
Scanning the human genome at kilobase resolution
- Jun Chen1,6,7,
- Yeong C. Kim1,6,
- Yong-Chul Jung1,
- Zhenyu Xuan2,
- Geoff Dworkin3,
- Yanming Zhang4,
- Michael Q. Zhang2, and
- San Ming Wang1,5,8
- 1 Center for Functional Genomics, Division of Medical Genetics, Department of Medicine, ENH Research Institute, Northwestern University, Evanston, Illinois 60201, USA;
- 2 Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724, USA;
- 3 Glenbrook High School, Northbrook, Illinois 60062, USA;
- 4 Section of Hematology/Oncology, University of Chicago Medical Center, Chicago, Illinois 60637, USA;
- 5 Robert H. Lurie Comprehensive Cancer Center, Northwestern University, Chicago, Illinois 60611, USA
-
↵6 These authors contributed equally to this work.
Abstract
Normal genome variation and pathogenic genome alteration frequently affect small regions in the genome. Identifying those genomic changes remains a technical challenge. We report here the development of the DGS (Ditag Genome Scanning) technique for high-resolution analysis of genome structure. The basic features of DGS include (1) use of high-frequent restriction enzymes to fractionate the genome into small fragments; (2) collection of two tags from two ends of a given DNA fragment to form a ditag to represent the fragment; (3) application of the 454 sequencing system to reach a comprehensive ditag sequence collection; (4) determination of the genome origin of ditags by mapping to reference ditags from known genome sequences; (5) use of ditag sequences directly as the sense and antisense PCR primers to amplify the original DNA fragment. To study the relationship between ditags and genome structure, we performed a computational study by using the human genome reference sequences as a model, and analyzed the ditags experimentally collected from the well-characterized normal human DNA GM15510 and the leukemic human DNA of Kasumi-1 cells. Our studies show that DGS provides a kilobase resolution for studying genome structure with high specificity and high genome coverage. DGS can be applied to validate genome assembly, to compare genome similarity and variation in normal populations, and to identify genomic abnormality including insertion, inversion, deletion, translocation, and amplification in pathological genomes such as cancer genomes.
Increasing evidence shows that the genome structure is highly variable within the normal human population (Iafrate et al. 2004; Sebat et al. 2004; Tuzun et al. 2005; Eichler 2006; Feuk et al. 2006; The Human Genome Structural Variation Working Group 2007). Recent studies also indicate that the genome structure in pathological situations, such as in cancer, is also highly altered, reflecting the heterogeneous and progressive nature of disease cells (Sjoblom et al. 2006; Greenman et al. 2007). Systematic analysis of normal genome variation will provide fundamental knowledge to understand the genetic basis for normal human diversity; comprehensive characterization of pathological genome alteration will identify genetic factors contributing to particular diseases, which could be used as diagnostic markers and therapeutic targets.
Genome structural changes range from whole chromosome gain or loss, to subchromosomal changes of translocation, amplification, inversion, insertion, and deletion, to single nucleotide changes including haplotypes and SNPs. Traditional cytogenetic approaches are widely used to successfully identify the changes at the whole chromosome to subchromosome levels (Huret et al. 2003). CGH and array-CGH are becoming commonly used to identify the changes at megabase to submegabase levels (Kallioniemi et al. 1992; Pinkel et al. 1998; Cai et al. 2002). SNP and haplotype studies are enabling the detection of the variation at the single nucleotide level (Sachidanandam et al. 2001; McCarroll et al. 2006). However, detecting the structural changes at kilobase to subkilobase levels remains a technical challenge. Although the genome-tiling array may detect such changes (Bertone et al. 2004; Cheng et al. 2005; Kim et al. 2005; Kapranov et al. 2007), its use in genome structural study is restricted by the higher cost, inability to accurately detect the balanced variations of inversion, insertion and translocation, and inability to detect the genomic contents not present in the reference genome sequences. Alternatively, the DNA sequencing-based approach can detect genome structural changes; its power has been well demonstrated by the fosmid pair-end sequencing study that detected hundreds of genome variations at 40 kb resolution (Tuzun et al. 2005) and the BAC end-sequencing profiling study that identified many genetic abnormalities in cancer cells at 200 kb resolution (Volik et al. 2006). However, the high cost, the complicated process, and the limited throughput capacity of the conventional Sanger sequencing system restrict its routine use for studying genome structure.
Several next-generation DNA sequencing technologies have recently been developed, such as the 454 pyrosequencing technology (Margulies et al. 2005). These technologies are less expensive, simplify the sequencing process, and increase the throughput capacity compared with the Sanger sequencing system, making them potentially useful for sequencing-based genome study (Bentley 2006; Green et al. 2006; Barski et al. 2007; Mikkelsen et al. 2007; Robertson et al. 2007). To explore the potential of using next-generation DNA sequencing technologies to study genome structure, we designed the DGS (Ditag Genome Scanning) technique aiming to analyze the genome structure at kilobase resolution. The basic concept of DGS is to fragment the whole genome into small restriction fragments, to collect two end tags to form ditags to represent the fragments, to use the 454 pyrosequencing system to sequence the ditag population, and to determine the genome origin of each detected fragment by referring to known genome sequences. By using the human genome reference sequences as a model, we performed computational analyses to study the relationship between ditags and genome structure. We also analyzed the ditags experimentally collected from GM15510 DNA, which was characterized by fosmid pair-end sequencing, and the ditags from the genomic DNA of Kasumi-1, a human leukemic cell line that has highly altered genome structure. Our evaluation indicates that DGS provides a powerful tool for genome structural study.
