompatible species concepts can be simultaneously valid (Ereshefsky 1992; Dupre 1993). Ereshefsky (1998) contends that to be a species pluralist of either sort is to give up being a realist as far as the overall category species is concerned. Individual species such as Homo sapiens or Pan troglodytes may be real enough (may have an existence outside the minds of biologists). But if different species differ in the nature of the processes that created them and/or the features that define them, then the word “species” itself has only a nominal significance, describing an arbitrary collection of entities. We accept and build on that argument here, but note that other philosophers question this obligatory coupling of realism and monism (Wilson 1996).
Such seemingly academic quibbles between monists and pluralists, and realists and nominalists, actually matter when we want to know something involving the species category in general, such as “how many species are there (at this site, or in that region, or in the world)?” or “what is the relationship between species richness and ecosystem stability?” These are interesting and genuinely scientific questions only in a realist context. If “species” is a real category, we learn something about nature when we find the answers. If it is not, we only learn something about the habits and beliefs of systematists.
Some authors have come to think that they can identify a common underlying principle, adopting a modernized version of George Gaylord Simpson's evolutionary species concept, by which “[a] species is a lineage (an ancestral-descendant sequence of populations) evolving separately from others and with its own evolutionary role and tendencies” (Simpson 1951). De Querioz's “general lineage concept of species” (de Queiroz 2005, 2007) calls these “metapopulation lineages,” or more specifically “segments of such lineages,” and rejects the imposition of various contingent operational criteria for recognizing them—interbreeding, phenotypic or genotypic clustering, or ecological role, for instance. He sees it as advantageous to thus separate “the conceptual problem of defining the species category (species conceptualization) from the methodological problem of inferring the boundaries and numbers of species.”
Indeed, it may provide some relief to give up arguments about what species truly are. But with a species concept whose general applicability actually relies on forsaking any real-world criteria for recognizing species, we are no better off in answering any of the interesting questions about the species category posed above. Indeed, we could be worse off, as shown in Figure 1. First (left panel), at any instant in time most metapopulations consist of multiple subpopulations, whose membership in a common lineage is in principle unknowable without reference to criteria of the sort the general lineage concept eschews (ability to interbreed or phenotypic similarity, for example). Not only can we not enumerate all species, we can't for sure identify any! Second, without such a way of telling which subpopulations are “separately evolving,” we cannot know where to draw the limits around a species, to distinguish it from a genus, for instance (Fig. 1, middle panel). These are problems in applicability, but the conundrum posed by the right panel of Figure 1 is still a deeper one, one of meaning. If evolution is reticulate and organismal history chimeric, we can no longer speak meaningfully of lineages because the lineage concept is inextricably part of “tree-thinking” (O'Hara 1997). This may not be an issue for most animals, but it can be for plants (Abbott et al. 2008), and it is at the heart of the prokaryotic species problem. Indeed, reticulate evolution through lateral gene transfer (LGT) is the elephant in the room of prokaryotic systematics. But before arguing that this elephant renders the “problem” unsolvable and trumps prevailing prokaryotic species concepts, we must define both the problem and the concepts, as currently construed.

The problematics of any metapopulation lineage-based general species concept. Arrowheads represent populations or subpopulations that might or might not comprise a single species. Often, phylogenetic relationships between such clusters of individuals will be unknown or ambiguous: Common memberships in a “metapopulation lineage” cannot be established (left panel). As well, there is in principle no way of knowing at what degree of divergence subpopulations assume independent “evolutionary roles and tendencies,” and thus no way of recognizing minimally inclusive groupings (that is, of distinguishing species from higher taxonomic groupings) (middle panel). And, when individuals are the product of extensive gene exchange, the very notion of lineage becomes problematic (right panel).
The prokaryotic species problem
Bacteriologists have wrestled independently with species for half a century, paying little attention to their nonmicrobial colleagues, and even less to philosophers (Franklin 2007). But for them, too, the crux of the issue is the difficulty of coupling generalizable prokaryotic species concepts (theories about genetic and ecological processes and cohesive forces that might give rise to discrete clusters of phenotypically and genomically similar individuals) with practicable prokaryotic species definitions (criteria or methods by which such clusters might be delimited and recognized, in the lab or in nature.)
A consensus on definitions emerged in the 1980s (Gevers et al. 2005, 2006; Staley 2006). According to this consensus, assignments to species should be made primarily on the basis of overall genotypic similarity, although phenotypic difference (pathogenicity for instance) should play a role in fine-scale differentiation. The gold standard for assigning two isolates to one species is a value of ≥70% in a standardized DNA–DNA hybridization experiment. A simpler measure, small subunit (SSU, or 16S) rRNA sequence identity, can be used to determine what are not species (strains with <97% identity). Individual species defined by these values often map to species taxa as traditionally defined by a battery of microscopic, biochemical, and physiological tests, which is how these cutoffs were set in the first place.
There are serious issues of comparability with eukaryotic species definitions, however: By the DNA–DNA hybridization measure, a single bacterial species can be as diverse as an entire vertebrate order. This must be partly why the total number of bacterial species recognized is only in the thousands versus millions for animals. Common sense tells us that minimally each animal species should provide its own substantial collection of unique bacterial parasites and commensals, not to mention the many many more prokaryotes that live free. Thus, there are efforts afoot to refine the species definition (Stackebrandt and Ebers 2006). Konstantinidis and Tiedje (2005) (see also Konstantinidis et al. 2006) suggest another measure, average nucleotide identity (ANI) as determined with shared orthologous genes. An ANI value of 95% corresponds roughly to traditionally defined bacterial species (or the 70% DNA hybridization value), and strains as distant as that can vary by up to 35% (or more, see below) in actual gene content, which must bear directly on phenotype. These investigators think that an ANI of 99% would match more closely to phenotypic diversity among species of animals and plants (Konstantinidis and Tiedje 2005), and perhaps even this is not stringent enough (Fig. 2).
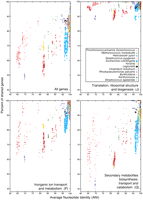
Comparison of average nucleotide identities (ANI) with gene content. 773 genomes available in NCBI's RefSeq database were initially clustered using 16S rRNA identity of at least 97% as a guide to form groups. A dozen clusters were selected (list of genomes within each cluster is available in Supplemental Table 1). For genomes within each cluster, pairwise ANI was calculated essentially as described in Konstantinidis and Tiedje (2005). Shared genes for each pair of genomes were identified as reciprocal top-scoring BLASTP matches (E-value < 0.001, z = 20,000,000). The proportion of shared genes was calculated as a ratio of the number of shared genes over the average number of genes in two genomes. Each ORF in a genome was assigned to a functional category according to the Clusters of Orthologous Groups (COG) database (August 2005 release), and three selected categories are depicted in this figure: categories J, P, and Q in COG category one-letter designation. Note that genomes of the E. coli/Shigella group have similar ANI values, but dramatically varying gene content. Some groups form tight clusters (e.g., Legionella spp.), while others exhibit a continuum of ANI/shared genes values (e.g., Burkholderia spp.). The clustering also exhibits a large variability in the number of shared genes if genes are considered by functional category.
