
Allelic variation and heterosis in maize: How do two halves make more than a whole?
Abstract
In this review, we discuss the recent research on allelic variation in maize and possible implications of this work toward our understanding of heterosis. Heterosis, or hybrid vigor, is the increased performance of a hybrid relative to the parents, and is a result of the variation that is present within a species. Intraspecific comparisons of sequence and expression levels in maize have documented a surprisingly high level of allelic variation, which includes variation for the content of genic fragments, variation in repetitive elements surrounding genes, and variation in gene expression levels. There is evidence that transposons and repetitive DNA play a major role in the generation of this allelic diversity. The combination of allelic variants provides a more comprehensive suite of alleles in the hybrid that may be involved in novel allelic interactions. A major unresolved question is how the combined allelic variation and interactions in a hybrid give rise to heterotic phenotypes. An understanding of allelic variation present in maize provides an opportunity to speculate on mechanisms that might lead to heterosis. Variation for the presence of genes, the presence of novel beneficial alleles, and modified levels of gene expression in hybrids may all contribute to the heterotic phenotypes.
It is often argued that a basic understanding of a process is critical to its use and manipulation. Arguably, one of the most notable exceptions to this idea is the phenomenon of heterosis. Heterosis, or hybrid vigor, has been the subject of intense research and speculation for well over a century; however, the basic mechanisms that cause or contribute to heterosis remain unclear (Coors and Pandey 1999). Despite this lack of understanding, breeders have quite successfully manipulated heterosis to increase the vigor of many domesticated species. In maize, it is estimated that the use of hybrids and heterosis increases yields ∼15% per annum (Duvick 1999).
Heterosis refers to the phenomenon in which the hybrid F1 offspring exhibit phenotypic characteristics that are superior to the mean of the two parents (mid-parent heterosis), or the better of the two parents (better parent heterosis) (Fig. 1). While mid-parent heterosis is scientifically interesting, it has relatively little economic importance. Better parent heterosis is the underlying rationale for the widespread use of hybrids in many agricultural species, and we focus our discussion on this phenomenon. The level of heterosis can be quantified for specific traits. “Heterosis per se” for a specific trait is quantified as the phenotype of the hybrid minus the phenotype of the better parent; subsequently, the “percentage of heterosis” is calculated as the heterosis per se divided by the better of the parental phenotypes. Often, the parents of heterotic offspring are inbred. In this case, the quantification of heterosis reflects both hybrid vigor and a recovery from inbreeding depression.

Phenotypic heterosis in the B73 × Mo17 hybrid. Representative B73, Mo17, and F1 hybrid ears and plants are shown. Note the increased size of the two hybrid ears and three hybrid rows relative to the two inbred parents (left, Mo17; right, B73).
Heterosis has been used in the breeding and production of many crop and animal species (Janick 1998; Melchinger and Gumber 1998). An early application of heterosis was breeding mules, which are derived from crossing a female horse and a male donkey (Goldman 1998). As pointed out by Troyer (2006), there are useful similarities in the examples of mules and hybrid corn. Both mules and hybrid corn exhibit superior phenotypes and stress tolerance relative to their parents. Additionally, farmers showed a willingness to purchase hybrid corn and mules despite obvious drawbacks: these products are added expenses and neither produce useful offspring, thus requiring the farmer to purchase a new organism each generation.
It is important to note that the application and use of hybrids in different species are influenced by several factors. For instance, if the degree of heterosis is relatively low in a given species, then it may not be cost-effective to use hybrids for commercial production. The efficient development of hybrids may be limited by the species mating system, as self-pollination and/or controlled crosses are difficult or impossible to conduct in many species. An efficient F1 hybrid production method is essential for hybrid commercialization. In fact, the early maize inbred varieties had extremely low seed yield, leading to the use of double cross hybrids until inbred varieties with higher seed yield could be developed (Duvick 2001).
Heterosis is evident not only in artificially selected populations, but also may be observed in natural populations (Mitton 1998; Hansson and Westerberg 2002). Allelic frequencies in a random sample of coniferous trees were in agreement with Hardy-Weinberg expectations; however, an excess of heterozygotes was observed when only the mature, oldest, or largest trees were sampled (Mitton and Jeffers 1989). Furthermore, a study of Pinyon pines suggested that heterozygotes are more resistant to herbivory pressure (Mopper et al. 1991). The exact mechanism that leads to the enhanced performance of the heterozygotes in these tree species has not been determined, but it is possible that heterosis is an important factor in fitness for many organisms.
Maize provides an excellent system for the study and application of heterosis. A wide range of natural genetic diversity has been captured in the current maize germplasm (Flint-Garcia et al. 2005; Wright et al. 2005; Troyer 2006). Maize is relatively easy to self- or cross-pollinate, which has enabled the development of both diverse inbreds and many hybrids for evaluation. Much of this review focuses on the heterosis derived from crossing maize inbred line B73 with inbred line Mo17 (Fig. 1). The B73 × Mo17 hybrid was widely grown commercially in the United States during the 1970s, with >15 million bags of seed sold, sufficient to plant ∼45 million acres (Troyer 2006). While there are many maize hybrids that exhibit high levels of heterosis, this particular hybrid provides an excellent example of heterosis involving two lines that are relatively well-characterized in terms of genetic mapping, gene expression, and genome organization.