Results
Design of the DGS technique
The DGS process includes the following major steps (Fig. 1): a genomic DNA sample is first fragmented by restriction digestion. The DNA fragments are cloned into plasmid vectors to generate a genomic DNA library. The library is then digested by MmeI to retain two short tags on each site of the cloned fragment in the same vector. The tag-vector-tag fragments are self-ligated to form a ditag. Ditags are released from the vectors, concatemerized, and massively sequenced by using the 454 DNA sequencing system. Ditags are extracted from the sequences based on the restriction sites. The genome origin of ditags is identified by mapping the ditags to a reference ditag database preconstructed based on virtual restriction fragments of known genome sequences. Based on the mapping results, each experimental ditag is classified either as the mapped ditag representing the DNA fragments with normal structure, or the trouble-mapped ditag representing the DNA fragments with different structure (Supplemental Fig. 1).
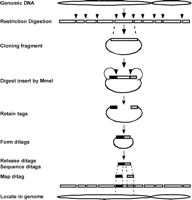
Schematics of the DGS process. See Results section for detailed explanation.
Computational analysis of DGS ditags
Using the human genome reference sequences (HG18) as a model, we studied the relationship between ditag and genome structure.
The total number of ditag-derived bases from 6-base restriction fragments matches the capacity of the 454 sequencing system
The number of restriction fragments from a genome determines the number of ditags, and, therefore, the number of total bases to be sequenced. To control the sequencing cost, we planned to analyze one genome through one 454 sequencing run that provides up to 20 Mbs per run (454 GS20 sequencing system). We analyzed various types of virtual restriction fragments in HG18 to find the range of the total bases from the corresponding ditags. The total number of ditag-derived bases from the 6-base restriction fragments is between 2 and 45 Mb (Table 1), a range that matches the capacity of the 454 sequencing system per run. Since the total bases from 8-base restriction fragments are far lower than the range and the 4-base restriction fragments are far higher than this range (data not shown), the 6-base restriction fragments are suitable for the designed system.
Number of fragments and ditag bases by 6-base restriction enzymes in HG18
Ditags from 6-base restriction fragments provide high resolution and high genome coverage
The size of the restriction DNA fragments represents the resolution. To determine the resolution, we analyzed the size distribution of virtual 6-base restriction fragments in HG18. The result shows that the size distribution varies widely, depending on the type of restriction fragments (Table 1). For example, the total number of Asp130I fragments is 85,897, but the number increases to 1,306,835 for the PstI fragments, due mainly to the changes in the number of smaller fragments. At a 6-kb cut-off, the number of fragments shorter than 6 kb varies over 75-fold between Asp130I fragments and PstI fragments (15,454 for Asp130I fragments vs. 1,169,283 for PstI fragments). In contrast, the number of fragments longer than 6 kb is rather constant between different types of restriction fragments, e.g., the number of fragments longer than 6 kb differs less than twofold between Asp130I and PstI fragments (70,443 for Asp130I fragments vs. 137,552 for PstI fragments). Although the absolute number of longer fragments remains stable in different types of 6-bp restriction fragments, its proportion decreases substantially in higher frequency restriction fragments. Therefore, the resolution of detection can be predetermined by selecting different types of 6-base restriction fragments. By targeting higher frequency restriction fragments, higher resolution and higher genome coverage can be attained. For example, SacI generates 599,852 fragments, of which 71% are shorter than 6 kb and 23% are shorter than 1 kb (Fig. 2).
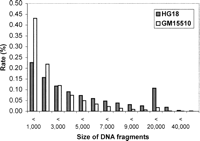
Size distribution of DNA fragments represented by ditags. The empty columns show the size distribution of the total virtual SacI DNA fragments from HG18, and the filled columns represents the size distribution of the virtual DNA fragments in HG18 mapped by GM1510. SacI ditags are shown.
Ditags provide high specificity to represent the original DNA fragments
Ditags have short sequences (on average 34 bp/ditag). We sought to determine whether the ditag population is highly specific in representing their original DNA fragments at the genome level. Our study shows that this is indeed the case. Taking the ditags from SacI fragments as an example. For the 599,805 ditags extracted from these fragments, 94% (565,472) map back specifically to their original fragments. The high specificity is consistent across different chromosomes except chromosome Y (Table 2A). Furthermore, not only ditags from the nonrepetitive sequences, but ditags from the repetitive sequences are also highly specific. Half of the human genome is composed of repetitive DNA. Consequently, 27% of ditags are from the purely repetitive DNA fragments and 40% of ditags are from the fragments across the nonrepetitive and the repetitive DNA (a ditag whose one single tag is from the nonrepetitive region and the other is from the repetitive region). For the ditags from the purely repetitive DNA fragments, 88% remain specific; for the ditags across the repetitive and nonrepetitive regions, 97% are specific (Table 2B). The high specificity of ditags for the repetitive DNA fragments enables use of ditag to analyze the structure in the repetitive regions of the genome.