Whatever species definition we adopt, there remains the problem of coupling to some underlying species concept(s) that rationalizes its methods and cut-off values. As Gevers et al. (2006) lament, “any effort to produce a robust species definition is hindered by the lack of a solid theoretical basis explaining the effect of biological processes on cohesion within and divergence between species.” Possible cohesive forces are addressed in the next sections, but it is worth mentioning here that two recent formulations of prokaryotic species concepts appear to be (deliberately) so general that, like de Querioz's general lineage concept, they finesse the concept–definition coupling. The first is Staley's “genomic-phylogenetic species concept” (Staley 2006), and the second is a “metapopulation lineage” formulation endorsed by Achtman and Wagner (2008). These latter authors, acknowledging a debt to and quoting de Querioz, claim that “unlike other species concepts, metapopulation lineages do not have to be phenotypically distinguishable, or diagnosable, or monophyletic, or reproductively isolated, or ecologically divergent, to be species. They only have to be evolving separately from other lineages. Microbes that form distinct groups owing to a cohesive force are metapopulation lineages and thus form species, whereas microbes without limits imposed by a cohesive force do not.”
This way of thinking embodies the spirit of what one hopes to capture with a species concept. But we must again point out that by giving up all methods of detecting or quantifying “cohesive forces,” such bare bones species concepts cannot be used to answer any questions we might have about species in general—such as how many there are, what their populations sizes are, and whether they are cosmopolitan or endemic.
Clustered diversity and its meaning for species
Basic to any notion of species is that in nature they comprise discrete clusters of organisms, defined genomically and phenomically—that genome/phenome space is not uniformly filled by a seamless spectrum of intergrading types. As Konstantinidis et al. (2006) note, “an important issue that remains unresolved is whether bacteria exhibit a genetic continuum in nature…”
It is necessary to recall here that even the simplest random birth and death model of replicating lineages will produce clusters of related individuals separated by gaps (Zhaxybayeva and Gogarten 2004; Mes 2008). Presumably, any biologically interesting species concept (one involving “cohesive forces”) will result in a clustering pattern distinguishable from (probably “gappier” than) such a stochastic process. It also seems certain that additional (and arguably “artifactual”) gaps disrupting any imagined seamless spectrum will arise simply because of sampling bias. Many populations intermediate in their phenotypes and genotypes might exist between two well-delineated species and yet not be known because they are of much lower abundance, for instance, or are found in different environments, or lack a specific marker that is selected for during isolation.
This latter sort of sampling error could easily result from the famous noncultivability of most microbes (Staley and Konopka 1985; Staley 2006). Only 1% are said to be readily brought into culture, and the trait of culturability might be patchily distributed even among close relatives. This problem could be especially severe for isolates of pathogens, whose virulence and drug resistance (traits that would be the basis for their inclusion in culture collections) can often be determined by a single plasmid, easily gained and lost. Thus, many factors could collude to convince us, falsely, that genomic/phenotypic clustering is the dominant pattern in nature.
This said, some bacterial species as traditionally recognized do seem to comprise cohesive (and exclusive) clusters of closely related organisms, as defined by ANI values in the vicinity of 95%–99% in all possible strain-to-strain comparisons of orthologous genes, together with lower values obtained with strains of what are thought to be sister species (Fig. 2; Konstantinidis et al. 2006). Similarly, MLSA (multi-locus-sequence-analysis) studies will—though not always—produce tight clusters with isolates thought for other reasons to represent a single species (see below, and Gevers et al. 2005; Hanage et al. 2005, 2006a).
Moreover, sequence data derived directly from environmental and metagenomic samples also frequently reveal the presence of “microdiverse clusters” of sequences for many genes (Rocap et al. 2002; Ward et al. 2006; Allen et al. 2007; Roesch et al. 2007; Pham et al. 2008; Woebken et al. 2008; Zo et al. 2008), and are taken as evidence of the existence at the sampled site of more or less discrete clusters of phenotypically and genotypically similar individuals—populations of a single “ecotype” or “species” (Gevers et al. 2005; Hanage et al. 2006a, b; Cohan and Perry 2007; Ward et al. 2008). Environmental and metagenomic methods employed include Sanger sequencing of cloned libraries of PCR-amplified rRNA genes or of the internal transcribed spacers (ITS) between them and of small random clones and fosmid ends, or massive pyrosequencing of uncloned DNA. With such methods, biases in cultivation are eliminated, but others remain: PCR amplification bias, the formation of artifactual chimeras, the presence of multiple slightly different rRNA genes in some species, errors in sequencing (creating an excess of very similar sequences), and, in very many studies, inadequate sampling (Schloss and Handelsman 2007).
Martin Polz's group has provided, in a series of studies of marine bacteria from a sampling site north of Boston, a multidimensional perspective on microdiversity and its potential significance for the species question. In an initial paper (Acinas et al. 2004), they showed that even when the above sources of error were excluded, >50% of ribotypes (rRNA sequence variants) from this site fell into clusters showing <1% sequence variation. This is a considerable excess of close sequences (that is, tighter clustering) over what would be expected with a random birth–death process (Martin 2002). Acinas et al. (2004) and, in an accompanying commentary, Giovannoni (2004) explained this in terms of the “ecotype model” (see below), according to which individuals whose ribotypes fall within a single cluster are all descendants of a lucky winner selected in the previous sweep of its ecotype.
In a subsequent paper, Thompson et al. (2005) examined genomic diversity among Vibrio splendidus isolates from this same site, all within the 99% rRNA identity cluster, by pulsed-field gel electrophoresis. They found astonishing differences in genome size, concluding that “this group consists of at least a thousand distinct genotypes, each occurring at extremely low environmental concentrations (on average, less than one cell per milliliter).” They speculated that “some proportion of the observed genotypic diversity may reflect the differentiation of (sub)populations that are specialized to particular environmental conditions in the complex life-style of vibrios (including free-living and animal- or particle-associated states).” To test this, Hunt et al. (2008) examined particle-associated (three sizes) and free vibrios from samples taken in different seasons and showed that different microdiverse clusters (defined by hsp60 gene sequences, which are more sensitive than SSU rRNA) do apportion themselves differently among fractions and seasons “consistent with our previous suggestion that rRNA gene clusters, which are roughly congruent with the deeply divergent protein-coding gene clusters…represent ecological populations.”
