
Abstract
Chlamydia trachomatis is the most common cause of sexually transmitted infections in the UK, a statistic that is also reflected globally. There are three biovariants of C. trachomatis: trachoma (serotypes A–C) and two sexually transmitted pathovars; serotypes D–K and lyphogranuloma venereum (LGV). Trachoma isolates and the sexually transmitted serotypes D–K are noninvasive, whereas the LGV strains are invasive, causing a disseminating infection of the local draining lymph nodes. Genome sequences are available for single isolates from the trachoma (serotype A) and sexually transmitted (serotype D) biotypes. We sequenced two isolates from the remaining biotype, LGV, a long-term laboratory passaged strain and the recent “epidemic” LGV isolate-causing proctitis. Although the genome of the LGV strain shows no additional genes that could account for the differences in disease outcome, we found evidence of functional gene loss and identified regions of heightened sequence variation that have previously been shown to be important sites for interstrain recombination. We have used new sequencing technologies to show that the recent clinical LGV isolate causing proctitis is unlikely to be a newly emerged strain but is most probably an old strain with relatively new clinical manifestations.
Chlamydia trachomatis is the major cause of sexually transmitted infections (STIs) globally with an estimated 89 million cases in 1995 (Peeling and Brunham 1996). C. trachomatis isolates are classified serologically with 15 serovariants, based on the major outer membrane protein (OmpA). Trachoma is caused by serovars A–C, whereas serovars D–K and L1, L2, and L3 are associated with sexually transmitted infections. Serovars D–K cause cervicitis in women and urogenital infections in men, and L1, L2, and L3 represent the three different serovars causing lymphogranuloma vernerum (LGV).
C. trachomatis-causing LGV are much more invasive than serovars A–K; they cause systemic infections, infect monocytes, and disseminate to the local lymph nodes, where they can cause large swellings characteristic of bubonic diseases. LGV serovars are endemic in parts of Africa, South East Asia, South America, and the Caribbean (Viravan et al. 1996; Behets et al. 1999; Mabey and Peeling 2002). The incidence of LGV in the West is low, and most of the reported cases are likely to have been imported following travel to endemic countries. However, a recent European “outbreak” of LGV in men who have sex with men (MSM) has been reported (Nieuwenhuis et al. 2003). These patients presented with proctitis rather than genital ulceration and the typical inguinal buboes, characteristic of LGV in the tropics, which were absent. These less obvious clinical features likely explain the initial difficulties encountered in describing the outbreak. It has been speculated that the European outbreak LGV strains causing proctitis represent a new emerging infection. This is supported by the observed variations in the ompA gene (ompA has been used to “genotype” chlamydial isolates), which indicates that these European proctitis isolates are a new variant of the LGV serovar L2, known as L2b. Conversely, it has also been suggested that rectal chlamydial infections are common, but their detection has not, until recently, become part of a routine and accredited testing protocol. This may infer, as has been suggested, that rectal LGV has been present for years and to attribute the newly observed proctitis infections to a new epidemic is incorrect (Schachter and Moncada 2005). In support of this, L2b was identifed in San Francisco in the 1980s (Spaargaren et al. 2005).
Complete genome sequences are only available for a single ocular and a single genital-tract isolate of C. trachomatis, Har-13 and UW-3, respectively (Stephens et al. 1998; Carlson et al. 2005). Comparison of these genomes revealed a very high level of synteny and genome-sequence conservation, although some gene differences were identified that may account for the differing disease outcomes associated with the different biovars (Caldwell et al. 2003; Carlson et al. 2004). Thus, comparison of an LGV isolate with these related strains is likely to provide further insights into the mechanisms that may influence disease outcome and the underlying genetic differences that differentiate these biovariants.
The aim of this study was to complete the genomic analysis of the remaining C. trachomatis biovariant, LGV. The LGV isolate sequenced, strain L2/434/Bu (referred to as C. trachomatis strain L2) was originally isolated from an inguinal bubo of a LGV case 40 yr ago in California (Schachter and Moncada 2005). In addition, we also obtained a recent clinical LGV isolate from a patient with proctitis in London (Table 1). Comparison of the genomes of these two strains should indicate the evolutionary origins of these isolates and answer the question of whether the proctitis “outbreak” is due to a new emerging C. trachomatis LGV biovariant or part of an ongoing, but until recently, undetected disease.
General properties of C. trachomatis genomes
Results
General features of the C. trachomatis strain L2 genome and comparisons with ocular and genital isolates
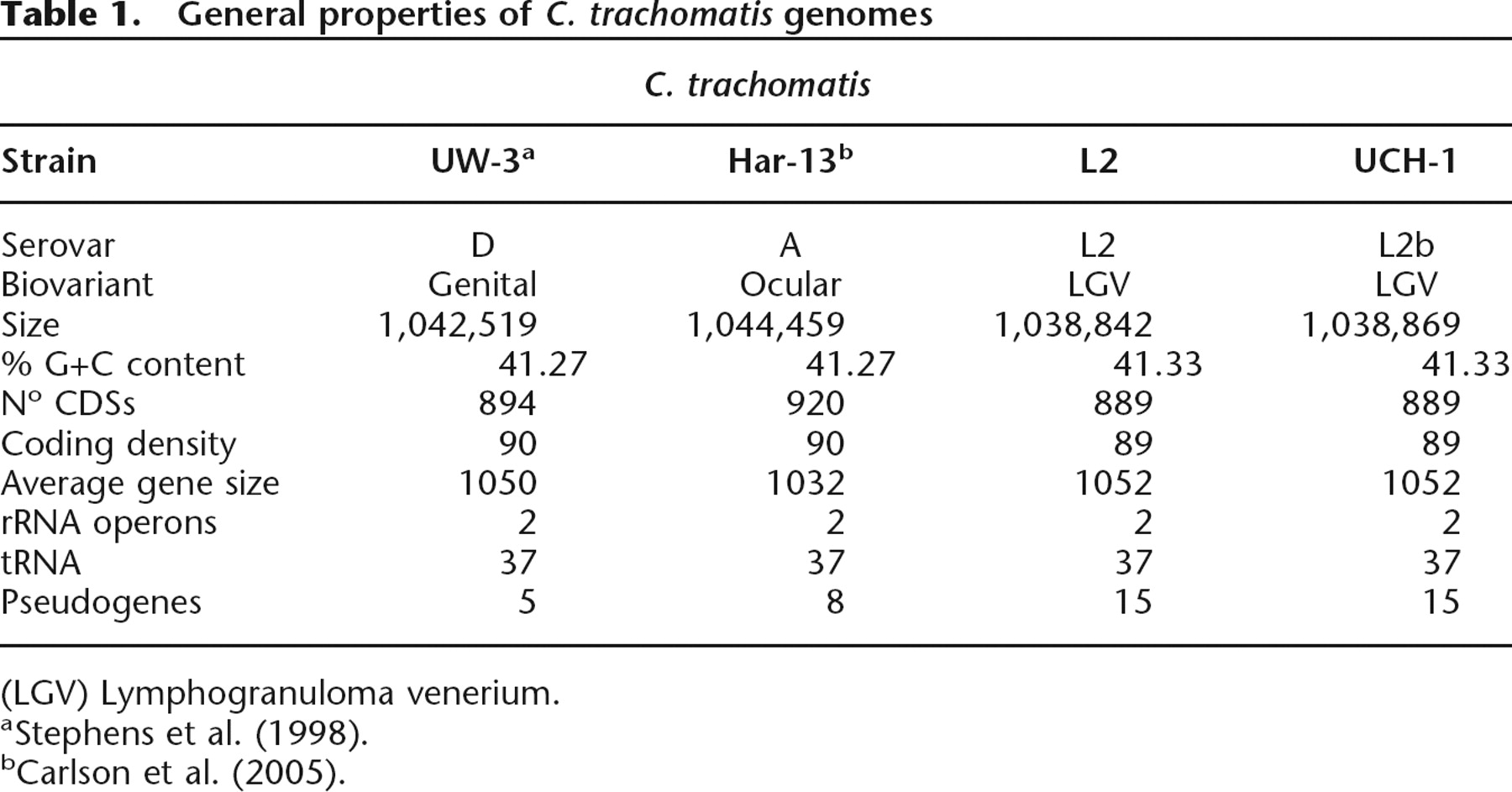
The genome of C. trachomatis strain L2 is composed of a single circular chromosome of 1.039 Mb and a plasmid of 7499 bp, predicted to encode 889 and eight coding sequences (CDS), respectively (Fig. 1; Table 1). The plasmid was identical in sequence and gene content to that previously published for this strain (Comanducci et al. 1988).
Circular representation of the C. trachomatis strain L2 chromosome. The outer scale shows the size in base pairs. From the outside in, circles 1 and 2 show the position of CDSs transcribed in a clockwise and anticlockwise direction. Using the published gene predictions for C. trachomatis strains UW-3 and Har-13, the strain L2 CDSs have been color-coded depending on whether they are: (blue) predicted and intact in all isolates; (pink) predicted and intact in L2 and UW-3; (green) predicted and intact in L2 and Har-13; (orange) defunct in L2, predicted and intact in Har-13 and UW-3; (red) unique to L2; (brown) defunct in all isolates. (Circles 3–10) C. trachomatis strain L2 CDSs that are present/absent in: C. muridarum (circles 3 and 4; present gray; absent yellow), Cp. felis (circles 5 and 6; present green; absent pink), Cp. caviae (circles 7 and 8; present pink; absent blue), and Cp. pneumoniae (circles 9 and 10; present red; absent blue) by reciprocal FASTA analysis. Circle 11 shows a plot of G+C content (in a 0.5-kb window); circle 12 shows a plot of GC skew ([G-C]/[G+C]; in a 0.5-kb window). The position of the PZ (pink) and the PLD CDSs in locus 1 and 3 are numbered accordingly and marked (red). Since the gene content of strain L2 and UCH-1 are essentially identical, these data apply equally to both isolates.