Specificity of SacI ditags in the human genome sequences (HG18)
Ditag allows for comparison of the similarity and differences between different genomes
In addition to the human genome reference sequences generated by the Human Genome Project, several individual human genomes have recently been sequenced and are publicly available. Using Venter’s genome sequences and HG18 as models, we evaluated the utility of ditags from individual genomes for comparing structural similarities and differences between different genomes. Comparison of the SacI ditags from the two genome sequences shows that 95.9% ditags from Venter’s genome are the same as that from HG18, implying that the DNA fragments represented by these ditags in Venter’s genome have the same structure as the corresponding ones in the HG18. However, 4.1% ditags from Venter’s genome cannot be mapped to HG18 ditags. These ditags represent the DNA fragments that are different between Venter’s genome and HG18 (Table 3). The comparison also shows that HG18 provides higher genome coverage than Venter’s genome sequences, as reflected by its higher number of total ditags, unique ditags, and unmapped ditags that represent the DNA fragments not included in Venter’s genome sequences. Therefore, the HG18 should be used as the optimal genome reference for ditag mapping study.
Comparison between Venter genome ditags and HG18 ditags
Experimental analysis of DGS ditags
To evaluate DGS experimentally, we collected ditags from GM15510 DNA. The same DNA was used for the construction of a fosmid library. This library was pair-end sequenced extensively, with the collection of 1.7 Gb (International Human Genome Study Consortium 2004). These sequences were used for studying genome variation with the identification of 297 variations in the GM15510 genome that differ from the human genome reference sequences (Tuzun et al. 2005). Ditags collected from the same DNA sample are evaluated with the existing rich genomic information, which serves as a control to evaluate DGS for detecting genome structural changes.
Experimental ditag collection
We analyzed two types of restriction fragments from GM15510 DNA: the SacI fragment that has a modest restriction frequency, and the HindIII fragment that has higher restriction frequency (Table 1). By using one 454 GS20 sequencing run, we collected 160,537 raw sequences of 14 Mb from SacI and HindIII ditags. From those sequences, we identified 331,010 ditag copies and 81,890 unique ditags including 46,354 SacI ditags and 35,536 HindIII ditags (Table 4; Supplemental Table 1). The coverage is about 10% for SacI ditags and 5% for HindIII ditags for fragments <6 kb that are clonable by plasmid vector, or 8% for SacI ditags and 4% for HindIII ditags for all fragments of the genome (Table 1). The ratio between the total collected ditag copies and the total unique ditags is about 4:1. In general, the SacI and HindIII data collections are consistent.
Mapping summary for the ditags collected from GM15510 and Kasumi-1 genomes
Constructing a comprehensive ditag reference database
We developed a comprehensive reference ditag database to determine the genome origin of the detected ditags. This database contains virtual ditags extracted from virtual restriction fragments in HG18. In addition, the database also includes reference ditags containing known SNP to identify the experimental ditags containing SNP. Taking advantage of the high-sequence similarity between the human genome and the chimpanzee genome (Li and Saunders 2005), reference ditags were also extracted from the chimpanzee genome reference sequences to identify the ditag whose original fragment is not included in the human genome reference sequences, but which has a homologous counterpart in the chimpanzee genome sequences. The reference database also includes the reference ditags extracted from the variation sequences determined by the GM15510-derived fosmid pair-end sequencing. To identify the ditags from the variations in the available individual human genome sequences, ditags were also extracted from the Celera human genome sequences, the Venter genome sequences, and the unassembled Watson 454 genome sequences. Supplemental Table 2 summarizes the reference ditag information.
The majority of ditags detected in GM15510 maps to the human genome reference sequences
The experimental ditags were mapped first to the reference ditags of HG18. Two-base mismatch was allowed for the ditags not mapped to the reference ditag in order to cover the ditags containing potential sequencing error or SNP. 454 Sequencing cannot determine the precise number of homobases in the homopolymer region (Goldberg et al. 2006). To address this issue, the unmapped ditags with homopolymer bases were identified and mapped to the reference ditags by allowing multiple mismatches for the homobases (Ng et al. 2006). Through these processes, 86.7% of ditags were identified as the mapped ditags, including 78% as the perfectly mapped ditags, 4.8% as the ditags containing sequencing errors or unknown SNP, 0.3% as known SNP-containing ditags, and 2.1% as homopolymer ditags (Table 4). The mapping specificity is equivalent to that of the computational analysis (Supplemental Tables 2, 3). In addition, the ditags mapped solely to the chimpanzee genome sequences account for 0.6% of the total ditags (Table 4; Supplemental Table 4). These ditags likely represent the human DNA fragments missed in the human genome reference sequences. The high mapping rate indicates that, under the given resolution, most of the DNA fragments in the GM15510 genome detected by ditags have the same structure as their corresponding fragments in HG18.
Ditag detects shorter DNA fragments
Detection of shorter DNA fragments implies the high resolution for analyzing genome structure. Computational analysis shows that the proportion of the fragments shorter than 6 kb is dominant among the total fragments generated by many high frequent 6-base restriction enzymes (Table 1). To verify this feature, we analyzed the size distribution of the virtual DNA fragments in HG18 that were detected by the experimental ditags (Fig. 2). Setting 6 kb as the cut-off, 93% of the detected DNA fragments are shorter than 6 kb, and 43% are shorter than 1 kb. These rates are even higher than those found for HG18, in which 72% of the fragments are shorter than 6 kb and 23% of the fragments are shorter than 1 kb. The higher rate of shorter DNA fragments is mostly due to the use of plasmid vector for the cloning that preferably clones shorter fragments. Such size distribution ensures the kilobase resolution for analyzing genome structure.