Several other model systems with which we can ask this same basic question—what does sequence microdiversity tell us about genomic and functional cohesiveness of prokaryotic populations at what might traditionally be called the species level?—are currently under intense scrutiny. The results have been interpreted variously by their different investigators, reflecting differing species ontologies. Ward and collaborators (Ward et al. 2006, 2008) have focused on the morphologically identical thermophilic Synechococcus strains growing in hotspring mats in Yellowstone, distinguishable only at the SSU rRNA sequence level or when identical (isoribotypic) there, by ITS sequence differences. Such differences correlate with depth in the mat and with temperature, and this research group, influenced by the theories of Cohan (see below), suggests that such ITS variations delineate separate thermally adapted ecotypes, which for them are the prokaryotic equivalent of eukaryotic species.
At the other end of the spatiotemporal scale are the studies of Chisholm's group with marine Synechococcus and Prochlorococcus (Johnson et al. 2006). Although all strains of the latter exhibit a species-defining 97% identity in SSU rRNA, niche partitioning by temperature, light intensity, and many other parameters can be readily shown (Johnson et al. 2006), and, as with Polz's vibrios, there are substantial differences in gene content (Kettler et al. 2007) between them (see Fig. 2). The latter investigators conclude that because of such genomic flexibility, “we have barely begun to observe the extent of micro-diversity among Prochlorococcus in the ocean. In particular, it will be enlightening to understand the complete genome diversity of the 105 cells in a milliliter of ocean water, and conversely, how widely separated in space two cells with identical genomes might be.”
Banfield and her collaborators have focused much more narrowly in their analyses of natural genomic/phenomic clustering, on DNA extracted from structured acid mine drainage biofilms communities floating in the Richmond Mine (California). Chosen as a low-diversity environment, this system is dominated by four to six “organism types,” both bacterial (Leptospirillum) and archaeal (Ferroplasma), and has proven dauntingly complex and dynamic from many perspectives and types of analysis (geographic, micro-spatial, genomic, and proteomic). Its species-like populations show substantial sequence heterogeneity (sequence microdiversity) and recombination both within and between them (Allen et al. 2007; Simmons et al. 2008).
Our own work bearing on microdiversity and genomic clustering has examined still another extreme environment, solar salterns, dominated by haloarchaea and (as has been recently discovered) Salinibacter (Mongodin et al. 2005). Papke et al. (2003) demonstrated the presence of microdiverse sequence clusters for SSU rRNA and the characteristic haloarchaeal rhodopsins, and documented by MLSA homologous recombination (avid within and substantial between) three “phylogroups” of Halorubrum (Papke et al. 2004, 2007). In these studies, and arguably in all systems discussed above, whether or not such phylogroups (isolates or genomes that cluster in phylogenies based on concatenated gene sequences)—or comparable collections of genomically similar individuals—should be described as species depends on the degree of clustering one requires before making such designation. No one's model of prokaryotic genome evolution would predict gene sequence data that lack all structure, so without some agreement in advance as to the extent of structuring expected, there is no principled way to claim that the existence of species has or has not been shown.
Similarly, no one's model of prokaryotic genome evolution entails that there should be no mapping of genotype to phenotype, so niche partitioning data cannot be taken as evidence for species without some prior understanding as to how finely grained such correlation is expected to be. Models in which each cell of the thousand different Vibrio genotypes Thompson et al. (2005) find in a single small seawater sample has its own nano-niche and thus is its own species cannot be discounted; nor can any particular model for how species are formed be excluded. As Ward et al. (2008) admit, “it is important that we keep an open mind as to how different forces that generate and/or act upon variation are involved in speciation. Also, differing patterns of population genetics highlight the fact that we cannot expect all microbes to evolve in the same way.”
It appears to us that in their quest for order and ways to describe it, the environmental microbiologists are embracing species realism, but in their recognition of the complex genetic and ecological processes and forces at play, are endorsing species pluralism. That this is a problematic combination (Ereshefsky 1998) is evidenced by the fact that many of the papers cited above use the word “species” in introductory or final concluding paragraphs, but in the presentation and interpretation of results the word does no apparent work.
Periodic selection of ecotypes: Cohan's theory
Of the two general sorts of models for prokaryotic speciation, the ecotype model, vigorously championed by Fred Cohan, and the Biological Species Concept, the former has firmer roots in the history of microbiology. It treats bacteria (and by extension archaea) as what they were traditionally thought to be—asexual clones.
How clonal organisms might be grouped into species has long seemed an awkward problem in bookkeeping. As Coyne and Orr (2004) admit, “if one is willing to regard completely asexual clones as distinct units, then one can indeed define and group them into ‘species.’ However, as more complete DNA sequences become available, such species will break down. For one must then delimit species based on differences at single nucleotide sites. Such a practice makes each individual, with its own unique mutations, a distinct species.” How independent clones manage to continue to look pretty much alike is an associated mystery, to be explained somehow by selection. Cohan's ecotype theory (Cohan 2006; Cohan and Perry 2007; Ward et al. 2008) solves both problems—in principle for asexual eukaryotes as well as prokaryotes—by invoking periodic selection, a phenomenon whose discovery long ago by Atwood et al. (1951) played a key role in the early development of bacterial genetics.
In Cohan's model, an asexual clone occupies a finite niche, its numbers kept in check by an environment in which there is limited living space. A mutant type arising within the clonal population that is more fit for whatever reason (generally, more efficient utilization of resources) will outcompete its sisters, and the mutation conferring the advantage will carry the mutant's descendants to fixation. In the process, diversity that has accumulated neutrally in the genomes of the clone's members will be “purged”: There is (by definition) no recombination, and so the genome in which the mutation first occurred sweeps to fixation along with the mutant cells that house it. Thus, within this lineage, which is what Cohan calls an ecotype, diversity stays limited, accumulating only to the extent that it can in the intervals between these sweeps. This is the “microdiversity” commonly observed in environmental sequence data for SSU rRNA or other marker genes. For instance, the data of Acinas et al. (2004), discussed above, which showed an excess of SSU rRNA sequences that were only about 1% different from each other (over expectation from a random birth and death model), can be taken to mean that there is only on average enough time for clones to accumulate that much diversity before the next clonal sweep.
So the cohesive force that allows such clones—ecotypes—to maintain relative homogeneity and evolve together in sync with environmental changes of all sorts is periodic selection. The ecotypes it defines will be more restricted than traditionally recognized bacterial species, but will satisfy many of the criteria enshrined in many species concepts developed for animals: cohesiveness, separation, and ecological differentiation. Existing named bacterial species are in Cohan's view (and in line with the thinking of Konstantinidis and Tiedje [2005]) most often analogous to eukaryotic genera.