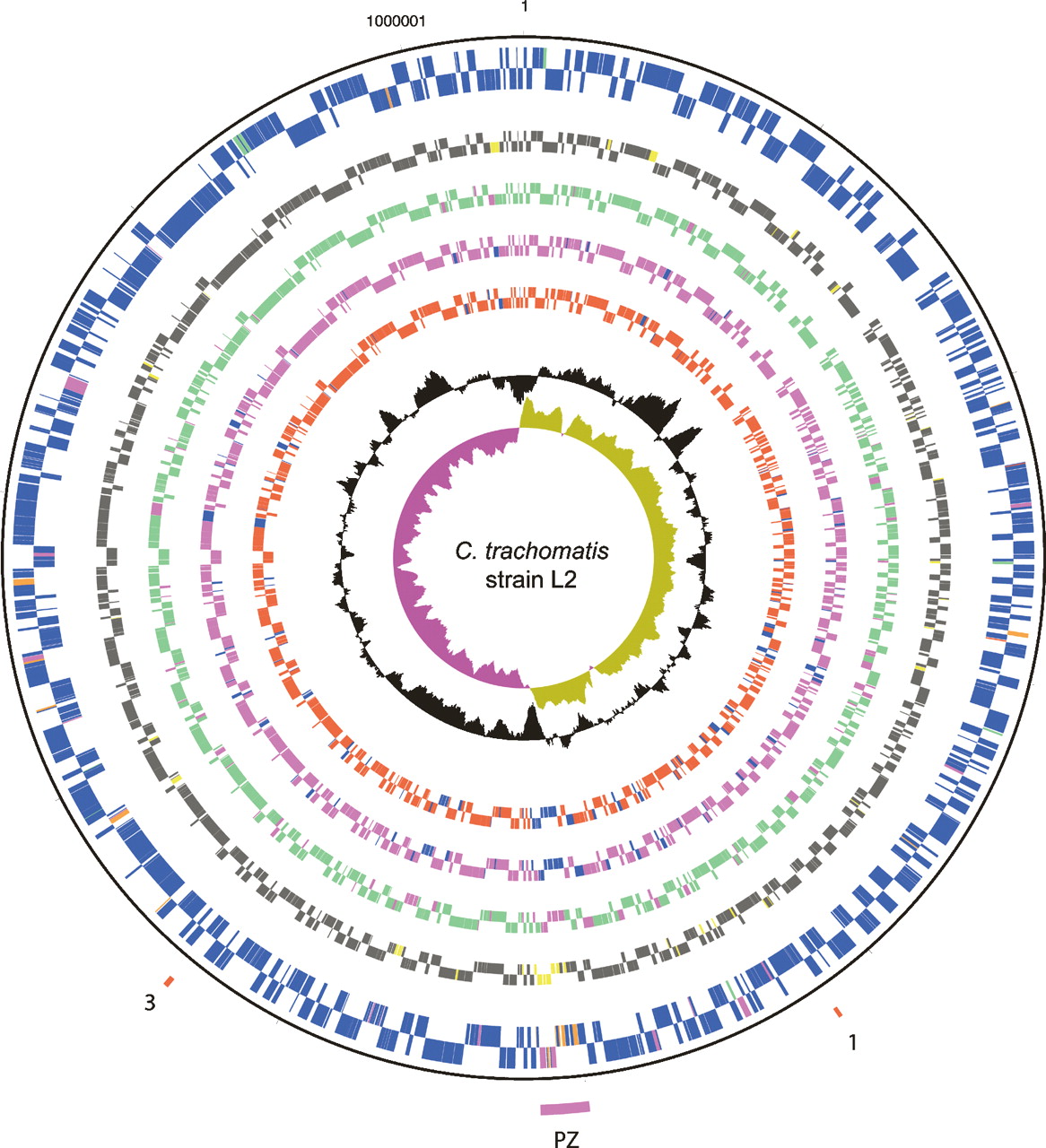
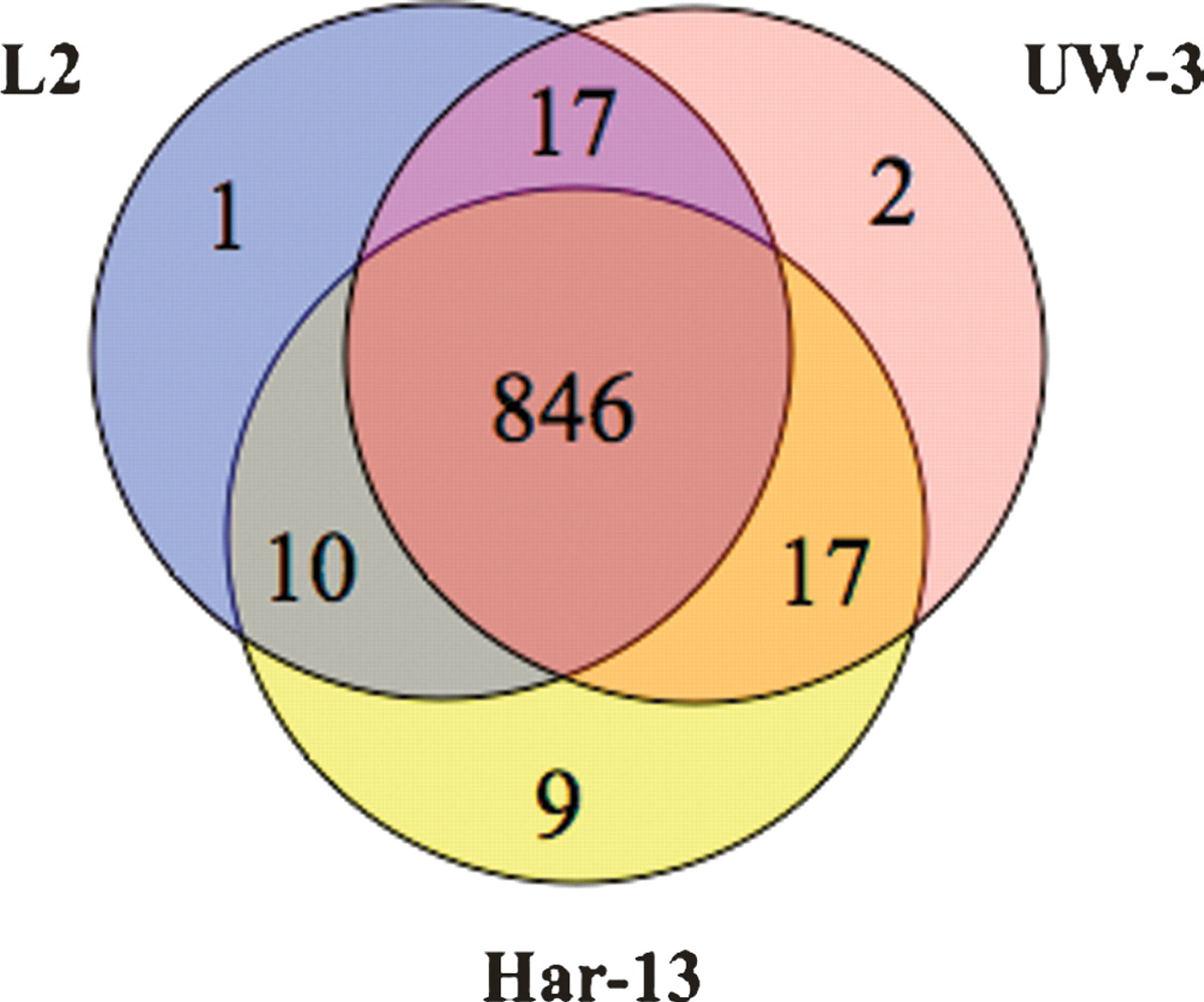
Comparison of the strain L2 genome sequence with those of strains C. trachomatis strain UW-3 (strain UW-3) and C. trachomatis strain Har-13 (strain Har-13) (Stephens et al. 1998; Carlson et al. 2005) showed that the genomes of all three isolates are similar in size, the number of CDSs, and nucleotide composition (Table 1). A detailed comparison of the predicted CDSs showed that 846 CDSs were apparently intact and common to all three genomes (Fig. 2). Moreover, the median nucleotide identity of CDS shared between strain L2 and either Har-13 or UW-3 was 99.53% and 99.55%, respectively. CDSs that were present in two or less of the three genomes could be entirely accounted for by either in silico differences in gene prediction, or as a consequence of functional gene loss (deletion and pseudogene formation) (Table 2), indicating that recent gene acquisition has not played a role in changes in disease causation in this group of organisms. To highlight possible phenotypic differences in the coding capacity of these C. trachomatis strains, pseudogenes were not included in our comparisons described by Figure 2, but they are described in detail in Supplemental Table 1 and discussed below.
