
A new way to explore the world of extracellular protein interactions
Abstract
Eukaryotic genomes encode large numbers of proteins that are either secreted or have exposed extracellular domains. It is highly likely that these proteins facilitate many important biological processes: however, as yet, most remain uncharacterized. Progress in this area of research has been impaired by the lack of a robust screening system that can be used to investigate interactions between large numbers of different extracellular proteins. In this issue, Bushell et al. introduce AVEXIS (avidity-based extracellular interaction screen), a high-throughput screening procedure, which can be used to identify even weak extracellular protein interactions with extremely high confidence. This assay represents an important development in the field of network biology. By combining data from the AVEXIS system with data produced by classical or variant yeast two-hybrid methods, it will be possible to assemble binary protein interaction networks that connect extracellular and intracellular processes. This information will dramatically increase our ability to understand a wide range of physiological processes and facilitate the development of better therapeutic strategies.
The challenge
The availability of genomic sequence for several eukaryotic organisms has provided a valuable insight into the potential complexity and composition of different proteomes. Bioinformatic studies predict that ∼20%–30% of all encoded proteins may be extracellular or membrane associated (Auerbach et al. 2002a). Given the diversity of different eukaryotic cell types and the vast number of biological processes that are in some way dependent upon cell surface interactions, it is perhaps not surprising that a large proportion of eukaryotic genes may encode proteins that are either secreted or have the potential to be exposed on the outer surface of cells. Therefore, one of the greatest challenges of the post-genomic era will be to define the full range of functions for each of the proposed extracellular proteins. For secreted proteins, the goal will be to identify a comprehensive profile of target receptors. However, in the case of transmembrane proteins, the challenge is more complex. This class of proteins provides a vital means of communication between extracellular and intracellular events. As such, these proteins participate in a range of interactions with secreted proteins, proteins on the surface of other cells, proteins within the same membrane, and intracellular proteins (Fig. 1). Despite significant increases in the amount of protein interaction data that is now available for yeast (Fromont-Racine et al. 1997; Ito et al. 2000; Uetz et al. 2000; Gavin et al. 2002; Ho et al. 2002), worm (Walhout et al. 2000; Li et al. 2004), fly (Giot et al. 2003; Stanyon et al. 2004; Formstecher et al. 2005), and humans (Colland et al. 2004; Rual et al. 2005; Stelzl et al. 2005), interactions involving transmembrane or secreted proteins are still significantly underrepresented in most available databases. This lack of data is perhaps not surprising as many of these interactions are intractable to investigation by standard biochemical purification methods, or conventional yeast two-hybrid approaches (Fields and Song 1989). In addition, interactions between extracellular proteins can be weak and transient (van der Merwe and Barclay 1994, 1996). They may also be dependent upon post-translational modifications that are acquired during exocytic transport. These inherent characteristics present severe technical challenges that must be overcome before physiologically relevant interactions can be detected in vitro. In light of these problems, it is not surprising that few people have taken up the challenge of investigating this class of protein interactions on a large scale. In this issue, Bushell et al. (2008) present clear proof of principle that the AVEXIS system can be used to detect even very transient (half-lives ≤ 0.1 sec) interactions between extracellular proteins in a large-scale/high-throughput format.
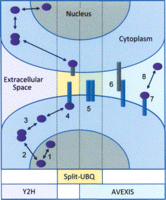
Schematic showing the range of protein interactions that occur in different physiological environments, together with examples of the complementary techniques that can be use to investigate binary protein interactions in each location. While conventional yeast two hybrid (Y2H) methods can be used to investigate many forms of intracellular interactions (1–3), analysis of interactions involving transmembrane or extracellular proteins requires different experimental strategies. In particular, the split ubiquitin system (Split-UBQ) is proving to be a versatile and reliable method of investigating interactions between integral membrane proteins within the same membrane (5) or between the intracellular domains of transmembrane proteins and cytoplasmic proteins (4). In contrast, the AVEXIS and DSCAM assay systems provide important new tools to investigate large numbers of extracellular protein interactions (6–8). Therefore, the combination of these technologies will facilitate the construction of more comprehensive protein interaction networks which link intracellular and extracellular processes.