What keeps one ecotype from invading the space of another, purging diversity on a grander scale, is that they are different ecotypes. One can use a resource that the other cannot, for instance. So two sister ecotypes can evolve cohesively within their own populations and yet diverge from each other even when they are in the same physical location (“sympatric”). Say they occupy the same chemostat with two substrates, A, which can be used only by ecotype alpha, and B, which can be used only by ecotype beta, because of differences already fixed in their respective genomes. The chemostat can support only so many alphas and so many betas, and, as fitter types arise within each, alpha and beta ecotypes will diverge from each other cohesively at all their loci, through periodic selection events that are “private” to each—as long as recombination between them is precluded.
Cohan has described several variations to the above, his Stable Ecotype model, in which microdiverse gene clusters are the hallmark of ecotypes. With the “Genotype+Boeing” version, rapid trans-global transport by humans makes it possible to find (transiently) multiple sequence clusters within an ecotype. The “Genetic Drift” model recognizes that stochasticity itself is a cohesive force in small populations. In the “Speedy Speciation” and “Species-less” scenarios, selective sweeps come so fast and furiously that diversity has no time to accumulate, at least for the markers chosen. In the “Nano-niche” model, many transient and not completely separated ecotypes are invoked. Although homologous recombination and LGT are discounted as forces of long-term cohesion in all Cohan's periodic selection models, they can be the sources of the genetic novelty that initiates a sweep or establishes a new ecotype.
Following the reasoning of Martin (2002) and Acinas et al. (2004), Cohan has developed an “ecotype simulation” algorithm, which equates a “flair [sic] of diversity” with a potential ecotype (Cohan and Perry 2007), and has shown some correlation with niche differentiation as inferred in a meta-analysis of several microbial ecology studies. In a recent collaborative study of strains of Bacillus simplex and the Bacillus subtilis–Bacillus licheniformis complex from Israel's “Evolution Canyons” (Koeppel et al. 2008), many ecotypes detected by this method “were confirmed to be ecologically distinct, with specialization to different canyon slopes with different solar exposure.”
Periodic selection has enormous intuitive appeal as a process, as an explanation for sequence microdiversity where this is observed, and as a model for prokaryotic population genetics. There is no doubt of its role in generating some of the patterns that have begun to emerge in environmental genetic and metagenomic data sets, although we are not aware of any direct demonstrations of its occurrence outside the lab. But there are several reasons to be cautious about giving it explanatory pride of place. First, there are no meta-analyses or indeed analyses of massive datasets of which we are aware that show that microdiverse clustering is globally more frequent or tighter than stochastic models of birth and death would allow for (Martin 2002; Zhaxybayeva and Gogarten 2004). Achtman and Wagner (2008) cite examples of several extensively studied taxa to which ecotype theory seems not to apply, concluding that the “two main problems with the ecotype concept [are] our lack of understanding of how rapidly diversity is purged and the paucity of observations from nature that support complete purging.” Indeed, in realistically large natural populations, clonal interference might often prevent complete purging in any single sweep (Gerrish and Lenski 1998). Second, in the studies of Polz's group (see also a similar analysis by Jaspers and Overmann [2004]), even microdiverse clusters that are as tight as possible (identical in the sequences of the marker gene under consideration) may differ on an individual-cell-to-individual-cell basis in gene content and (hence) microniche. Third, Simmons et al. (2008), from what may be the most direct metagenomic test of the ecotype theory, conclude that an acid mine Leptospirillum population “does not fit the predictions of the stable ecotype model.” And fourth, there is experimental evidence for more complex evolutionary behavior even in systems much simpler than the soil or the ocean, for instance that of Maharjan et al. (2006) (see also de Visser and Rozen 2006) that followed divergence of Escherichia coli in a chemostat. To quote their summary, “this clonal population radiated into more than five phenotypic clusters within 26 days, with multiple variations in global regulation, metabolic strategies, surface properties, and nutrient permeability pathways. Most isolates belonged to a single ecotype, and neither periodic selection events nor ecological competition for a single niche prevented an adaptive radiation with a single resource. The multidirectional exploration of fitness space is an underestimated ingredient to bacterial success even in unstructured environments.” In a sense, Cohan's Nano-niche model may accommodate this, but at the expense of robbing periodic selection of any power in explaining real data. A one-cell-one-ecotype model may not be an outrageously radical alternative.
These concerns aside, what threatens all ecotype models is homologous recombination: If the rate at which this mixes up alleles in parts of the genome that are not driving selective sweeps exceeds that at which such sweeps are completed, then diversity will not be purged, except at the loci under selection. Periodic selection will have failed as a cohesive force. Similarly, homologous recombination at loci other than those that maintain two sympatric populations as ecologically distinct—such as the A- and B-substrate catabolizing genes that differentiate alpha and beta ecotypes in our example above—will render these ecotypes homogeneous over most of their genomes and prevent their cohesive divergence (or “speciation,” if Cohan's ecotypes are considered species). In fact, the current principal rival of ecotype models as a species concept for prokaryotes is based on homologous recombination.
Homologous recombination and a Biological Species Concept for prokaryotes
Although bacterial “sex” (conjugation) was discovered in the 1940s, microbiologists thought of prokaryotes as more-or-less exclusively asexual (clonal) life forms until shortly before the turn of the millennium. Studies by Milkman, Selander, and others in the 1970s and 1980s using multi-locus-enzyme-electrophoresis (MLEE), a technique borrowed from Drosophila population genetics, appeared to reinforce this notion by showing linkage disequilibrium (little recombination) between chosen markers for E. coli and several other bacteria.
Nevertheless, recombination could be detected at a finer scale by comparing aligned DNA sequences (for instance, in the trp operon; Milkman and Bridges 1990), and a reanalysis of much of the MLEE data by John Maynard Smith and collaborators in 1993 suggested that the case for clonality was not so compelling after all. Indeed, these researchers could “identify four types of bacterial populations: a fully sexual population such as Neisseria gonorrhoeae; a population such as Neiserria meningitidis, which is sexual but, because of its epidemic epidemiology, is superficially clonal; Rhizobium-like populations, which are sexual at the fine scale but do not recombine between populations; Salmonella-like populations that are clonal at all levels” (Smith et al. 1993).
Multi-locus sequence typing (MLST), first developed to characterize (“type”) strains of Neisseria meningitidis and other pathogens (Maiden et al. 1998), has now come to be the dominant method for analysis of population structure with prokaryotes of all kinds (in which context it is more commonly called MLSA). For the most part, MLSA has supported the sort of typology inferred by Maynard Smith et al. (1993) while continuing to surprise us by showing just how not asexual some bacteria (and archaea) can be! Typically, this approach involves PCR amplification of ∼500-bp fragments of a number (usually seven) of orthologous “housekeeping” genes from many (often hundreds of) isolates, their sequencing, and subsequent analyses by a number of specialized programs (available at http://www.mlst.net/). Its use has shown a great number of traditionally recognized bacterial species to be “recombinogenic,” sometimes highly so. In 2001, Spratt and collaborators (Spratt et al. 2001) concluded that “in many species, recombinational replacements contribute more greatly to clonal diversification than do point mutations and, in some species, recombination has been sufficient to eliminate any phylogenetic signal from gene trees.” Turner and Feil (2007) cite a five- to 10-fold excess of events of recombination over mutation for Neisseria mengitidis and Streptococcus pneumoniae, and note that recombination events in general entail more nucleotide substitutions than does mutation, so the ratio of recombination-derived to mutation-derived nucleotide changes will be several fold higher still. In Staphylococcus aureus, on the other hand, mutation outpaces recombination by about 10 to 1: This pathogen is essentially clonal.