Ditag detects DNA fragments that vary from the human genome reference sequences
A total of 2,298,774 fosmid end sequences were generated from the GM15510 fosmid library (International Human Genome Study Consortium 2004). The possible variations affecting smaller regions in the GM15510 genome could be present in the sequences. By comparing ditags that are not mapped to HG18 to these sequences, the variation could be identified. We investigated this possibility. Reference ditags were extracted from the sequences containing at least two SacI or HindIII sites detectable by ditags (Fig. 3A). The experimental ditags that do not map to HG18 were mapped against these reference ditags. A total of 58 experimental ditags was identified to map to the fosmid end sequences covering 289 bp on average. Comparing each mapped sequence to HG18 shows variations including novel sequence, deletion, insertion, and ditag sequence changes, including mutation in the restriction site and mismatch in the tag sequences (Fig. 3B,C; Supplemental Table 5). Although these variations were included in the original fosmid sequences, they were not identified as variations at the 40-kb resolution (Tuzun et al. 2005), but were detected by the ditags with the higher resolution. Only a few ditag-detected 55 variations overlap with the 297 variations detected in the GM15510 by the fosmid study. This is likely attributed to the factor that fosmid sequences target large variations covered by two end fosmid sequences, whereas ditags target small variations within a single fosmid sequence. Limited genome coverage by the ditags may also contribute to this issue (Fig. 6A, below).

New variations identified by GM15510 ditags. (A) Comparison between ditags and fosmid sequences. Reference ditags were extracted from fosmid sequences with two restriction sites. Experimental ditags not mapped to HG18 were compared with the fosmid reference ditags to identify the variations within the fosmid sequences. (B) Summary of the results. Comparing the GM15510 experimental ditags to the reference ditags of GM15510 fosmid sequences identified 55 variations. (C) Example of an insertion variation detected by a ditag. This insertion was identified in a fosmid sequence (TI number 146956937) by a ditag AAGCTTTAACACCGTG/GTTTCTTCAAAAGCTT (See Supplemental Table 4). The tag 1 was released from a SacI site in the fosmid sequence, which does not exist in HG18. The fosmid sequences covered by the ditag contain a 24-base insertion.
Recently, three sets of the human genome sequences have become publicly available, including the Celera human genome sequences, the Venter genome sequences, and the unassembled Watson genome sequences that are the raw 454 sequences of ∼250 bp per sequence. These sequences provide a rich source to identify the experimental ditags originated from the variations in individual human genomes. We extracted reference ditags from these three sets of human genome sequences. Comparing with these reference ditags shows that, of the experimental ditags not mapped to HG18, 317 ditags mapped to the Celera genome sequences, 552 ditags mapped to the Venter genome sequences, and 65 ditags mapped to the Watson genome sequence (Table 4; Supplemental Table 6). The relatively higher mapping rate to the Venter genome sequences is likely due to the unassembled nature of the used sequences that contributed more reference ditags than the assembled sequences; the lower mapping rate to the Watson genome sequences is due to the short length of the 454 sequences, of which many do not contribute reference ditags since they do not have two (SacI or HindIII) restriction sites for reference ditag extraction. Together with the GM15510 fosmid-sequence mapping results, of the ditags not mapped to HG18 and chimpanzee genome sequences, 664 ditags mapped to 1007 loci that contain variations across four individual genomes (Fig. 4A,B; Supplemental Tables 5, 6). The 664 ditags account for 0.8% of the total ditags (664/81,890), which approximates the 1% variation in the GM15510 genome determined at 40-kb resolution (Tuzun et al. 2005). Most of the 664 ditags map to more than one individual genome. For example, of the 169 SacI ditags mapped to the Celera genome, 149 also mapped to the Venter genome, 10 to the Watson genome, four to the GM15510 genome, and two mapped to all four individual genomes. The ditags mapped to more than one individual genome represent the genome variations common in these individual genomes.

Genome variations in four individual genomes detected by ditags. The GM15510 ditags not mapped to HG18 were mapped against the reference ditags from the four human genome sequences of GM15510 fosmid sequences, Celera genome sequences, Venter genome sequences, and Watson genome sequences.
Ditag detects unknown genome variation
The current knowledge of normal genome variations is still limited, and many potential genome variations in individual genomes remain to be identified. The ditags not mapped to existing genome sequences provide a resource to identify unknown genome variations. An alternative mapping approach was developed to explore this possibility. In this approach, an experimental ditag not mapped to the reference ditags was first separated into two single ditags, and each single tag was then mapped individually to reference ditags. Using this approach, the majority of the ditags previously not mapped to the reference ditags becomes mapped (Table 4), including ditags with both single tags being mapped and with only one single tag being mapped. Because of the decreased specificity of single tag mapping, however, a single tag could map to multiple location in the genome. For the ditags with both single tags mapped, we divided these into three groups, including group one, in which both mapped to single location, group two, in which one mapped to multiple locations, and group three, in which both mapped to multiple locations. Of these three groups, group one provides the highest mapping accuracy, and group three the lowest mapping accuracy. We predicted the variations of insertion, deletion, inversion, and translocations for ditags in group one and group two. No predictions were made for group three because of the low mapping specificity (Supplemental Tables 1, 8). Ditags with two unmapped single tags may represent the DNA fragments not included in the human genome reference sequences. However, each subgroup may also contain artificial ditags generated by cloning and sequencing errors (see Discussion).