A description of the C. trachomatis strain L2 pseudogenes identified by whole-genome comparisons with C. trachomatis strains UW-3 and Har-13
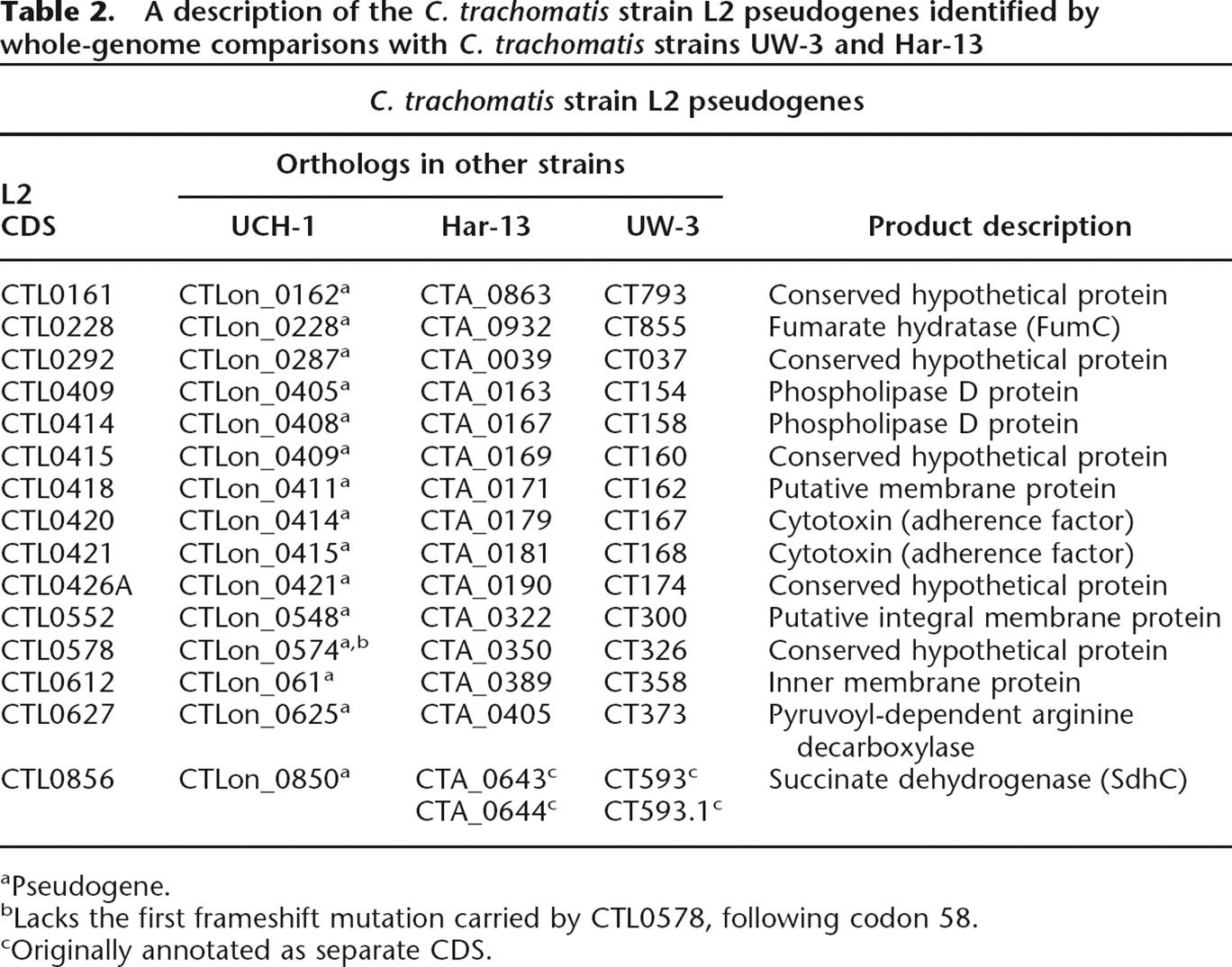
aPseudogene.
bLacks the first frameshift mutation carried by CTL0578, following codon 58.
cOriginally annotated as separate CDS.
Distribution of predicted and functional CDSs shared between C. trachomatis strains L2, UW-3, and Har-13. The Venn diagram shows the number of CDS predicted to be functional, that are unique or shared, between one or more C. trachomatis isolates (see Methods). Pseudogenes were counted as absent in this analysis.
Wider comparisons were also performed between strain L2 and more distant members of the Chlamydiaceae, including Chlamydophila pneumoniae (Read et al. 2000), Chlamydia muridarum (Read et al. 2000), Chlamydophila caviae (Read et al. 2003), and Chlamydophila felis (Azuma et al. 2006) (Fig. 1). These members of the Chlamydiaceae are associated with hosts as diverse as humans and frogs and can cause infections ranging from human adult-onset asthma to feline conjunctivitis (Saikku 1999; Longbottom and Coulter 2003). Despite the considerable variations in disease and host range, the chlamydial genomes are remarkably similar in content: 834, 784, 783, and 777 orthologs of strain L2 CDSs were detected in C. muridarum, Cp. felis, Cp. Caviae, and Cp. pneumoniae, respectively (Fig. 1).
These whole-genome comparisons agreed with previous findings showing that the Chlamydiaceae are extremely similar in gene content, even amongst more distantly related isolates (Read et al. 2003; Carlson et al. 2005; Thomson et al. 2005). Since there was no evidence of recent gene acquisition in strain L2, we concentrated our analysis on the apparent loss of gene function (Table 2).
Evidence of functional loss
The plasticity zone (PZ)
The plasticity zone (PZ) is the site of the most extensive variation in sequence and gene content between chlamydial genomes (Read et al. 2000, 2003). The variation in size of the C. trachomatis PZ is largely due to differential deletion of the cytotoxin gene(s), which have been almost entirely deleted from strain L2, leaving two gene remnants CTL0420 and CTL0421 (Belland et al. 2001; Carlson et al. 2004) (Fig. 3).
Comparison of the PZ locus of C. trachomatis strains L2, UW-3, and Har-13 ACT comparison (http://www.sanger.ac.uk/Software/ACT) of amino acid matches between the complete six-frame translations (computed using TBLASTX) of representatives of the PZ regions of the three sequenced C. trachomatis genomes: Har-13, C. trachomatis strain Har-13; UW-3, C. trachomatis strain UW-3; and L2, C. trachomatis strain L2. The red bars spanning between the genomes represent individual TBLASTX matches. Forward and reverse strands of DNA are shown for each genome (dark-gray lines). CDS are marked as colored boxes positioned on the three forward and three reverse translation reading frames (pale-gray lines). Regions mentioned in the text are marked and the CDSs labeled and color coded: Cytotoxin gene fragments (yellow), tryptophan biosynthetic genes (intact pale green; pseudogene orange), and phospholipase D (Locus 2; orthologous PLD CDSs are colored similarly). PZ regions are marked (purple box on DNA lines). The position of deletion events (*) and frameshift mutations (black arrowheads) within the PLD CDS are marked. Multiple systematic gene identifiers contained within parentheses indicates that the CDS was originally annotated as two CDSs.
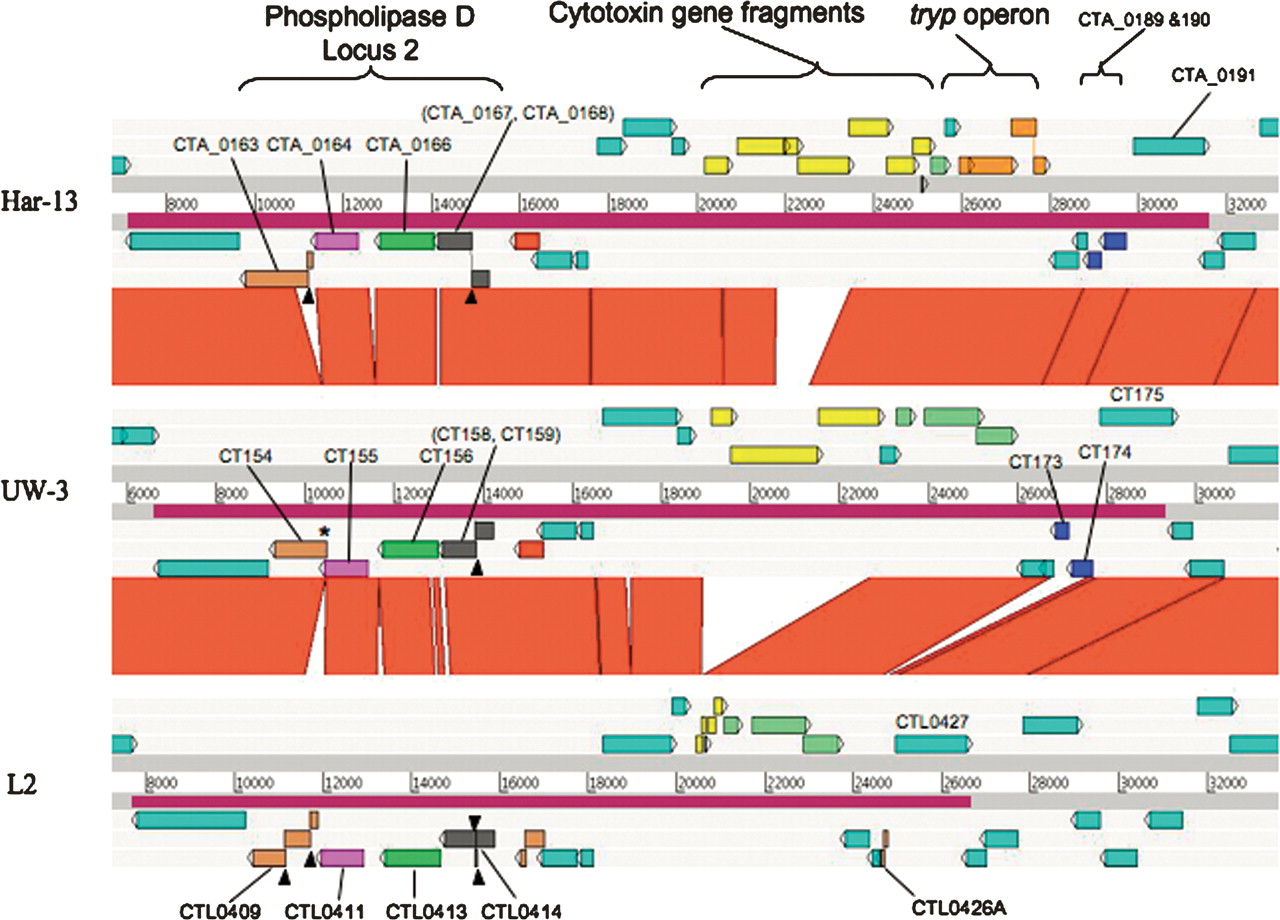
However, there is further evidence for genome decay in strain L2; CTL0426A is a remnant formed by the likely deletion of two CDS, orthologs of which (CT173/CTA_0189 and CT174/CTA_190; unknown function) remain present and intact in strains UW-3 and Har-13 (Fig. 3). Other CDSs within the PZ that show variation include those encoding Phospholipase D (PLD). The PZ encodes four PLD CDSs (CTL0409, CTL0411, CTL0413, and CTL0414; PLD locus 2), which show extensive variation in sequence between isolates. Whilst orthologs of CTL0411 and CTL0413 in C. trachomatis strains UW-3 and Har-13 appear intact and conserved in sequence, those for CTL0409 and CTL0414 have suffered multiple independently acquired frameshift mutations and deletion events (summarized in Fig. 3).