Helicobacter pylori, a cause of human ulcers, is likely the most highly recombinogenic bacterium known, although there is some quibbling over how rates should be measured (Falush et al. 2001; Perez-Losada et al. 2006; Suerbaum and Josenhans 2007). About half of all humans carry the organism in their stomachs, having been colonized by one or several strains early in life. Recombination occurs through transformation and incorporation of relatively short stretches of DNA, aided perhaps by the absence of a fully effective mismatch repair system (which functions normally to reduce recombination between sequences that are not highly similar.) Inter-strain, within-host recombination promotes diversification and may be essential for this pathogen to defeat host defenses (Suerbaum and Josenhans 2007).
Homologous recombination may similarly be instrumental in the evolution of pathogenesis in E. coli, once viewed as predominantly clonal. In a collaborative effort involving many of the key labs in the field, MLSA of new and previously characterized isolates from around the globe supports the conclusion that “the genetic structure of E. coli housekeeping genes does not fit a classical clonal framework,” and that “pathogenic strains have accelerated rates of mutation and recombination” (Wirth et al. 2006). By an independent method—mining the alignment of the completed genomes of six E. coli and Shigella flexneri strains for patches of recombinant sequence—Mau et al. (2006) also infer that “the rate of intraspecies recombination in E. coli is much higher than previously appreciated.”
In 1991, in one of the first studies to demonstrate incongruence of phylogenies of different genes from a single collection of strains, Dykhuizen and Green (1991) ventured that recombination might become the basis of a robust bacterial species concept, patterned after Mayr's BSC. They wrote, “Biological species are interbreeding groups of organisms, with each species separated from others through reproductive barriers. This definition implies that the phylogenies of different genes from individuals of the same species should be significantly different, whereas the phylogeny of genes from individuals of different species should not be significantly different. Thus, we have an operational criterion for the defining of bacterial species.” This reasoning informs many contemporary attempts to develop a concept-based species definition for prokaryotes.
It is important to remember that the BSC is a coin with two sides. First, frequent recombination is its cohesive force: Members of a species so defined resemble each other in genotype and phenotype because they draw from a common evolving gene pool, from which diversity is purged on a gene-by-gene basis through selection and drift. Second, members of different (even sibling) species diverge in genotype and phenotype because they do not share a common gene pool: There exist “reproductive barriers.” It is in respect to the nature, strength, and universality of such barriers that the “BSC for prokaryotes” remains problematic.
What might barriers be? Geography per se may not matter much. Although the “everything is everywhere” rubric (for review, see O'Malley 2008) does need to be tempered with a realistic appreciation of relative rates of dispersal and divergence—“geotypes” (locally divergent populations) surely must exist (Papke et al. 2003; Whitaker et al. 2003)—prokaryotes do seem to get around pretty well. Identical ribotypes (and sequences for other genes) are not infrequently found on opposite sides of the globe (Glockner et al. 2000; Massana et al. 2000). Extreme physical or ecological sequestration should comprise a barrier, and there is, for instance, little recombination between populations of several maternally transmitted insect endosymbionts, with Wolbachia being a striking exception (Baldo et al. 2006). Restriction-modification systems may limit recombination, although that is perhaps not their evolutionary function (Kobayashi 2001). The widespread but patchy distribution of such systems among strains of H. pylori seems not to preclude avid recombination, although it may account for the small size of recombination patches (Aras et al. 2002).
More relevant may be the availability, avidity, and host ranges of the conjugative plasmids, phages, and DNA-uptake systems that bring heterologous DNA into cells. In many cases, we might expect that these elements are most active when uptake might be most beneficial (Sorensen et al. 2005), for instance during antibiotic stress or in biofilms, or as a specific example for Vibrio cholerae in the presence of chitin as an indicator of the proximity of its copepod host (Meibom et al. 2005). (Conceivably, the selected function of competence for DNA uptake might be nutritional, only coincidentally favoring genetic exchange [Cameron and Redfield 2006].) Importantly, the active agents of prokaryotic gene transfer themselves, within or between species, are themselves “selfish” genetic elements whose evolutionary interests are best served by frequent and promiscuous exchange. And even when “rampant” on an evolutionary scale, exchange is so infrequent in the lives of individual prokaryotic cells that there is no significant direct selection for decreasing exchange per se, simply to avoid nonproductive mating. (For sexual eukaryotes in which each reproduction requires a genetically compatible partner, there is such selection [Coyne and Orr 2004].) So we do not expect to find evolved barriers to interspecific mating in prokaryotes (except insofar as they result indirectly from selection for resistance to viruses.)
One seemingly insurmountable and intrinsic barrier to exchange by homologous recombination (but not to LGT processes via other pathways of integration) has been embraced by theorists. This is the log-linear decline in recombination frequency with divergence in sequence, demonstrated in several experimental systems by Cohan and collaborators (for instance, Majewski et al. 2000). It is appealing to imagine that cohesive clusters might spontaneously arise by the accumulation of neutral sequence divergences progressively subdividing an initially panmictic population into increasingly nonrecombining subpopulations, which nevertheless continue to maintain within-subpopulation cohesion by recombination. These would be species in the full sense of Ernst Mayr's BSC.
Falush et al. (2006), Fraser et al. (2007), and Hanage et al. (2006b) have developed in silico models that do just this, when the parameters are set appropriately. But, as Hanage et al. note, the successful parameters are not realistic biologically. They write that, “we therefore do not predict speciation in sympatric populations with high rates of recombination, unless the empirically determined reduction in recombination rate with sequence divergence is much steeper than that which has been reported. Based on the reported relationship, we expect distance-scaled recombination to reinforce and maintain genetic separations which are initially created by allopatry or niche differentiation, but not to generate them.”