Unearthing the roots of AVEXIS
As with many important advances in science, the technology and philosophy that underpin the AVEXIS system are in part rooted in a series of pioneering developments. In particular, work by Alan Williams (Williams and Barclay 1988) catalyzed the identification and characterization of the immunoglobulin superfamily (IgSF). In addition to providing the foundations for the large-scale analysis of IgSF proteins described by Bushell et al. (2008), these early studies also demonstrated the immense value of using systematic approaches to drive forward biological research. Subsequent analysis of IgSF proteins led to the identification of several novel interaction partners (including Brown et al. 1998; Wright et al. 2000; Barclay 2003) and the realization that interactions between cell surface receptors can have both low affinities (10–100 μM) and fast dissociation rates (van der Merwe and Barclay 1994, 1996). To circumvent this problem, researchers developed methods of generating oligomeric forms of the extracellular domains of membrane proteins in order to increase avidity and enhance assay sensitivity. A common method of increasing avidity is to fuse the ectodomains of membrane proteins to an immunoglobulin Fc region. This results in the production of recombinant dimeric proteins, which can facilitate the detection of some extracellular protein interactions (Fanslow et al. 1992; Wojtowicz et al. 2007). However, further increases in assay sensitivity were achieved by coupling monomeric fusion proteins to beads (Brown et al. 1998; Wright et al. 2000), or by generating fusions with the cartilage oligomeric matrix protein (COMP) (Tomschy et al.1996; Voulgaraki et al. 2005). This rich heritage of systematic analysis and technology development is clearly reflected in the AVEXIS system. However, the achievement of translating these pioneering results into a robust high-throughput assay that is capable of investigating thousands of potential interactions should not be underestimated. This is a very important contribution to the field of network biology.
Providing the missing link(s)
In brief, AVEXIS is essentially an ELISA style assay (Fig. 2 A,C) in which the complete ectodomains of membrane proteins are expressed as monomeric baits, which are biotinylated to facilitate oriented binding to streptavidin-coated plates. Adsorbed bait proteins are then probed with soluble pentameric prey complexes in which protein ectodomains are fused to the pentamerization domain of COMP (Tomschy et al. 1996; Voulgaraki et al. 2005). Prey constructs also contain the beta-lactamase protein to facilitate the detection of bait–prey interactions. To ensure correct folding and the addition of appropriate post-translational modifications, both bait and prey proteins were expressed as secreted proteins in human HEK293 cells. Finally, assay conditions were optimized to ensure that positive and negative interactions could be differentiated with minimal washing steps, thereby maximizing the potential to detect extremely low affinity or transient interactions. Using a set of gold standard known positive or known negative interactions, assay conditions were optimized to ensure that no false-positive interactions were detected. Once established, these stringent parameters were subsequently used to test interactions between an additional set of 110 proteins, most of which were derived from the zebrafish IgSF. In all, 6105 unique bait/prey combinations were tested in this preliminary study. While the deliberate policy of “zero tolerance” to false-positive data may have resulted in some true-positive interactions being missed, the high stringency of the AVEXIS assays ensures that confidence in positive interactions is extremely high. In fact all “high confidence” positive interactions were reconfirmed when validated and quantified by surface plasmon resonance (SPR). It is interesting, however, to note that Bushell et al. (2008, this issue) only consider interactions to be high confidence if they are reciprocally detected in both possible bait/prey conformations (i.e., bait-A + prey-B and bait-B + prey-A). Interactions detected in only one bait/prey orientation were categorized as “lower confidence” interactions. This level of stringency is indicative of the outstanding technical quality that is inherent throughout this study. However, it is important to note that eight out of nine lower confidence interactions detected in the AVEXIS screen were also reconfirmed when analyzed by SPR. Therefore, within the context of this study lower confidence may not necessarily mean “low confidence.” Fortunately, the investigators do provide a detailed explanation of the criteria used to assign confidence limits.
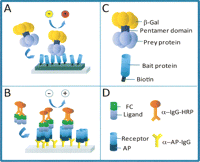
Diagrammatic representation of the AVEXIS (A) and DSCAM (B) assay. A description of the molecular reagents used in each assay system is shown in panels C and D.
While the AVEXIS system clearly offers great potential for the future investigation of extracellular protein interactions, very few homophilic interactions were detected in these initial studies. The reason for this trend is not yet clear. It is possible that some homophilic interactions may be excluded by the stringency of the assay conditions. Alternatively, as the investigators suggest, the lack of homophilic interactions detected by the AVEXIS assay could be due to the relatively high avidity of prey–prey interactions, which may form in preference to homophilic prey-bait interactions. Interestingly, Wojtowicz et al. (2007) recently published a large-scale investigation of extracellular protein interactions in which the vast majority of positive interactions were homophilic. Using a high-throughput ELISA-based assay, Wojtowicz et al. (2007) performed an elegant systematic analysis of the interaction preferences that are exhibited by different isoforms of the Drosophila DSCAM protein. Although AVEXIS and the DSCAM screens are both essentially ELISA-based, high-throughput assays which can be used to investigate thousands of potential extracellular protein interactions, there are significant differences between the two approaches. For example, in the DSCAM assay, bait or “receptor” proteins were fused to the human placental alkaline phosphatase protein (AP in Fig. 2), while prey or “ligand” proteins were fused to the FC region of the human IgG1 protein (Fig. 2 B,D). Receptor and ligand constructs were then incubated together with culture media containing HRP conjugated α-IgG in plates precoated with antibodies against AP. After repeated wash cycles, a colorimetric reporter system was used to quantify HRP activity. Using this system Wojtowicz et al. (2007) analyzed the selective binding properties of thousands of different DSCAM isoform pairs. These studies demonstrate a striking trend toward homophilic interactions among protein isoforms. In fact, the investigators present evidence that >18,000 DSCAM ectodomain isoforms could exhibit homophilic binding specificity. This diversity of homophilic interactions could actually be sufficient to confer unique identities to individual neurons, thereby enabling neurons to distinguish self and non-self interactions (Wojtowicz et al. 2007).