Ditag detects chromosome Y fragments
GM15510 is from a female and, therefore, it has no Y chromosome. However, a group of ditags mapped to the reference ditags from the Y chromosome (Supplemental Table 7). We searched the ditag-covered Y chromosome fragment sequences in the entire genome. Eight loci were identified in chromosomes 1, 8, 9, 10, and 13, but none in chromosome X. Each mapped autosome locus is located within a functional genes, but this is not the case for its counterpart in Y chromosome (Table 5A). Four of these autosome loci, including three SacI fragments and one HindIII fragment, are clustered in chromosome 10 within the first intron of the FANK1 gene (Fibronectin type 3 and ankyrin repeat domains protein 1), a gene containing six ANK repeats and a fibronectin type-III domain (Table 5B). Two of the chromosome 10 fragments were also detected in Kasumi-1 genome, which also lacks Y chromosome (see below).
Mapping ditag-detected chromosome Y fragments to autosomes
Ditag detects cancer genome alterations
Cancer genome structure can be substantially altered from normal genome. We used Kasumi-1 cells as a model to test the power of DGS in detecting genome alterations. Kasumi-1 is a leukemic cell line whose genome varies greatly from the normal genome, as reflected by its complicated karyotype (Asou et al. 1991; Horsley et al. 2006; Supplemental Fig. 1). We collected ditags from Kasumi-1 SacI DNA fragments by using a single 454 sequencing run that doubled the ditag detection over the GM15510 SacI restriction fragments (Table 4; Supplemental Table 8). The ditags collected provide 39% coverage for the fragments <6 kb in HG18, or 28% for the total genome fragments in HG18. The experimental ditags were processed by using the established ditag mapping procedure. The results show the following features.
-
Large genome size. Under a defined scale of ditag sequencing, the ratio between the number of total ditag copies and the number of total unique ditags reflects the relative size of different genomes. The lower ratio represents the larger size and the higher ratio represents the smaller size of the genome. In Kasumi-1, the ratio is 2:1 (350,005 SacI ditag copies generate 168,281 unique ditags), whereas in GM15510 ditags, the ratio is 6:1 (280,481 SacI ditag copies generate 46,354 unique ditags). Consistent with the results from Kasumi-1 karyotyping that show many extra genome contents such as the trisomy 3 and trisomy 8, the size of the Kasumi-1 genome is substantially larger than the GM15510 genome.
-
High frequent genome structural alteration. This is reflected by the high rate of Kasumi-1 ditags not mapped to the human genome reference sequences. Compared with the 86.7% in GM15510, only 73.2% are the mapped ditags in Kasumi-1 ditags. The difference is due largely to the lower rate of perfectly mapped ditags: only 65.2% of Kasumi-1 ditags are perfectly mapped to HG18 in contrast to 78% in GM15510. The lower mapping rate leads to a higher incidence of trouble-mapped ditags: 26.8% of Kasumi-1 ditags are the trouble-mapped ditags, compared with 13.3% of the GM15510 ditags.
-
Presence of normal genome variations. Mapping the ditags to the four individual human genome sequences identified 1198 ditags that represent the variations in normal human genomes (Fig. 4C; Supplemental Table 9). The rate (0.7%) is similar to the one observed in GM15510 ditag-mapped variations (0.8%). Considering that the scale of Kasumi-1 SacI ditag collection doubled that of GM15510, we tested whether the increased ditag detection could detect more variations in GM15510 genome identified by fosmid sequencing. We compared all Kasumi-1 ditags with the reference ditags extracted from the 33 fully sequenced fosmid clones containing the 297 variations. Of the 307 SacI reference ditags from these clones, 123 are mapped by the Kasumi-1 ditags, of which 116 ditags are common to the HG18, whereas seven ditags are located in the variations of four insertions and one deletion in five fosmid clones (Fig. 5A; Supplemental Table 10A). For example, the fosmid variation AC153461 contains an 8002-bp insertion that does not map to HG18. Ten reference ditags are present in this sequence and six are located within the insertion. Of these six reference ditags, five were detected by Kasumi-1 ditags, two of which are across the junctions between the normal sequences and the insertion, and three are purely located within the insertion (Fig. 5B; Supplemental Table 10B). The mapping of Kasumi-1 ditags to the normal variation ditags indicates that Kasumi-1 genome contains the genome variations present in the normal individual genomes.
-
Chromosome Y fragments remaining in Kasumi-1 genome. Kasumi-1 cells originated from a male, but karyotype analyses consistently show that the whole Y chromosome is lost from the cell (Asou et al. 1991; Supplemental Fig. 1). However, 11 ditags map specifically to the reference ditags of chromosome Y (Supplemental Table 7), of which three were also detected in GM15510, and their covered sequences were located in chromosomes 9 and 10 (Table 5). The presence of eight Y chromosome-specific ditags indicates that these chromosome Y fragments did not disappear but integrated into other chromosome(s) in the Kasumi-1 genome.