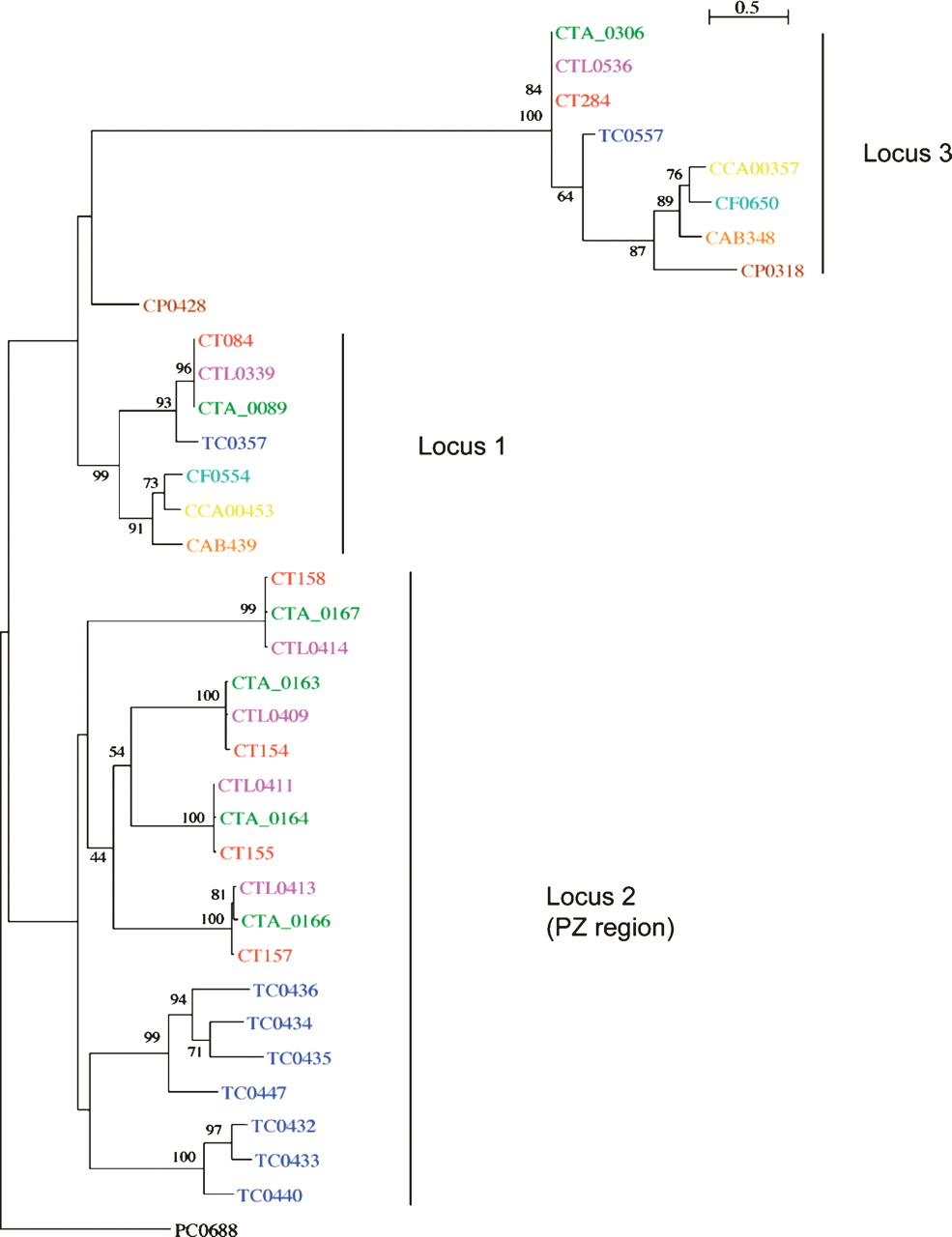
The PZ PLDs are known to be unique to C. trachomatis and the related rodent pathogen, C. muridarum (Nelson et al. 2006). However, there are two other non-PZ-located PLDs in strain L2: CTL0339 and CTL0536 located in locus 1 and 3, respectively (Fig. 1; Nelson et al. 2006; this study). The PLDs from locus 1 and 3 are conserved in Chlamydiaceae, with orthologs present in most other sequenced chlamydial genomes. Phylogenetic analysis of all of the C. trachomatis PLDs showed that the PLDs from the different loci are phylogenetically distinct (Fig. 4). Interestingly, the topology of the phylogenetic tree for the PZ PLDs suggests that they have arisen by paralogous expansion since the divergence from a common ancestor.
Phylogenetic relationships of chlamydial phospholipase D proteins. The protein names have been colored to indicate chlamydial strains: C. trachomatis strains: L2 (pink), UW-3 (red), and Har-13 (green); C. muridarum (dark blue); Cp. felis (light blue); Cp. caviae (yellow); Cp. abortus (orange); Cp. pneumoniae (brown); Candidatus Protochlamydia amoebophila UWE25 (black). Maximum likelihood tree built from protein sequences, using ClustalX, Phylip (Version 3.6), and NJplot. The numbers at the tree branches are percentage bootstrap values indicating the confident levels at that node where congruent. The bar indicates the genetic distance between species (one substitution per 100) as displayed in the branch lengths.
Metabolism
There is evidence of functional gene loss outside of the PZ region of strain L2. CDSs CTL0228 and CTL0856 are pseudogenes of fumarate hydratase (fumC; fumarase C) and succinate dehydrogenase (sdhC; cytochrome B subunit), respectively. Fumarases are involved in the citric acid cycle, either reversibly converting fumarate into L-malate or converting oxaloacetate into succinate, depending on oxygen availability (Guest and Roberts 1983; Guest et al. 1985). Whilst fumC appears intact in strains UW-3 and Har-13 (CTA_0932 and CT855, respectively), the strain L2 ortholog, CTL0228, carries two frameshift mutations. Our findings further confirm the previous observations, in which a truncated FumC was detected in strain L2 during 2D gel/proteomic analysis (Shaw et al. 2002).
Like FumC, succinate dehydrogenase is also required for the metabolism of fumarate by facilitating the aerobic interconversion of fumarate and succinate. In Escherichia coli, succinate dehydrogenase is encoded by four genes, sdhA–D. sdhA and sdhB encode the succinate dehydrogenase flavoprotein and iron–sulphur protein subunits, respectively. SdhA and SdhB are both anchored to the cytoplasmic membrane by SdhC and SdhD. SdhC also possesses a cytochrome b556 domain involved in electron transport.
Analysis of the C. trachomatis genome sequences showed that all carried orthologs of the genes sdhA, sdhB, and sdhC (CTL0854–CTL0856), but none possessed sdhD homologs. However, it is evident that whilst the sdhA and sdhB remain intact, the orthologs of sdhC in all of the sequenced C. trachomatis carry the same frameshift mutation (following codon 103), as well as other additional point mutations (Table 2). This appears to be specific to C. trachomatis, since these genes are intact in the other sequenced Chlamydiaceae (data not shown).
Other potentially important pseudogenes lie within a cluster of two CDSs encoding a pyruvoyl-dependent arginine decarboxylase (CTL0627; PvlArgDC) and an arginine/ornithine antiporter (CTL0628/ArcD) (Graham et al. 2002). PvlArgDC and arcD are present in all of the three C. trachomatis genomes, and both CDSs are intact in strain UW-3 (CT373 and CT374, respectively). However, the CTL0627 (PvlArgDC) carries a mutation in strain L2 and the ortholog of arcD (encompassing CDS CTA_0406-CTA_0408) carries multiple mutations in strain Har-13.
The PvlArgDC family of enzymes are involved in the biosynthesis of putrescine from L-arginine. Since Chlamydia spp. lack any other genes for arginine catabolism (Graham et al. 2002), it has been suggested that these genes may be involved in uptake of host L-arginine, along with a proton. Arginine is predicted to be degraded to agmatine, which is subsequently exported from the cell. The process of uptake and degradation of arginine is thought to play a role in pH homeostasis, both in Chlamydia and other bacterial pathogens such as the salmonellae (Graham et al. 2002; Kieboom and Abee 2006). This effect is mediated by the uptake of protons and concomitant depletion of acidic products made from carbohydrate fermentation by the host.
Recent clinical C. trachomatis LGV isolate causing proctitis: Biological properties and sequence
The newly described epidemics of LGV proctitis afforded a unique opportunity to compare a recent clinical LGV strain, with atypical clinical manifestations, with that of the classical LGV strain L2. We isolated and sequenced an LGV strain from a patient with proctitis in London in 2006: strain L2/UCH-1/proctitis (referred to as strain UCH-1; see Methods). Comparisons of the growth rate and inclusion formation of strain UCH-1 with the classical LGV strain L2, passaged in tissue culture over many decades, showed no morphological differences or increase/decrease in replication proficiency (data not shown).