The ability of the DSCAM assay to identify very specific homophilic interactions may be due to the relative strength of interactions between DSCAM isoforms or to subtle differences between the avidity of bait/prey constructs used in this assay compared to the AVEXIS system. However, while the DSCAM assay is clearly excellent at identifying precise homophilic interactions, this approach involves multiple wash cycles and may be less suited to the detection of very weak or transient interactions. Clearly, both systems offer great potential for the future analysis of large numbers of extracellular protein interactions. Indeed, the complementary features of each assay system may well broaden the spectrum of extracellular protein interactions that can be investigated in a high-throughput format.
To date, the AVEXIS system has predominantly been used to investigate interactions between members of the zebrafish IgSF. This is undoubtedly a very powerful system that provides the potential to combine high confidence protein interaction data with genetic perturbation phenotypes, in situ hybridization patterns, and protein expression profiles. Using this approach, Bushell et al. (2008) have investigated the distribution of novel high confidence interaction partners during the early stages of zebra- fish development. Significantly, these data show that several high confidence interaction partners were expressed within the same cells, or on discrete populations of cells that are juxtaposed during development. No doubt future studies in this model organism will provide valuable insights into the potential function of orthologous proteins in other eukaryotic organisms, which are far less tractable to this form of in vivo analysis.
Future directions
As current human protein interaction networks are predominantly composed of intracellular interactions (Futschik et al. 2007), the development of two assays that are capable of screening thousands of potential extracellular protein interactions is a major contribution to the field of network biology. In order to provide maximum utility to both biological and medical research, it will be important to use these new approaches to generate protein interaction maps that depict routes of connectivity between extracellular and intracellular networks (Fig. 1). Given that we now have the ability to investigate large numbers of extracellular protein interactions, and the fantastic contributions that are currently being made by the split ubiquitin system (Johnsson and Varshavsky 1994; Auerbach et al. 2002b; Thaminy et al. 2003), it should eventually be possible to construct protein interaction networks that extend seamlessly beyond the boundaries of the cell to provide a detailed picture of receptor–ligand interactions and intercellular communication networks. Clearly, this would be a major achievement that would significantly advance the development of new therapeutic strategies.
With respect to the analysis of interactions between human extracellular proteins, it is important to emphasize that the AVEXIS system is perfectly suited to this challenge. Not only are bait and prey constructs routinely generated in human cells, interactions between a series of known human interaction partners were replicated in the AVEXIS system in order to facilitate assay optimization. Therefore, with the expected availability of large numbers of expression clones from the human ORFeome initiative, the AVEXIS system should provide an excellent method for performing a large-scale investigation of interactions between human extracellular proteins. Although this is a tempting prospect, it is also important to remember that there is an absolute need to build high-quality binary interaction maps for both humans and model organisms. Indeed, our ability to fully exploit human protein interaction networks will be directly dependent upon our ability to accurately extrapolate functional and genetic information from model organisms. This will require a detailed understanding of the similarities and differences that exist between the different interactomes. This information will be essential when considering the identification of novel therapeutic targets where efficacy, functional redundancy, and potential side effects may all depend on subtle variations in proteome composition and network structure. Clearly, model organisms will always provide opportunities for network perturbation and in vivo investigation that are not possible in humans. Therefore, without high density maps for model organisms, progress in human biology will be seriously impaired. As the AVEXIS system can be applied to many systems, it should prove to be an invaluable tool, which is capable of driving forward research into the relatively unexplored world of extracellular protein interactions and cell–cell communication networks.
Footnotes
-
↵1 Corresponding author.
↵1 E-mail c.sanderson{at}liv.ac.uk; Fax 44-(0)-151-7944434.
-
Article is online at http://www.genome.org/cgi/doi/10.1101/gr.074583.107.
- Copyright © 2008, Cold Spring Harbor Laboratory Press