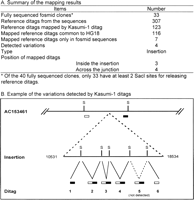
Kasumi-1 genome contains the variations in the GM15510 genome detected by fosmid sequences. (A) Summary of the mapping between the Kasumi-1 ditags and the reference ditags extracted from the variations in the 33 fully sequenced fosmid clones. Four insertion variations were detected by Kasumi-1 ditags that detected the junction and the insertion sequences. (B) Example of the insertion confirmed by ditags. Variation AC153461 contains an 8002-bp insertion. Six ditags detected this insertion, four of which were within the insertion, and two crossed the junctions between the normal sequences and the insertion. (S) SacI restriction site.
Amplifying ditag-detected DNA fragment by using ditag sequences
Although informatics analysis can identify the ditags representing DNA fragments with normal structure or with abnormal structure, physically isolating and sequencing the ditag-detected DNA fragments will be useful to confirm the mapping results and for further study. A ditag is from two ends of a short DNA fragment within a kilobase(s). With 16-bases in each single tag, the two single tags in a ditag can readily serve as the sense and antisense PCR primers to amplify its original DNA fragment. Using this approach, we analyzed 16 GM15510 SacI ditags from the 55 ditag-mapped fosmid end sequences (Fig. 3A) by using the GM15510 DNA as the templates, and 11 ditags were confirmed (Supplemental Table 11A). Five SacI ditags that mapped to fosmid sequences were further tested in a panel of DNA samples of 10 individuals. The result shows that two represent the DNA fragments commonly present in all 10 individuals, and three represent the DNA fragments only existing in five individuals (Fig. 6; Supplemental Table 11B). Further testing of five HindIII ditags in the same DNA panel shows that nearly all are present in each of the 10 individual genomes (Supplemental Fig. 4; Supplemental Table 11B). Sequencing the amplified DNA using different types of ditags including the mapped ditags, the SNP ditags, and the trouble-mapped ditags shows that 60% are consistent with the ditag mapping results (Supplemental Table 12; Supplemental Fig. 2).
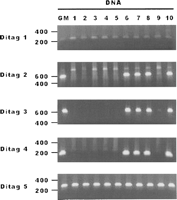
Ditag-detected genome variations in multiple individual genomes. Variations in GM15510 detected by five SacI ditags (Supplemental Table 12B) were tested in a panel of 10 DNA samples (Coriell). (GM) GM15510 DNA was used as the positive control. The results show that two variations are present in all 10 individual genomes and three variations exist in only four individual genomes.
Discussion
Genomic changes affecting small regions exist widely across the human genome (Mills et al. 2006). These genomic changes may play critical roles in various biological processes, such as genome stability, phenotypes, and diseases. While comprehensive identification of these genomic changes is essential, it remains a technical difficulty. Aiming to provide a tool for high-resolution analysis of genome structure, we developed the DGS technology. DGS integrates multiple concepts into a linear system, including restriction digestion for breaking the genome into small fragments, pair-end sequencing used in BAC and fosmid sequencing for structural analysis (International Human Genome Study Consortium 2004; Volik et al. 2006), ditag sequencing used in ChIP-PET for detecting protein-bound DNA fragments (Wei et al. 2006; Dunn et al. 2007), 454 sequencing technology for massive sequencing collection (Margulies et al. 2005), and the known genome sequences as the references for determining the genome origin of the detected ditags. Compared with fosmid pair-end sequencing, DGS increases the resolution toward the kilobase level, thus significantly increasing the power of detecting the variation affecting smaller loci in the genome.
Recently, a technology termed Paired-End Mapping (PEM) was reported (Korbel et al. 2007). While both PEM and DGS aim to analyze genome structure at high-resolution by targeting the two end sequences of the DNA fragments, significant differences exist between these two approaches:
-
Different sources of the end sequences. DGS tags are from a specific type of restriction fragments, whereas PEM tags are from randomly generated fragments.
-
Different resolutions. By selecting highly frequent restriction fragments, DGS provides higher resolution toward subkilobases, whereas PEM provides >3 kb resolution.
-
Different lengths of tag sequences. DGS only sequences 32 bp for each detected fragment, whereas PEM sequences over 100 bases for each detected fragment.
-
Adaptation to different next-generation sequencing systems. The short ditag (32 bp) enables DGS to use all three types of next-generation DNA sequencers for ditag collection (the 454 system [100–200 bp/read], Illumina’s Genome Analyzer [35–50 bp/read], or AB’s SOLiD system [35 bp/read]); only the 454 system can be used for PEM. The much higher sequencing productions of Genome Analyzer (>1 Gb/run) and SOLiD (>1 Gb/run) over 454 system (100 Mb/run) allow DGS to target higher frequent restriction fragments to reach higher genome coverage and resolution.
-
Different mapping processes. To determine the genome origin, DGS ditags map to a preconstructed reference ditag database based on the same type of restriction fragments. The ditag mapping is a simple process that takes minutes to complete the mapping analysis for a full set of ditags in a regular desktop computer. The PEM mapping process is challenging. Because PEM sequences are from randomly generated DNA fragments, each sequence must search against the entire genome. Mapping millions of PEM tag sequences in one data set requires the exhaustive use of large computational power. For example, 200,000 cpu hrs on 440 processors were used to map one set of PEM data to the human genome reference sequences (Korbel et al. 2007). The same issue exists for mapping CHIP-PET ditags to the reference genome sequences, as these ditags are also from the randomly generated DNA fragments (Chiu et al. 2006).