Strain UCH-1 was sequenced using a pyrosequencing approach (454/Roche GS20). In total, 691 single base-pair differences were detected using MUMmer (Delcher et al. 1999) in strain UCH-1 compared with strain L2. Of these, 123 were found within homopolymeric tracts and 568 were either base substitutions or indels. All of the bases that were predicted to cause premature termination or frameshifts in CDSs otherwise conserved between strain L2 and strain UCH-1 were resequenced using PCR amplification and capillary sequencing. In total, we resequenced 71 single nucleotide polymorphisms (SNPs) that lay within homopolymeric tracts, 66/71 of which were incorrect 454 base calls. Conversely, we resequenced 48 SNPs and/or indels, all of which were correctly called. This is consistent with substitution errors being relatively rare in 454/Roche pyrosequencing data, while errors in base calls extending or contracting the length of homopolymeric tracts being more frequent (Margulies et al. 2005). We also resequenced all of the mutations in all of the strain UCH-1 pseudogenes (see below).
However, since we did not resequence all of the remaining called differences between strains, the sequence should be considered to be a contiguous draft. A summary of all of the base-pair differences can be found in Supplemental Table 2, along with a comment on whether the base has been confirmed by capillary resequencing, lies within a homopolymer, or represents a low-confidence base call.
Analysis of the coding capacity of strain UCH-1 showed that it was identical to strain L2 (Table 1). This extended to the complement of pseudogenes; with only one exception, CTLon_0574 (CTLon_0574 carries 2/3 mutations found in its strain L2 pseudogene ortholog CTL0578); all of the strain UCH-1 pseudogenes exhibited the same defining mutation(s) carried by their defunct strain L2 orthologs (Table 2).
Based on these data, we estimate that there are 573 high-confidence base differences that distinguish strain UCH-1 from strain L2. Of these bases, 457 occur within CDSs, which is slightly fewer than would be expected assuming a random distribution (data not shown). The most notable sequence variation (because of its possible utility for diagnostics) is found within a CDS that encodes the translocated actin recruiting phosphoprotein (Tarp; CTLon_0712). C. trachomatis entry into the host cell is a result of chlamydial reconfiguration of the host cell’s actin skeleton, and it is proposed that Tarp contributes to the pathogen-directed phagocytosis, i.e., the uptake of elementary bodies (EBs) (Jewett et al. 2006). The differences between Tarp in C. trachomatis strains L2, UW-3, and Har-13 (i.e., two large deletions and a high number of SNPs) has already been noted (Carlson et al. 2005), and may be significant in accounting for the difference in tropism between LGV, the trachoma (serovars A–C), and sexually transmitted serovars D–K. The Tarp sequences of strain UCH-1 and strain L2 are more similar to each other than the C. trachomatis serovars A–K Tarp sequences. However, there are a number of notable differences in sequence between Tarp from strain L2 and UCH-1: a deletion of 12 bases between nucleotides 718 and 731 of strain L2, resulting in the loss of the four amino acids: asparagine, isoleucine, tyrosine, and glutamic acid. There is a three-base in-frame insertion between bases 480 and 481 of strain L2, resulting in an extra alanine at this point, and a three-base in-frame deletion between bases 933 and 937, resulting in the loss of an alanine. The latter occur in one of the six repeat units in the 5′ insertion/deletion-coding region of Tarp (Carlson et al. 2005). Apart from the aforementioned differences, there are three SNPs between strains UCH-1 and L2, which are actually conserved in the corresponding nucleotide positions in C. trachomatis strains UW-3 and Har-13, suggesting a role for intra-serovar recombination in the diversity of these genes, as has been reported for the Chlamydia (Gomes et al. 2007). There is only one SNP in strain UCH-1 (from a “C” residue in C. trachomatis strains L2, UW-3, and Har-13 to “T” in strain UCH-1) that appears to be unique. This, in addition to the 12-base deletion mentioned earlier, are potential targets for differential molecular diagnostic analysis, for example, by use of FRET-probe based real-time PCR, for the discrimination of L1, L2, and L3 from the proctitis strain UCH-1.
These data show that when comparing the classical LGV isolate and the recent clinical isolate causing proctitis, there is no evidence of gene gain or obvious mutations that would indicate further functional loss. Therefore, the only differences between these two isolates that could explain the differing disease outcomes include altered levels of gene expression brought about, for example, by mutations in the promoter regions or through SNPs that lead to amino acid substitutions that impact on protein function. Whilst it is not possible to accurately predict the former, the possible functional impact of amino acid substitutions can be investigated by comparing the level of conservative and nonconservative sequence changes.
Distribution of SNPs and dN/dS analysis of the strain L2 genome
Figure 5 shows that the distribution of SNPs across the genome of strain L2 is not uniform, and that there are discrete regions of high-sequence variation when compared with C. trachomatis strains UW-3, Har-13, and UCH-1 (Fig. 5A). The most parsimonious explanation for this is that these regions are hot spots for strong diversifying selection and, therefore, may encode CDSs important for lifestyle. Alternatively, as has been previously suggested for C. trachomatis, these candidate loci may act as break points for DNA exchange, thereby introducing the higher levels of sequence variation by recombination (Gomes et al. 2007).
(A) Distribution of SNPs over the C. trachomatis strain L2 genome compared with strains Har-13, UW-3, and UCH-1. (i) The G-C skew (G+C/G-C). The position of the origin and terminus are marked. (ii) Shows the sum of all the SNPs in C. trachomatis strain L2 compared with strains Har-13, UW-3, and UCH-1, plotted as a frequency over a given window size. (iii) Regions with a high SNP density are numbered and the CDSs within this region are described in Supplemental Table 3. The base-pair positions are given in Mbp (bottom) and the window sizes are marked. The maximum, minimum, and average frequencies for each plot are given, where appropriate (right). (B) The distribution of C. trachomatis strain L2 CDSs with a dN/dS ratio >1. The dN/dS values for all of the C. trachomatis strain L2 CDSs (x axis) compared with their orthologs in strains Har-13, UW-3, and UCH-1 are shown (dN/dS >1 only). CDS displaying a high dN/dS ratio are described in Table 3.
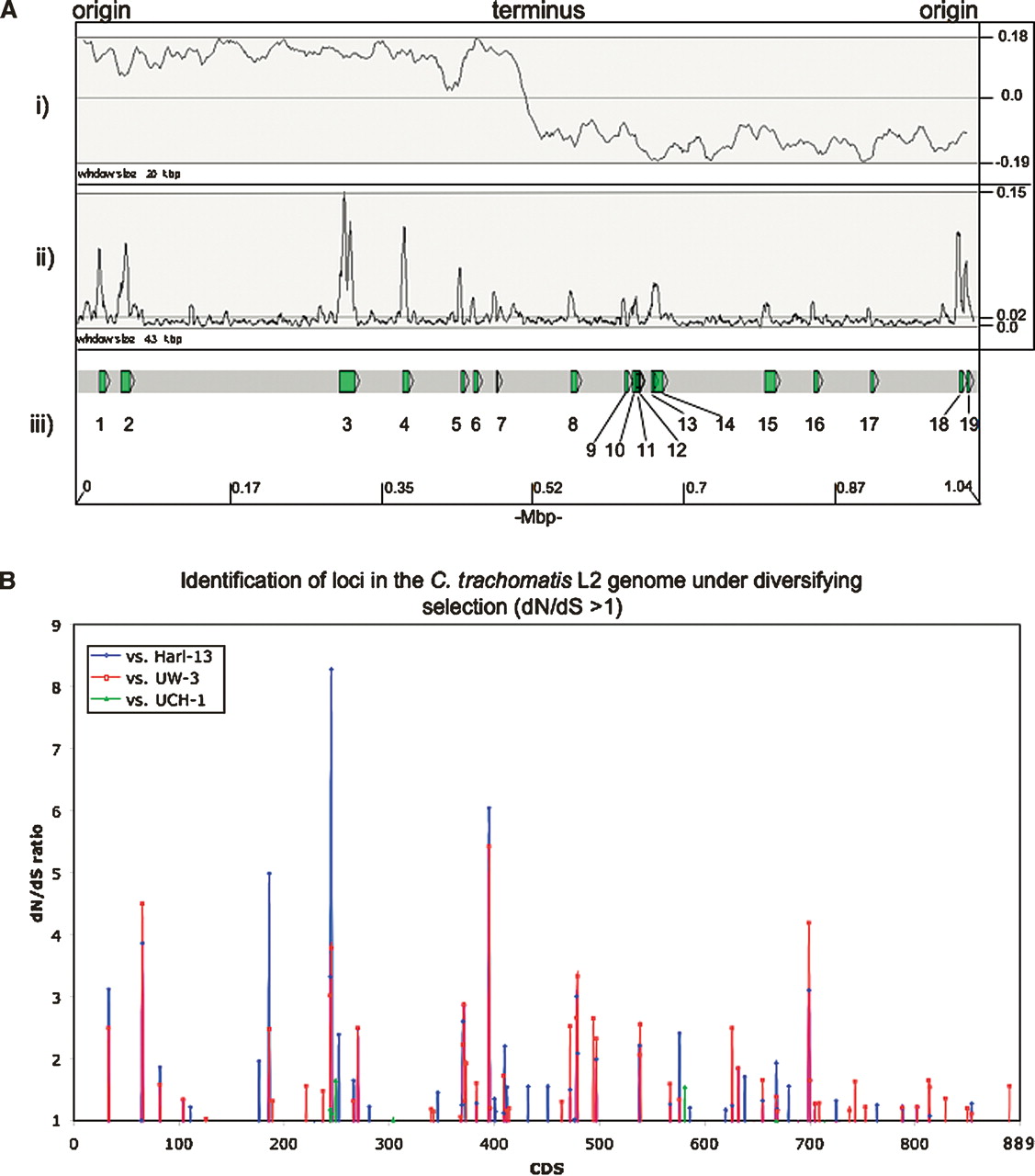
To investigate the functional and evolutionary significance of the identified SNPs, nonsynonymous (dN) and synonymous (dS) substitution rates were calculated for strain L2 CDSs in comparison to their orthologs in strains Har-13, UW-3, or UCH-1 (Fig. 5B; Table 3). Comparison of dN and dS can be used to assess the influence of selection on protein evolution. A low dN/dS ratio (dN/dS <<1) indicates strong stabilizing selection, whereas a high ratio (dN/dS >1) indicates positive selection and diversification. Since the majority of CDS shared between strain L2 and either Har-13 or UW-3 share >99% sequence identity, it was not possible to accurately identify those CDSs that were truly under strong stabilizing selection. However, the calculation of dN/dS was useful to identify genes or domains subject to diversifying selection, such as putative virulence factors or candidate vaccine targets that modulate the host immune response (Smith et al. 1995). Table 3 shows strain L2 CDSs found to have a dN/dS ratio >1 in this analysis. The number of CDSs with a dN/dS >1 was greatest when strain L2 was compared with for strain UW-3 (63), followed closely by the comparison strain Har-13 (55), and then strain UCH-1 (5).