There are four even more compelling reasons to question the relevance of such idealized neutral models of bacterial speciation, all of which might have the effect of making the reduction in recombination rate with sequence divergence even shallower, and the concept of “reproductive barriers” even more onerous:
-
Genes and parts of genes diverge in sequence at very different rates, and thus will “speciate” at different rates, even in the absence of selection. (That is to say, if (1) recombination falls off as sequences diverge, (2) different genes diverge at different rates, and (3) species are defined by [reduced] rates of between-population recombination, then a population that behaves like two species at one locus could behave as only one at another.) As Retchless and Lawrence (2007) point out, genes that differentiate the niches of two incipient species (and loci closely linked to them) will stop recombining and begin to diverge early in their separation, while at unlinked loci the two populations can behave as one for much longer. Thus, they calculate that “different regions of the Escherichia coli and Salmonella enterica chromosomes diverged [at different times] over a ca. 70 million year period.”
-
Lateral gene transfer can introduce substantial lengths of DNA from quite unrelated sources. Subsequent recombination between the introduced stretch and its source population can mean that different regions of the same genome “belong” to different species (Nesbø et al. 2006). And, minimally, such stretches introduced by LGT will reduce recombination and thus speed divergence in adjacent regions of the recipient genome (Lawrence 2002).
-
More divergent sequences are more likely to encode differently adapted proteins, and thus will more often offer some selective advantage over the sequence they replace. The population genetic modeling of Townsend et al. (2003) led them to conclude that most DNA conferring an advantage will more often come from genomes diverged by 7%–21% (well outside the species, by any definition), in spite of the reduced recombinational integration.
-
With “mutator” mutants affecting the mismatch repair system, these investigators further calculated that the accepted divergence range is increased to 13%–30%, because such mutants have less stringent requirements for sequence matching in recombination. Indeed, there is a well-elaborated theory that much evolution proceeds via “mis-match repair intermezzos” (Denamur et al. 2000). During such episodes, recombination with more divergent versions of resident genes introduces pre-tested variants for evolution to try out, after which the repair deficiency is itself repaired by recombinational replacement of the mutant mutator genes.
Determining whether (in spite of these theoretical objections) homologous recombination really is constrained by “reproductive barriers” poses its own problems, discussed in our next section. But in any case, the factors affecting homologous recombination are so various and contingent, and the conditions under which it alone will create species so constrained, that we believe it cannot be relied on as the fundamental principle of any universally applicable prokaryotic species concept. And, like periodic selection, it is powerless in the face of LGT.
Lateral gene transfer: Acknowledging the elephant
When, scarcely a decade ago, there were only a handful of sequenced prokaryotic genomes, it was already apparent that many would boast a sizeable fraction of genes that were detectably the result of LGT from genomes outside their immediate phylogenetic vicinity (Ochman et al. 2000). But, we think, most microbiologists then thought of such transfers as ancient events, and that there would be little need to sequence more than one genome for each species: the K12 genome would be the E. coli genome.
The comparison of three E. coli genomes (K12, the enterohemorrhagic “hamburger disease” pathogen O157:H7, and the uropathogenic CFT073) reported by Welch et al. (2002) thus gave us a shock. In a since then frequently reproduced Venn diagram, these investigators showed that of the 7638 genes present in at least one of the three strains, only 2998 (39.2%) were present in all. (A new comparison by Rasko et al. [2008], based on 17 genomes predicts a “core” of about 2200 genes, out of a total of about 13,000 as plateau values when this mine has been exhausted.)
Now (November 2008) there are at least 380 groups of two or more (up to 22) bacterial or archaeal genome sequences that could be called conspecific by virtue of their SSU rRNA sequences being >97% identical, and many of these “species” show comparable gene content variability (Fig. 2). Similar conclusions about gene content variation can of course be drawn from unsequenced genomes by comparative genome hybridization analyses (Dorrell et al. 2005; Hotopp et al. 2006; Earl et al. 2008), and as “next generation” methods drop further in cost, we can expect to see more and more within-species comparative datasets.
To reconcile gene content variability with a workable systematics, Lan and Reeves (2000) proposed the “species genome concept,” where the species genome comprises all genes found in all representatives of the species. In this formulation, “the genes of any individual will include two components: the core set of genes and the auxiliary genes. Genes found in most individuals, which we can call the core set of genes for that species, are the genes that determine those properties characteristic of all members of the species. Additionally, each strain will have some auxiliary genes, which determine properties found in some but not all members of the species.” These elements are also basic to the “pangenome” construction of Tettelin et al. (2005) (see also Medini et al. 2005; Ward and Fraser 2005), which in addition embraces the notion that some pangenomes are “closed” (so that a complete genome census might be obtained from a handful of genomes) while others are “open” (new genes appear with each new genome sequence).
Species (however defined or designated) are thus expected to vary in two ways. The first variable, which is the focus of both the ecotype and BSC species concepts, is the genotypic “cohesiveness” of their core genomes—quantifiable, for instance, as the range of ANI values observed in pairwise comparisons between strains. The second variable, poorly addressed by either model, is the similarity of their complement of auxiliary (or “dispensible”) genes, which presumably relates to phenotypic cohesiveness. Konstantinidis and Tiedje (2005) have developed an extremely useful and graphic way of comparing these two measures, and four plots we have prepared using their method are displayed as Figure 2. Although ORFans and prophage-related material comprise a non-negligible fraction of “auxiliary” genes in our “all genes” comparison (Fig. 2, upper left panel), when these values are recalculated using only genes in the Clusters of Orthologous Groups (COG) database, the plots look remarkably similar. And among COGs, functional categories differ in degree of variability in largely predictable ways, which in itself is good evidence that many auxiliary genes play a role in strain biology—they are not all just transient DNA detritus picked up by agents of gene transfer.
To a remarkable degree, genotypic cohesion may be uncoupled from phenotypic cohesion. This undermines the predictive value of ANI or other measures of genotypic similarity, and suggests that populations defined by microdiverse marker gene sequence clusters (or even identical marker gene sequences as in the case of the vibrios of Thompson et al. [2005]) may consist of individuals with a range of phenotypes, even, perhaps, with every cell being its own ecotype. Neither the ecotype nor BSC models can readily accommodate this possibility without seriously compromising their claims to underwrite a meaningful species concept.
The presence of a gene in some but not all strains can reflect either gain or loss since their divergence from a species ancestor (but see below for reservations about the concept of species ancestors). There is a logical argument that gain (through LGT, paralog creation by duplication and divergence, and, rarely, de novo creation) must roughly balance loss. This argument is that, otherwise, genomes would continue to become larger or smaller, untenable as a general proposition. Moreover, in the context of a believable within-species strain phylogeny, gains and losses can in principle be distinguished and enumerated by parsimony based on presence/absence, although there will always be a problem in mistaking loss—which proceeds through slow mutational erosion of detectability—for primitive absence (Zhaxybayeva et al. 2007). Such parsimony-based assessments of rates of gain and loss have recently been made for strains of E. coli, Bacillus, Corynebacterium, and Prochlorococcus (Kettler et al. 2007; Marri et al. 2007; Hao and Golding 2008; van Passel et al. 2008). In each case, hundreds of genes have been gained (by LGT, not duplication or de novo creation) and/or lost in the evolution of the species' lineages.