-
Different mapping rates. The mapping rate of DGS ditag is much higher (78% of GM15510 ditags, but only 63% of PEM sequences mapped perfectly to HG18). This is largely due to the fact that a DGS ditag is used as a single unit for mapping, whereas the PEM maps the two end sequences separately (Korbel et al. 2007).
-
Different size of DNA fragments for PCR amplification. The smaller size of fragment represented by ditags than that detected by PEM makes it easier to be amplified by regular PCR.
-
Different lengths of sequences for PCR primer design. DGS ditags provide only 16 bp for primer design, resulting in limitations; PEM provides longer sequences, facilitating better primer design.
Determining genome origin of experimental ditags depends on the reference ditag database. Although they provide the majority of the reference ditags representing the common structure in different individual genomes, the human genome reference sequences were derived from a few individual genomes that lack genome variation information in the human population (Feuk et al. 2006). Therefore, including other genome sequences as the reference ditag sources should help to identify the genome origin of ditags that represent genome variations in different individual genomes. In our study, we constructed a reference ditag database that contains reference ditags extracted from the human genome reference sequences, the SNP sequences, the GM15510 genome fosmid sequences, the Celera genome sequences, the Venter genome sequences, and the Watson genome sequences. This comprehensive reference ditag database has been very useful for identifying the ditags originated from not only the common parts, but also the variable parts among the human genomes. For example, 249 ditags from GM15510 were identified as SNP-containing ditags, and 1010 ditags were identified as the ditags representing human genome variations. The reference ditag database can be continuously expanded to include new genome information, such as these from the database of genome variants (http://projects.tcag.ca/variation/), new genome variations soon to be identified by the Human Genome Variation Project (The Human Genome Structural Variation Working Group 2007), and the newly initiated 1000 Human Genome Project (Hayden 2008).
The detection of Y chromosome ditags in both GM15510 genome and Kasumi-1 genome raises an interesting issue for the origin of Y chromosome. GM15510 is from a female origin, therefore, its genome does not have Y chromosome; the Y chromosome is absent in Kasumi-1 genome, although it was from male origin. Both GM15510 and Kasumi-1 therefore provide a model to identify the origin of Y chromosome. It is considered that autosomes are the origin of sex chromosomes, including X and Y chromosomes, and X contributed a substantial portion of Y chromosome (Bishop et al. 1984; Skaletsky et al. 2003). Therefore, it would be expected that many ditag-detected Y fragments should map to X chromosome. However, all eight Y ditag-detected Y fragments map to autosomes (Table 5). This suggests that the autosomes might directly contribute more contents of Y chromosome than previously thought. In addition, six of the eight detected Y chromosome ditags contain repetitive Y sequences, confirming that ditag can be used for analyzing the structure of repetitive regions that are difficult to analyze so far (Table 2B).
It is unclear whether the normal genome variation is also widely preserved in pathological genomes. In the Kasumi-1 ditag mapping study, 1% of ditags map to normal genome variations distributed in the four individual human genomes, which is close to the 1.2% observed in the GM15510 DNA. This result clearly shows that a pathological genome can also contain normal genome variations. Therefore, when using the genetic changes identified in a pathological genome as potential disease markers, they must be distinguished from normal genome variation.
The trouble-mapped ditags contain information reflecting genetic variations, but may also include experimental artifacts generated during cloning and sequencing processes. For example, many trouble-mapped Kasumi-1 ditags can be classified to represent “translocation,” as each of their single tags mapped to different chromosomes (Supplemental Table 8). However, certain “translocation” ditags are likely from cloning artifacts in which two fragments of different chromosome origin were ligated together and cloning into a single vector that contributes to an artificial “translocation” ditag. Similar results were also observed in mRNA ditags used in fusion transcript detection, in which a substantial number of ditags representing “fusion transcripts” could not be verified (Ruan et al. 2007) and in PEM tags that were estimated to account for ∼2% of total PEM tag sequences (Korbel et al. 2007). Because the probability of forming the same false “translocation” ditag will be rare among the total fragments, these artificial ditags could be largely excluded by eliminating the trouble-mapped ditags with single copy. In addition, the use of ditag sequences as PCR primers has its limitation. Each tag consists of only 16 bases, and not all tags provide ideal sequence composition as PCR primers. These factors could affect the efficiency and specificity of PCR amplification.
In summary, DGS technology provides a useful tool for studying genome structure. It can be applied to validate genome assembly, to compare the genome similarity and variation in normal populations, and to identify genomic abnormalities including insertion, inversion, deletion, and translocation in pathological genomes, such as cancer genomes.
Methods
Computational ditag analysis
The human genome sequences (HG18, http://genome.ucsc.edu/) were used for this study. Virtual restriction fragments from different restriction sites were generated from the sequences. A 17-bp tag was extracted from the virtual fragment of both 5′ end and 3′ end. The two 17-bp tags were then connected to form a virtual ditag to represent the original virtual DNA fragment. The genomic location of each virtual ditag and its original virtual DNA fragment were recorded. Various programs written in Perl were used to study the correlation between the ditags and the genome sequences.