C. trachomatis strain L2 CDSs with a dN/dS ratio >1 when compared to either C. trachomatis strains Har-13 or UW-3
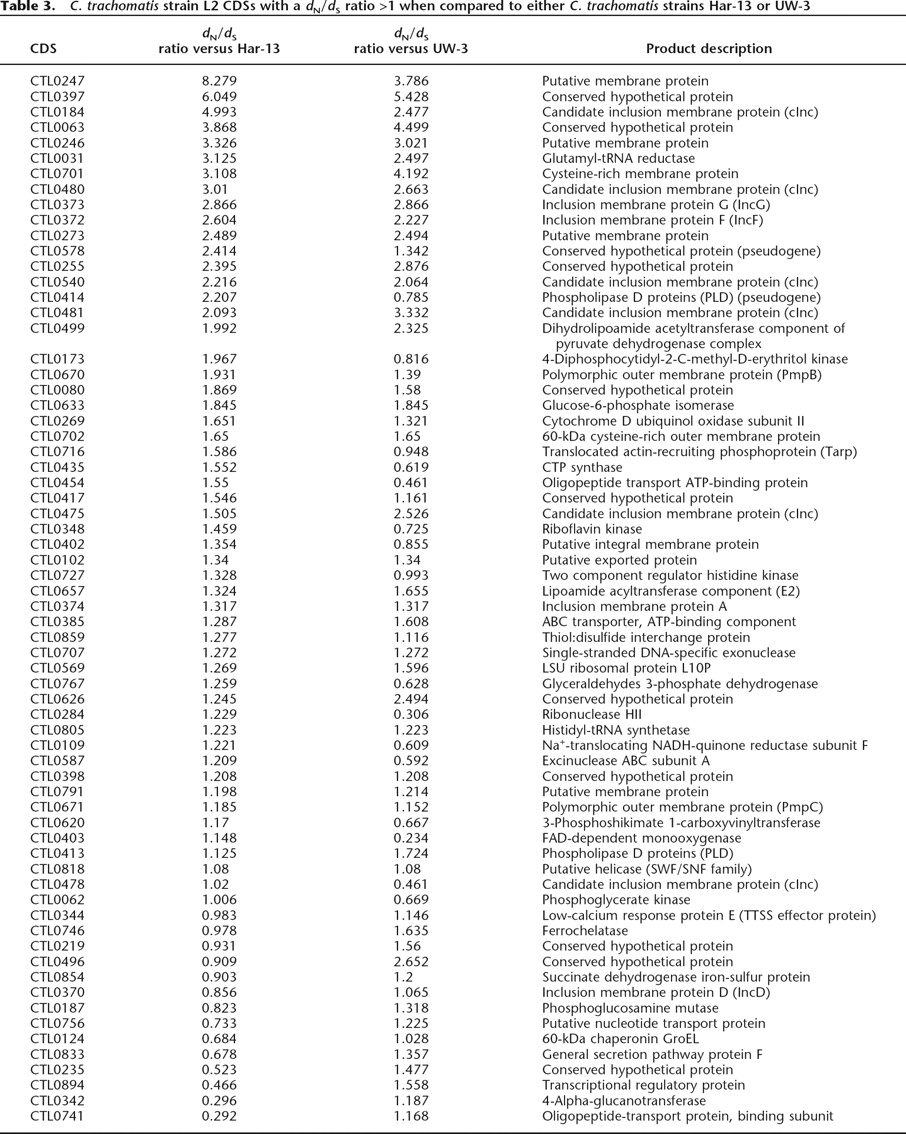
CDSs, with a dN/dS >1, taken from the strains L2 and UCH-1 comparison included pmpH (Pmp; polymorphic outer membrane protein), three other membrane proteins, and exodeoxyribonuclease (CTL0583) (data not shown). Similarly, hypothetical and membrane proteins were highly represented in CDSs with a dN/dS >1 or both strain Har-13 and UW-3 comparisons (38%–60% depending on the comparison; Table 3). But, also included within proteins showing the highest dN/dS >1 were pseudogenes, which would be expected to show a higher level of divergence (these genes are not expressed and therefore not subject to purifying selection), and those known to be important for interactions with the host: Pmp proteins (Grimwood and Stephens 1999), inclusion membrane proteins (Inc) (Bannantine et al. 1998a, b, 2000; Rockey et al. 2002) and Tarp. Interestingly, some of the other membrane proteins with a dN/dS >1 were predicted to have features of their secondary structure in common with Inc proteins. These have all been denoted candidate Inc (cInc) proteins (Table 3).
The interpretation of dN/dS data has to be tempered by the observation of Rocha et al. (2006), who identified a higher preponderance of nonsynonymous substitution in the genomes of closely related strains, as opposed to more distantly related strains. This discrepancy in the rate of substitution is thought to result from the short period of time over which purifying selection has been able to act on the genomes of closely related strains since diversification from a common ancestor. Where nonsynonymous mutations result in amino acid changes that have a mild effect on the fitness of the organism, purifying selection takes longer to exert its evolutionary influence. The observed high dN/dS ratio for CDS encoding core metabolic and housekeeping functions (Table 3), which are not obvious targets for diversifying selections, may be due to genetic “noise” rather than a strong selective pressure. To this end, it is worth noting that for some of the CDSs, such as CTL0348 (encoding riboflavin kinase; Table 3), in one comparison the dN/dS >1 and in another dN/dS <1 (dN/dS ratio = 0.725). However, the results shows that many of the CDSs that have high dN/dS ratios for both comparisons are from those known to interact with the host, suggesting that this analysis has identified an expanded set of candidate proteins that modulate host–cell interactions.
Discussion
Whole genome sequence analysis of the LGV biovariants completes the data set that now samples the complete range of diversity found in C. trachomatis. This work shows that the LGV genome is remarkably similar to the previously sequenced ocular and genital C. trachomatis isolates. The low variation in gene content between these biovariants is in agreement with previous microarray data, and rules out the possibility that additional acquired DNA present in C. trachomatis strain L2 could explain differences in tissue tropism and disease outcome. It is clear from the comparisons that gene loss and/or small-scale mutational change are the major driving forces shaping host adaptation and tissue tropism of C. trachomatis. The major region of variation is the PZ and variation here is primarily due to the loss/degeneration of the cytotoxin gene(s) as previously described (Belland et al. 2001).
In addition, PLDs are important sites of sequence variation, showing both multiple deletions and frameshift mutations. Phospholipases are known to play important roles in pathogenesis in a wide range of bacterial pathogens (Schmiel and Miller 1999; McDermott et al. 2004; McKean et al. 2007). C. trachomatis carries three PLD gene clusters, two of which are present in all of the sequenced Chlamydia and Chlamydophila, and the third cluster, found in the PZ, is specific to C. trachomatis and C. muridarum and is phylogenetically distinct from the two other PLD clusters. This analysis showed evidence of the PZ PLDs having expanded separately in C. trachomatis and C. muridarum, suggesting that this family of enzymes may have played, and continue to play, an important role in species-specific adaptation.
These genome comparisons have highlighted evidence for further loss of metabolic capacity. C. trachomatis has lost the genes encoding citrate synthase, aconitase, and isocitrate dehydrogenase (McClarty 1999); thus, additional blocks at the level of conversion of succinate to fumarate (sdhC pseudogene) and fumC in C. trachomatis strain L2 would indicate that the TCA cycle does not function in LGV isolates. Interestingly, Chlamydia appear to have recently acquired a dicarboxylate transporter (sodTi); the role of this gene product seems to be to transport 2-oxoglutarate from the cytoplasm of the host eukaryotic cell. If both fumarase and succinate dehydrogenase are inactive, then 2-oxoglutarate cannot be converted to oxaloacetate, but must be used directly for succinate and succinyl CoA synthesis in the process generating GTP.
The significance of finding mutations in arcD and PvlArgDC in strains L2 and Har-13, respectively, whilst they are apparently intact in the genital isolate strain UW-3, is unclear. Studies on the pH of C. trachomatis inclusions indicates that either a similar homeostatic balance to the host cell (Dautry-Varsat et al. 2005) or a pH close to 6.0 occurs; thus, it seems unlikely that the presence of intact arginine transport and metabolism alone accounts for the differences in disease outcomes associated with the different genital tract biovariants (Schramm et al. 1996).
Whilst it is clear from experimental data that loss of a particular pathway (e.g., trp biosynthesis) accounts for a single, important, metabolically defined difference between chlamydial biovariants (Stephens et al. 1998; Fehlner-Gardiner et al. 2002; Carlson et al. 2005), this is likely to be a unique observation, as there are no other single genetic features that correlate with a particular biotype and for which there is also phenotypic evidence. Thus, the new genome sequence data shows that the determinants of tropism and invasiveness of LGV isolates are likely to be multifactorial and complex. Without a genetic system to allow testing of the properties of “single gene” mutations, it is not possible to assign pathogenic properties to single factors.