Hao and Golding (2006) infer that for Bacillus cereus and relatives, rates of gain/loss are “comparable to or greater than the rate of nucleotide substitution.” It is important, we think, to realize that a patchy distribution of “auxiliary” genes among the lineages of a putative species, while compelling evidence for multiple “acquisitions,” need not entail that number of independent events of importation of the gene from outside the group (LGT). Often, the novel gene will have been inserted into one lineage by some homology-independent transposition mechanism early in the group's history, and then “passed back and forth” among lineages by homologous recombination between regions flanking the site of insertion. Gains result when the donor genome has the gene, and loss by precise excision happens when it doesn't, and either might be favored under different conditions: Such auxiliary gene-containing or -deficient loci are simply alternative alleles. Thus, the evidence (for review, see Achtman and Wagner 2008) for an ancient origin of many of a group's auxiliary genes is not inconsistent with rapid gain and loss by lineages within the group.
Many transferred genes are, it is clear, only transient residents of the genomes that receive them. This was predicted by Berg and Kurland (2002) in a modeling study which concluded that “genome size is maintained in microorganisms by a quasi-steady state mechanism in which random fluctuations in the effective acquisition and deletion rates result in genome sizes that vary from patch to patch,” leading to their claim that few acquisitions are retained long enough to be counted in the comparative genomic analyses used in constructing large scale phylogenies, or the universal Tree of Life. This might be so, but the adaptive value of a transferred gene plays out in the very local microecological context of the cell in which it is found. A model in which a “metapopulation” corresponding more or less to a species as recognized by the BSC (or even, in terms of marker gene microdiversity, one of Cohan's ecotypes) actually comprises a myriad of subpopulations (micro-ecotypes) transiently exploiting a myriad of microniches by virtue of genes that they have transiently acquired has considerable appeal.
And, of course, the literature of the last three decades is replete with examples of undeniable or at least highly probable adaptations produced by individual LGT events, although seldom if ever have the effects of such adaptations on fitness been comparatively evaluated. We choose three examples from the recent literature that also bear specifically on some of the issues raised above.
-
Methicillin resistance in the much-feared methicillin-resistant Staphylococcus aureus (MRSA) is encoded by one of several types of a mobile genomic island, SCCmec (Deurenberg and Stobberingh 2008). Although rapid global spread of antibiotic-resistant clones is a widely accepted phenomenon, it appears that MRSA has arisen hundreds of times around the world through independent acquisitions of SCCmec in local MSSA (sensitive) S. aureus populations (Nubel et al. 2008). A population genetic study limited to only resistant isolates will, by under-representing a global population of MSSA from which they have independently arisen, give a false impression of clustering.
-
Streptococcus agalactiae was the first named species described as having an ”open pangenome,” each new sequence revealing about 30 new genes (Tettelin et al. 2005). Brochet et al. (2008), from a combined experimental and computational analysis, concluded that much of the variability among strains of S. agalactiae is the result of introduction—through conjugation and integration by homologous recombination between flanking regions—of large (up to 334 kb) DNA segments. As a result, “each chromosome is a mosaic of large chromosomal fragments from different ancestors suggesting that large DNA exchanges have contributed to the genome dynamics in the natural population.” Thus, patchy distributions of even large segments of “foreign” DNA, which might be difficult to explain as the result of multiple rare independent between-species LGT events, could (as we surmised above) be the result of relatively more frequent within-species homologous recombination. And, such large segments could easily engage in smaller recombinational events, creating genomes whose different parts effectively belong to different species, as suggested by Nesbø et al. (2006).
-
Earl et al. (2008) have been using microarray-based comparative genomic hybrization among strains of Bacillus subtillis all showing >99.8% identity. They note that there is “variability in nearly all ‘functional’ categories of genes, some of which could prove ecologically relevant by changing (expanding or limiting) the environments in which these strains can live. Divergence was observed in genes that encode proteins involved in the uptake and breakdown of carbohydrates (e.g., xylose) and amino acids (e.g., glutamine) in addition to several cell surface-associated proteins, including those involved in environmental sensing. The observed variability among these loci, and others like them, indicates that certain metabolic and environmental-monitoring capabilities might not be required for the life of B. subtilis in all environments.” Although not unique, this “species” should prove a good model for demonstrating just how much gene-content and phenotypic/ecological diversity can be accommodated within groups that form single microdiverse clusters with phylogenetic marker genes.
Clustering of cores: A possible recourse for species monism and realism
With either ecotype or BSC concepts, it seems to make sense to many investigators to define genealogical relationships within and between “species” on the basis of the genes that are shared, the so-called species “core.” And the technique in play that seems most appropriate is MLST (MLSA) applied to “housekeeping genes” that by their conserved and essential functions might be least prone to LGT. In most of the many recent such studies, undertaken primarily for undeniably useful classificatory (“species definition”) purposes, but sometimes more problematically to test species “concepts,” sequences of PCR amplification products for five to 10 such genes from many individual isolates are concatenated (combined as if a single gene) and used to construct trees. Resulting clusters (or clades, when the trees are taken as phylogenies) are evaluated on the basis of their separation from one another, and agreement with species or subspecies is recognized in traditional phenotype-based (or minimally SSU rRNA tree-based) classifications. Although this seems a far safer classification procedure than taking microdiverse clustering of single marker gene sequences as indicators of natural organismal groupings, results are mixed.
Hanage et al. (2005, 2006a) note that concatenated gene sequences support tight and separate clustering of strains corresponding to named Burkholderia pseudomallei and B. thailandensis, and no alleles are shared between them. Because there is frequent recombination between various Neisseria species, on the other hand, clusters based on concatenates are “fuzzy,” and assignment of individuals can be ambiguous. Similarly, there is between-species exchange among Streptococcus pneumoniae, S. pseudopneumoniae, S. mitis, and S. oralis, such that “the individual gene trees completely fail to resolve the streptococcal species clusters identified using the concatenated sequences,” although these latter seem reasonably discrete. It should be noted then, that by the initial criteria of Dykhuizen and Green (1991) cited above, these four “species” would comprise only one. Hanage et al. (2005) themselves muse that “species clusters are not ideal entities with sharp and unambiguous boundaries: instead they come in multiple forms and their fringes, especially in recombinogenic bacteria, may be fuzzy and indistinct.”