DGS process
Human DNA samples of GM15510 (Coriell) and Kasumi-1 were used for DGS ditag collection. The detailed DGS protocol is provided in the Supplemental material. Briefly, genomic DNA was fractionated by SacI or HindIII restriction digestion. The pZErO vector (Invitrogen) was modified, whereby four wild-type MmeI sites were mutated and two MmeI sites were introduced into the polylinker region next to the SacI or HindIII site. The digested DNA sample was dephosphorylated by phospatase to prevent fragment–fragment ligation, and cloned into the vector to generate a genomic DNA library. The library was then digested by MmeI. The tag-vector-tag fragments were purified and religated to form a ditag library. Ditags from the propagated ditag library were released by SacI or HindIII digestion, purified, and concatemerized by using T4 DNA ligase. The concatermers at 200–500 bp were purified and cloned into the p454-SacI or HindIII vector containing the 454 adaptor sequences (5′tGCCTCCCTCGCGC CATCAG-3′, 5′tGCCTTGCCAGCCCGCTCAG-3′) to form a ditag concatemer library. After library propagation, the concatemers were released from the library by EcoRI and HindIII digestion for the SacI ditag library or NotI digestion for the HindIII ditag library, and gel purified for 454 DNA sequencing collection.
Construction of reference ditag database
The following sequences were used for the construction of the ditag reference database: Human genome reference sequences HG18: http://hgdownload.cse.ucsc.edu/goldenPath/hg18/bigZips/; human dbSNP 126: http://www.ncbi.nlm.nih.gov/SNP/) ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606/; Chimpanzee genome reference sequences: PanTro2, March 2006: http://hgdownload.cse.ucsc.edu/goldenPath/panTro2/bigZips/; GM15510 fosmid paired-end sequences: http://www.ncbi.nlm.nih.gov/Traces/trace.cgi?&cmd=retrieve&val=CENTER_PROJECT%20%3D%20%22G248%22&size=0&retrieve=Submit; Celera genome sequences: http://www.ncbi.nlm.nih.gov/sites/entrez?db=genomeprj&cmd=Link&LinkName=genomeprj_nuccore_wgs&from_uid=1431; Venter genome sequences: ftp://ftp.ncbi.nih.gov/pub/TraceDB/Personal_Genomics/Venter/; Watson raw 454 genome sequences: ftp://ftp.ncbi.nih.gov/pub/TraceDB/Personal_Genomics/Watson/.
Reference ditags were extracted from virtual restriction fragments of each type of DNA sequence, following the same approach described above (Computational ditag analysis). Ditags containing SNPs within the 10 bases were identified by referring to the SNP information and the SNP-ditags were generated by replacing the bases with the SNPs at the corresponding positions. The reference ditags extracted from each type of genome sequences are listed in a Web site (http://projects.bioinformatics. northwestern.edu/sanmingwang/).
Ditag mapping
Ditags were extracted from raw 454 sequences based on SacI or HindIII restriction sites. Each ditag was extracted as 32 bases with 16 bases from each single tag. Ditags containing less than 32 bases were discarded. For mapping to the reference ditags, all experimental ditag data were imported into the reference ditag database using Microsoft Access. Initial mapping was done with exact matches between the experimental ditags and HG18 reference ditags. For these unmapped experimental ditags, 1 base mismatch in each single tag was allowed for the mapping. Possible homopolymer ditags were identified from the nonmapped ditags by searching ditags with more than two homobases. One base of these ditags was stretched, e.g., AAA → AAAA, or shortened, e.g., AAA → AA, and one base at the end of a single stretched tag was removed to retain the normal length of the regular ditag, or one base of the shortened tag was added at the end of a single tag to create four possible cases of A, C, G, and T. These ditags were mapped to the reference ditags again. Exact match was done using SQL commands in Microsoft Access. Based on mapping results, ditags were classified as the mapped ditags or trouble-mapped ditags.
Amplifying the DNA fragments detected by ditags
Supplemental Material provides a detailed protocol. In brief, each single tag of 16 bases in ditag sequences was used to design a sense primer and an antisense (reverse/complementary) primer, with four extra bases (ATTC) added to the 5′ end of the sense primer and TTAG to the 5′ end of the antisense primer to increase the primer length to 20 bp. PCR was performed for 30 cycles at 95°C for 30 sec, 60°C for 30 sec, and 72°C for 60 sec. PCR products were checked on 2% agarose gels, or cloned into the pGEM-T vector (Promega) for sequencing confirmation. The resulting sequence was mapped to the human genome reference sequences through the UCSC genome browser (http://genome.ucsc.edu/cgi-bin/hgGateway). To determine whether the genome variations detected by ditags are present in different individual genomes, a Coriell human DNA panel (Human Variation Panel–Caribbean [GM17350–GM17359]) was used as the templates (http://ccr.coriell.org/).
Acknowledgments
The study was partially supported by NIH grant R01HG002600, the Daniel F. and Ada L. Rice Foundation, the Chicago Biomedical Consortium supported by The Searle Funds at The Chicago Community Trust, and the Mazza Foundation (S.M.W.).
Footnotes
-
↵7 Present address: Department of Marine Technology and Engineering, Xiamen University, China.
-
↵8 Corresponding author.
↵8 E-mail swang1{at}northwestern.edu; fax (224) 364-5003.
-
[Supplemental material is available online at www.genome.org.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.068304.107.
-
- Received September 4, 2007.
- Accepted February 14, 2008.
- Copyright © 2008, Cold Spring Harbor Laboratory Press