The most overwhelming impression of the C. trachomatis strain L2 genome is the lack of variation in terms of coding capacity when compared with the other serovars. This is further reinforced by the comparison of strain L2 with the recent proctitis strain UCH-1 (isolated in 2006). It has been proposed that the LGV proctitis strain is a new epidemic isolate that is rapidly spreading worldwide. Whilst the symptoms caused by the proctitis strains are atypical, the genome of UCH-1 is almost identical to that of strain L2, which was isolated in 1969 and has been adopted as a “model” strain by many laboratories for routine studies. Although it is impossible to rule out that SNPs do not subtly change the function or the level of expression of key genes within the genome, we can say that there is no additional coding capacity to explain the differences in clinical manifestations. We favor the views of Schachter and Monocada (2005) that this strain, far from being a newly introduced and rapidly spreading infectious strain, is simply a classical LGV isolate and has been circulating in the human population for a long time: an old strain causing a new disease.
Methods
Bacterial strains
We sequenced the widely studied C. trachomatis strain L2/434/Bu (L2; ATCC VR902B) and a recent clinical isolate strain L2/UCH-1/proctitis (UCH-1). C. trachomatis strain UCH-1 was isolated from a rectal swab of a 49-yr-old MSM who was HIV positive and Hepatitis C negative. He attended a London Genital and Urinary Medicine (GUM) clinic in January 2006 showing clinical signs of proctitis. The rectal sample tested positive for C. trachomatis (negative for Neisseria gonorrhoeae) using a conventional C. trachomatis Nucleic Acid Amplifcation Test (CT NAAT). This sample was sent to the Health Protection Agency (Colindale) for routine LGV analysis. Confirmation that this strain was a LGV biovariant was ascertained using the primers CT1 and CT5 as described in Ngandjio et al. (2003). To determine whether this isolate was part of the wider European outbreak of the newly described L2b group of LGV isolates, we sequenced the ompA gene and identified the definitive base change at nucleotide 485. No other variations in the ompA gene were detected, indicating that the sample contained a single isolate and, hence, it was not considered necessary to perform plaque purification. This minimized the amount of passaging in vitro and the likelihood that attenuating mutations would arise during culture.
Growth of C. trachomatis strains L2 and UCH-1
C. trachomatis strain L2 was propagated in L929 cells in suspension cultures and the DNA purified from EBs as previously described (Birkelund and Stephens 1992).
C. trachomatis strain UCH-1 was initially isolated in BGMK cells in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 0.03 M glucose, vancomycin at 10 μg/mL−1, Gentamicin at 10 μg/mL−1, and Cycloheximide at 1 μg/mL−1.
Purification of chromosomal DNA for genome sequencing
Mycoplasma-free BGMK cells were used to amplify C. trachomatis strain UCH-1. Six passages in vitro were sufficient to yield 14 × T-175 tissue culture flasks infected to confluence with this strain. EBs, purified as described by Skipp et al. (2005), were incubated with protease K (200 μg/mL) for 1 h at 60°C prior to extraction of chromosomal DNA using a Promega Wizard Genomic Purification kit according to the manufacturer’s protocol. The C. trachomatis strain UCH-1 genomic DNA was quantified spectrophotometrically and analyzed by gel electrophoresis prior to genome sequence analysis.
Sequencing C. trachomatis strains L2 and UCH-1
The genome of strain L2 was obtained from 21,573 end sequences (giving 8× coverage) derived from pUC18 (insert size 1.4–2.2 kb) small insert libraries using dye terminator chemistry on ABI3700 automated sequencers. End sequences from larger insert plasmid (pMAQ1 9–12 kb, 9–12-kb insert size) libraries were used as a scaffold. The sequence was assembled, finished, and annotated as described previously (Parkhill et al. 2000) using the program Artemis (Berriman and Rutherford 2003) to collate data and facilitate annotation.
C. trachomatis strain UCH-1 chromosomal DNA was sequenced using a 454/Roche GS20 machine, according to the manufacturer’s protocols, producing 304,953 reads with an average length of 105 bp, representing a theoretical 30.8-fold coverage of the genome. The 454 sequence reads were assembled de novo into 18 nonredundant contigs with an average of 29.8-fold coverage, using the 454/Roche Newbler assembly program. These contigs were reordered based on BLAST alignments with strain L2. The gaps between these contigs were closed by directed PCR and the products sequenced with BigDye terminator chemistry on ABI3730 capillary sequencers.
The genomes of C. trachomatis strains L2 and UCH-1 have been submitted to the public database.
In silico genome analysis
The genome sequences of C. trachomatis strains L2, UW-3, and Har-13 were compared pairwise using the Artemis Comparison Tool (ACT) (Carver et al. 2005). Pseudogenes had one or more mutations that would ablate expression; each of the inactivating mutations was subsequently checked against the original sequencing data.
Orthologous gene sets were identified by reciprocal FASTA searches. Only those pairs of homologous CDSs were retained for further analysis, where the predicted amino acid identity was ≥40% over 80% of the protein length. These genes were then subject to manual curation using gene synteny to increase the accuracy of this analysis. This strategy was applied to pairwise comparisons of the genomes of C. trachomatis strains L2, UCH-1, UW-3, and Har-13, as well as C. muridarum (strain Nigg), Cp. felis (strain Fe/C-56), Cp. caviae (strain GPIC), and Cp. pneumoniae (strain AR39).
Synonymous and nonsynonymous substitution-rate calculations were determined for orthologous gene sets generated by reciprocal best-match FASTA analysis and aligned using Needle from the EMBOSS suit of software (Rice et al. 2000). dN and dS values were calculated using the PAML software suit (Yang 1997) by the method of Nei and Gojobori (1986), as implemented by the yn00 program (part of the PAML software suit). The method developed by Nei and Gojobori uses the Jukes and Cantor (1969) formula to estimate the number of synonymous and nonsynonymous substitutions per site.
Acknowledgments
We thank the core-sequencing and informatics teams at the Sanger Institute for their assistance and The Wellcome Trust for its support of the Sanger Institute Pathogen Sequencing Unit. This work was supported by The Wellcome Trust grants 080348 to I.N.C. We acknowledge the expert technical support of Lesley Cutcliffe, Rachel Skilton, S. Kalman, and R.W. Davis.
References
- ↵Y. Azuma,H. Hirakawa,A. Yamashita,Y. Cai,M.A. Rahman,H. Suzuki,S. Mitaku,H. Toh,S. Goto,T. Murakami,(2006) Genome sequence of the cat pathogen, Chlamydophila felis. DNA Res. 13:15–23.
- ↵J.P. Bannantine,D.D. Rockey,T. Hackstadt,(1998a) Tandem genes of Chlamydia psittaci that encode proteins localized to the inclusion membrane. Mol. Microbiol. 28:1017–1026.
- ↵J.P. Bannantine,W.E. Stamm,R.J. Suchland,D.D. Rockey,(1998b) Chlamydia trachomatis IncA is localized to the inclusion membrane and is recognized by antisera from infected humans and primates. Infect. Immun. 66:6017–6021.
- ↵J.P. Bannantine,R.S. Griffiths,W. Viratyosin,W.J. Brown,D.D. Rockey,(2000) A secondary structure motif predictive of protein localization to the chlamydial inclusion membrane. Cell. Microbiol. 2:35–47.
- ↵F.M. Behets,J. Andriamiadana,D. Randrianasolo,R. Randriamanga,D. Rasamilalao,C.Y. Chen,J.B. Weiss,S.A. Morse,G. Dallabetta,M.S. Cohen,(1999) Chancroid, primary syphilis, genital herpes, and lymphogranuloma venereum in Antananarivo, Madagascar. J. Infect. Dis. 180:1382–1385.
- ↵R.J. Belland,M.A. Scidmore,D.D. Crane,D.M. Hogan,W. Whitmire,G. McClarty,H.D. Caldwell,(2001) Chlamydia trachomatis cytotoxicity associated with complete and partial cytotoxin genes. Proc. Natl. Acad. Sci. 98:13984–13989.
- ↵M. Berriman,K. Rutherford,(2003) Viewing and annotating sequence data with Artemis. Brief. Bioinform. 4:124–132.
- ↵S. Birkelund,R.S. Stephens,(1992) Construction of physical and genetic maps of Chlamydia trachomatis serovar L2 by pulsed-field gel electrophoresis. J. Bacteriol. 174:2742–2747.
- ↵H.D. Caldwell,H. Wood,D. Crane,R. Bailey,R.B. Jones,D. Mabey,I. Maclean,Z. Mohammed,R. Peeling,C. Roshick,(2003) Polymorphisms in Chlamydia trachomatis tryptophan synthase genes differentiate between genital and ocular isolates. J. Clin. Invest. 111:1757–1769.
- ↵J.H. Carlson,S. Hughes,D. Hogan,G. Cieplak,D.E. Sturdevant,G. McClarty,H.D. Caldwell,R.J. Belland,(2004) Polymorphisms in the Chlamydia trachomatis cytotoxin locus associated with ocular and genital isolates. Infect. Immun. 72:7063–7072.
- ↵J.H. Carlson,S.F. Porcella,G. McClarty,H.D. Caldwell,(2005) Comparative genomic analysis of Chlamydia trachomatis oculotropic and genitotropic strains. Infect. Immun. 73:6407–6418.
- ↵T.J. Carver,K.M. Rutherford,M. Berriman,M.A. Rajandream,B.G. Barrell,J. Parkhill,(2005) ACT: The Artemis comparison tool. Bioinformatics 21:3422–3423.