Most such work has been done with pathogens, but the observations hold as well for “environmental” prokaryotes. In Papke et al. (2007) we showed that, although discrete “phylogroups” could be resolved with concatenated gene sequences for saltern haloarchaea, phylogenies made with individual genes were variously incongruent: There is substantial recombination between phylogroups. Whether or not phylogroups should be called species is not decidable by further experimentation: What's at issue is the species definition and how much fuzziness we want to accept within it. As another environmental example, Figure 3 shows a graphic summary of phylogenetic analysis of marine cyanobacterial data. Although a robust tree can be made from the collective signal from up to 19 Prochlorococcus and Synechococcus genomes (O. Zhaxybayeva, F. Doolittle, T. Papke, and P. Gogarten, in prep.), individual genes do disagree with this, with undeniable statistical support. This conflicting signal is not noise, but rather evidence of an important evolutionary process—the exchange of genetic information between clustered populations with interpopulation barriers of varying strength. There is for this group, dominant in our oceans, mounting evidence that phages play a key role in shuttling such information back and forth, and for local adaptation—to physical conditions, nutrient and light availability—mediated by gene acquisition and loss (Kettler et al. 2007). “Auxiliary” genes, which make up the majority of this assemblage's “species genome,” might better be seen as context-dependent; gene exchange is the process by which they are recruited into action. The genome core is not immune to such exchange, and so any strain or species phylogeny constructed from the concatenated sequences of shared genes must be considered a useful fiction, an oversimplification of a much more complex evolutionary history.

Phylogenetic relationships among selected genomes in the Prochlorococcus marinus/marine Synechococcus group. Each point in a triangle (simplex) represents a set of orthologous genes that contains at least four analyzed genomes (and as many as 19 genomes from this group). Position of the point in the barycentric coordinate system (triangle) depends on bootstrap support values for each of three possible tree topologies with which each vertex is associated. The closer the point to the vertex, the higher its bootstrap support for that tree topology. Poorly resolved relationships result in points located closer to the center of the triangle. Values at each vertex refer to the number of sets of orthologous genes that support the tree topology at the vertex overall, with at least 80% and at least 90% bootstrap support, respectively. For a full description of the methodology used to analyze embedded quartets, see Zhaxybayeva and Gogarten (2003) and Zhaxybayeva et al. (2006). Genomes are designated by their strain names. (Bold) Genomes of marine Synechococcus spp., (italics) low-light adapted Prochlorococcus marinus genomes, (plain font) Prochlorococcus marinus high-light adapted strains (all genomes are from NCBI's RefSeq database). Full analyses of the phylogenetic relationships within this group as well as details on the selection of sets of orthologous genes and phylogenetic analyses performed will be presented elsewhere (O. Zhaxybayeva, F. Doolittle, T. Papke, and P. Gogarten, in prep.).
Why should there be species?
There must be some degree of fuzziness that is too extreme for us to permit in groups we want to call species, but without prior agreement on this, even species nominalism is unworkable as scientific discourse. The ecotype and BSC concepts might each under some conditions produce clusters so tight that most microbiologists would call them species, but it is not sufficient to show that this is sometimes possible. If we are to be prokaryotic species monists (using one concept to fit all prokaryotic cells), then we must claim that it is always necessary—that whatever process we endorse (periodic selection or homologous recombination, or some as yet undescribed more complex third mechanism) must produce clusters of individuals with the agreed upon level of genomic and/or phenomic cohesion, under all conditions. But it should be transparently clear that under many conditions homologous recombination can frustrate periodic selection in this regard, while LGT and the contingent properties of the recombination system and of the selective forces operating can prevent the creation of the “reproductive barriers” essential to the BSC. Thus, species monism is not tenable.
If we abandon monism and accept (as pluralists) that some prokaryotes form species by periodic selection and some by following the tenets of the BSC (and perhaps some by other, still unknown, mechanisms), then it is very hard not to admit that some prokaryotes may not form species at all. So, “species” is demoted from a category with universal biological application to a descriptor of certain (different) types of population structure that we call by the same term only nominally, and to which not all organisms need belong. That is, species pluralism robs the category species of its claim on reality.
This is not to say that specific groups of organisms (for instance, Helicobacter pylori or Sulfolobus solfataricus) cannot be taken as real. Even for groups that are fuzzier than allowed by whatever criteria are collectively accepted as a definition (for example, Stackebrandt et al. 2002), it can still be useful to apply specific names in a host of practical clinical, environmental, and experimental contexts. This position was likely that of Darwin, who wrote, “I look at the term species as one arbitrarily given for the sake of convenience to a set of individuals resembling each other” (Darwin 1859). And it is certainly that of Ereshefsky (1998), who writes (in the nonmicrobial context): “Nothing I have said casts doubt on the existence of those taxa we call ‘species’. We remain confident that there are such taxa as Homo sapiens and Canis familiaris. Of course, it might be odd to call them ‘species’ in light of the heterogeneity argument [like that presented in this article for species pluralism]… The important point here is that the nonexistence of the species category does not imply that the taxa we call ‘species’ are mere artifacts.”
After species
There is a strong-felt need for a robust prokaryotic species ontology. Koeppel et al. (2008), for instance, state that “to fully understand any community's ecology, we need to identify its ecologically distinct populations and to determine their mutual interactions, because these are the units that contribute uniquely to community assembly, function, and dynamics.” Similarly, Ward et al. (2008) assert that there are “ecologically distinct, species-like groups of bacteria and we believe it is essential to identify and define these populations if we are to develop a predictive understanding of microbial community composition, structure and function.” And, indeed, if there is not such a general thing as prokaryotic species (as monists would have it), or if there are species but not all prokaryotes belong to them (as pluralists must allow), then there can be no satisfactory answers to questions such as (1) how many species of prokaryote are there, globally or even in any one place; (2) what is the typical population size of a prokaryotic species; or (3) are bacterial species generally cosmopolitan? This would be a disappointment perhaps, but it is no excuse for forcing a conceptual straitjacket on unruly data.
And such force may not be necessary. In the absence of species definitions or concepts we can still probe prokaryotic diversity at the sequence level (Huber et al. 2007), and we can still explore correlations between gene content, phenotype, and ecology at the level of cells and populations of cells (Hunt et al. 2008). We can still document recombination between genomes in a complex population (Allen et al. 2007) or reconstruct the metabolic activities of complex microbial communities (Hallam et al. 2004). Although “species” is a useful word in conceptualizing such experiments, it plays no role in collecting or analyzing the data, or making useful predictions from it, as the exercise of re-reading papers like those just cited with the word “species” excised will show. (Indeed, some do not use the word.) We anticipate that as metagenomics and the sophisticated computational environment needed to understand and represent metagenomic data evolve, the word will disappear from scientific literature.
Footnotes
-
↵1 Corresponding author.
E-mail ford{at}dal.ca; fax (902) 494-1355.
-
[Supplemental material is available online at www.genome.org.]
-
Article is online at http://www.genome.org/cgi/doi/10.1101/gr.086645.108.
- Copyright © 2009 by Cold Spring Harbor Laboratory Press