- ↵M. Comanducci,S. Ricci,G. Ratti,(1988) The structure of a plasmid of Chlamydia trachomatis believed to be required for growth within mammalian cells. Mol. Microbiol. 2:531–538.
- ↵A. Dautry-Varsat,A. Subtil,T. Hackstadt,(2005) Recent insights into the mechanisms of Chlamydia entry. Cell. Microbiol. 7:1714–1722.
- ↵A.L. Delcher,S. Kasif,R.D. Fleischmann,J. Peterson,O. White,S.L. Salzberg,(1999) Alignment of whole genomes. Nucleic Acids Res. 27:2369–2376, 10.1093/nar/27.11.2369.
- ↵C. Fehlner-Gardiner,C. Roshick,J.H. Carlson,S. Hughes,R.J. Belland,H.D. Caldwell,G. McClarty,(2002) Molecular basis defining human Chlamydia trachomatis tissue tropism. A possible role for tryptophan synthase. J. Biol. Chem. 277:26893–26903.
- ↵J.P. Gomes,W.J. Bruno,A. Nunes,N. Santos,C. Florindo,M.J. Borrego,D. Dean,(2007) Evolution of Chlamydia trachomatis diversity occurs by widespread interstrain recombination involving hotspots. Genome Res. 17:50–60.
- ↵D.E. Graham,H. Xu,R.H. White,(2002) Methanococcus jannaschii uses a pyruvoyl-dependent arginine decarboxylase in polyamine biosynthesis. J. Biol. Chem. 277:23500–23507.
- ↵J and Grimwood,(1999) Computational analysis of the polymorphic membrane protein superfamily of Chlamydia trachomatis and Chlamydia pneumoniae. Microb. Comp. Genomics 4:187–201.
- ↵J.R. Guest,R.E. Roberts,(1983) Cloning, mapping, and expression of the fumarase gene of Escherichia coli K-12. J. Bacteriol. 153:588–596.
- ↵J.R. Guest,J.S. Miles,R.E. Roberts,S.A. Woods,(1985) The fumarase genes of Escherichia coli: Location of the fumB gene and discovery of a new gene (fumC) J. Gen. Microbiol. 131:2971–2984.
- ↵T.J. Jewett,E.R. Fischer,D.J. Mead,T. Hackstadt,(2006) Chlamydial TARP is a bacterial nucleator of actin. Proc. Natl. Acad. Sci. 103:15599–15604.
- ↵T.H. Jukes,R.C. Cantor,(1969) in Mammalian protein metabolism III, Evolution of protein molecules, ed H.N. Munro, (Academic Press, New York), pp 21–132.
- ↵J. Kieboom,T. Abee,(2006) Arginine-dependent acid resistance in Salmonella enterica serovar Typhimurium. J. Bacteriol. 188:5650–5653.
- ↵D. Longbottom,L.J. Coulter,(2003) Animal chlamydioses and zoonotic implications. J. Comp. Pathol. 128:217–244.
- ↵D. Mabey,R.W. Peeling,(2002) Lymphogranuloma venereum. Sex. Transm. Infect. 78:90–92.
- ↵M. Margulies,M. Egholm,W.E. Altman,S. Attiya,J.S. Bader,L.A. Bemben,J. Berka,M.S. Braverman,Y.J. Chen,Z. Chen,(2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380.
- ↵G. McClarty,(1999) Chlamydia: Intracellular biology, pathogenesis, and immunity, Chlamydial metabolism as inferred from the complete genome sequence (Wiley, New York), pp 69–100.
- ↵M. McDermott,M.J. Wakelam,A.J. Morris,(2004) Phospholipase D. Biochem. Cell Biol. 82:225–253.
- ↵S.C. McKean,J.K. Davies,R.J. Moore,(2007) Expression of phospholipase D, the major virulence factor of Corynebacterium pseudotuberculosis, is regulated by multiple environmental factors and plays a role in macrophage death. Microbiol. 153:2203–2211.
- ↵M. Nei,T. Gojobori,(1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3:418–426.
- ↵D.E. Nelson,D.D. Crane,L.D. Taylor,D.W. Dorward,M.M. Goheen,H.D. Caldwell,(2006) Inhibition of chlamydiae by primary alcohols correlates with the strain-specific complement of plasticity zone phospholipase D genes. Infect. Immun. 74:73–80.
- ↵A. Ngandjio,M. Clerc,M.C. Fonkoua,J. Thonnon,F. Njock,R. Pouillot,F. Lunel,C. Bebear,B. De Barbeyrac,A. Bianchi,(2003) Screening of volunteer students in Yaounde (Cameroon, Central Africa) for Chlamydia trachomatis infection and genotyping of isolated C. trachomatis strains. J. Clin. Microbiol. 41:4404–4407.
- ↵R.F. Nieuwenhuis,J.M. Ossewaarde,W.I. van der Meijden,H.A. Neumann,(2003) Unusual presentation of early lymphogranuloma venereum in an HIV-1 infected patient: Effective treatment with 1 g azithromycin. Sex. Transm. Infect. 79:453–455.
- ↵J. Parkhill,B.W. Wren,K. Mungall,J.M. Ketley,C. Churcher,D. Basham,T. Chillingworth,R.M. Davies,T. Feltwell,S. Holroyd,(2000) The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403:665–668.
- ↵R.W. Peeling,R.C. Brunham,(1996) Chlamydiae as pathogens: New species and new issues. Emerg. Infect. Dis. 2:307–319.
- ↵T.D. Read,R.C. Brunham,C. Shen,S.R. Gill,J.F. Heidelberg,O. White,E.K. Hickey,J. Peterson,T. Utterback,K. Berry,(2000) Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 28:1397–1406, 10.1093/nar/28.6.1397.
- ↵T.D. Read,G.S. Myers,R.C. Brunham,W.C. Nelson,I.T. Paulsen,J. Heidelberg,E. Holtzapple,H. Khouri,N.B. Federova,H.A. Carty,(2003) Genome sequence of Chlamydophila caviae (Chlamydia psittaci GPIC): Examining the role of niche-specific genes in the evolution of the Chlamydiaceae. Nucleic Acids Res. 31:2134–2147, 10.1093/nar/gkg321.
- ↵P. Rice,I. Longden,A. Bleasby,(2000) EMBOSS: The european molecular biology open software suite. Trends Genet. 16:276–277.
- ↵E.P. Rocha,J.M. Smith,L.D. Hurst,M.T. Holden,J.E. Cooper,N.H. Smith,E.J. Feil,(2006) Comparisons of dN/dS are time dependent for closely related bacterial genomes. J. Theor. Biol. 239:226–235.
- ↵D.D. Rockey,M.A. Scidmore,J.P. Bannantine,W.J. Brown,(2002) Proteins in the chlamydial inclusion membrane. Microbes Infect. 4:333–340.
- ↵P. Saikku,(1999) Epidemiology of Chlamydia pneumoniae in atherosclerosis. Am. Heart J. 138:S500–S503, 10.1016/S0002-8703(99)70285-1.
- ↵J. Schachter,J. Moncada,(2005) Lymphogranuloma venereum: How to turn an endemic disease into an outbreak of a new disease? Start looking. Sex. Transm. Dis. 32:331–332.
- ↵D.H. Schmiel,V.L. Miller,(1999) Bacterial phospholipases and pathogenesis. Microbes Infect. 1:1103–1112.
- ↵N. Schramm,C.R. Bagnell,P.B. Wyrick,(1996) Vesicles containing Chlamydia trachomatis serovar L2 remain above pH 6 within HEC-1B cells. Infect. Immun. 64:1208–1214.
- ↵A.C. Shaw,K. Gevaert,H. Demol,B. Hoorelbeke,J. Vandekerckhove,M.R. Larsen,P. Roepstorff,A. Holm,G. Christiansen,S. Birkelund,(2002) Comparative proteome analysis of Chlamydia trachomatis serovar A, D and L2. Proteomics 2:164–186.
- ↵P. Skipp,J. Robinson,C.D. O’Connor,I.N. Clarke,(2005) Shotgun proteomic analysis of Chlamydia trachomatis. Proteomics 5:1558–1573.
- ↵N.H. Smith,J. Maynard Smith,B.G. Spratt,(1995) Sequence evolution of the porB gene of Neisseria gonorrhoeae and Neisseria meningitidis: Evidence of positive Darwinian selection. Mol. Biol. Evol. 12:363–370.
- ↵J. Spaargaren,H.S. Fennema,S.A. Morre,H.J. de Vries,R.A. Coutinho,(2005) New lymphogranuloma venereum Chlamydia trachomatis variant, Amsterdam. Emerg. Infect. Dis. 11:1090–1092.
- ↵R.S. Stephens,S. Kalman,C. Lammel,J. Fan,R. Marathe,L. Aravind,W. Mitchell,L. Olinger,R.L. Tatusov,Q. Zhao,(1998) Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282:754–759.
- ↵N.R. Thomson,C. Yeats,K. Bell,M.T. Holden,S.D. Bentley,M. Livingstone,A.M. Cerdeno-Tarraga,B. Harris,J. Doggett,D. Ormond,(2005) The Chlamydophila abortus genome sequence reveals an array of variable proteins that contribute to interspecies variation. Genome Res. 15:629–640.
- ↵C. Viravan,D.A. Dance,C. Ariyarit,S. Looareesuwan,Y. Wattanagoon,T.M. Davis,V. Wuthiekanun,S. Tantivanich,B.J. Angus,N.J. White,(1996) A prospective clinical and bacteriologic study of inguinal buboes in Thai men. Clin. Infect. Dis. 22:233–239.
- ↵Z. Yang,(1997) PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13:555–556.
Notes
[1] [Supplementary material is available online at www.genome.org. The genome sequence data from this study has been submitted to EMBL: AM884176, AM886278, AM884177, and AM886279.]
[2] Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.7020108