The relative measurements for several traits of the B73 and Mo17 parental inbred lines and the hybrid are listed in Table 1 (Zanoni and Dudley 1989; Auger et al. 2005b). Note that the quantitative measurements of heterosis vary significantly for different traits. The variation in levels of heterosis among traits leads to difficulties in accurately quantifying the amount of overall heterosis. There is also variation in the relative level of heterosis for different traits between different hybrids. For example, one maize hybrid may exhibit significant heterosis for plant height but have low heterosis for grain yield, while a second hybrid may exhibit high heterosis for grain yield but very low heterosis for height (Zanoni and Dudley 1989; data not shown). This variation suggests that the same set of genes does not control all heterotic responses. Additionally, heterosis does not simply result from the overall genetic diversity within a hybrid, but is likely a reflection of diversity at specific, important genes that contribute to a particular trait. This view is supported by the ability to map QTLs that contribute to heterosis for individual traits in maize and rice (Stuber et al. 1992; Xiao et al. 1995; Li et al. 2001; Luo et al. 2001; Lu et al. 2003).
Phenotypes and heterosis in B73 relative to Mo17
In the simplest view, the reverse of heterosis is inbreeding depression, in which progressive self-pollination or sibling matings reduce the genome-wide heterozygosity and overall fitness of an organism. Inbreeding depression is likely caused by the fixation of deleterious alleles within a lineage. Plant breeders have created an “unnatural” inbred state in maize, a naturally outcrossing species, which may help explain why maize exhibits relatively high heterosis when compared to other species. The inbreeding depression in inbred maize lines is alleviated when a hybrid is formed, and the plants exhibit superior characteristics. A fundamental question is whether the reversal of the effects of inbreeding in the F1 hybrid (via complementation of deleterious alleles with “good” alleles from the other parent) is sufficient to explain heterosis, or whether other mechanisms are at work. Two hypotheses, dominance and overdominance, have been proposed to explain heterosis (Text Box 1).
In this review, we focus on three key questions that address heterosis from a genomics perspective: (1) What allelic differences exist between heterotic parental lines in terms of types of variation and prevalence of variation? (2) How do these allelic variants interact in the F1 hybrid organism? (3) How does the interaction of allelic variants result in heterotic phenotypes?
The term “allelic variation” (broadly defined here as the sequence or regulatory differences found in different parental genotypes) is used in each of the three questions. While we discuss each of these questions in the context of maize, it is anticipated that similar mechanisms apply to other organisms.
Allelic variation in maize
The current era of genomic sequencing and global gene expression analysis has provided a wealth of information about intraspecific allelic variation. Intraspecific allelic variation can include sequence changes, structural changes in the genome, altered expression levels, and epigenetic changes. At some level, heterosis is the result of variation between the parental lines, since in the absence of variation (inbreds), there is no heterosis. Our assumptions of the nature and frequency of allelic variation can limit how we consider heterosis (Crow 1999; Phillips 1999).
Allelic diversity in maize genic sequences
One of the most common approaches toward documenting allelic diversity is to compare the sequence of genic regions (including coding regions, introns, untranslated regions, and single-copy DNA surrounding genes) from multiple strains or varieties in order to identify variation. This variation can then be used for mapping or association studies. An investigation of randomly selected sequences in the maize inbred B73 relative to inbred Mo17 found that, on average, indel polymorphisms occur every 309 bp and SNPs occur every 79 bp (Vroh Bi et al. 2005). The analysis of 300–500 bp amplicons found that 44% of the sequences contained at least one polymorphism in B73 relative to Mo17. In general, it is estimated that there is one polymorphism every 100 bp in any two randomly chosen maize inbred lines (Tenaillon et al. 2001; Ching et al. 2002). Sequence diversity data have been evaluated for several hundred diverse inbred lines of maize at >3000 genic sites (Zhao et al. 2006). These data provide extensive information about the SNP and indel frequencies of maize alleles. Collectively, these studies indicate that maize has a relatively high level of sequence polymorphism compared to many other species. For example, the level of sequence diversity in genic sequences within maize is estimated to be higher than the level of diversity between humans and chimpanzees (Buckler et al. 2006).
Structural diversity in maize
Structural genome diversity involves alterations in DNA sequence beyond SNPs or small indels. This would include large-scale chromosomal differences, altered location of genes or repetitive elements, or differences in the presence of sequences in one inbred relative to another. Large-scale genome differences between different maize inbred lines were first identified through cytogenetics studies. Barbara McClintock and others analyzed heterochromatic knob (highly condensed, tandem repeat regions) content and size to characterize genome variation in maize (Brown 1949; McClintock et al. 1981; Adawy et al. 2004). Recent studies have documented differences in the content for several classes of repetitive DNA between maize inbreds at the chromosomal level (Kato et al. 2004). Flow cytometry studies have also documented significant variation in the size of the maize genome between different inbred lines (Laurie and Bennett 1985; Lee et al. 2002).
Sequence-based methodologies have documented maize genome structural diversity at a higher resolution (for reviews, see Buckler et al. 2006; Messing and Dooner 2006). BAC-based studies focusing on the diversity of allelic regions among different maize inbred lines identified a surprising level of intraspecific structural diversity at several loci (Fig. 2). Fu and Dooner (2002) sequenced BAC contigs that contain the bz gene from the inbred B73 and McC genotypes and, as expected, documented numerous SNP and short indel polymorphisms within the genes present in this region. In addition, they found a surprising level of structural diversity in the two regions that resulted in extensive sequence nonhomologies. This structural diversity includes variation in repetitive elements and variation in the presence of genic fragments.

Potential examples of allelic variation in maize. A hypothetical small chromosomal region is diagrammed for B73 and Mo17. The genes (red) are separated by clusters of transposons (black) and several different classes of retrotransposons (various shades of green). There are likely to be numerous SNPs and small indels within this region. The composition of repetitive sequences also shows significant variation between the two inbreds. The specific location of a causative change that has resulted in functional allelic variation is indicated by the yellow lightening bolts. Gene A is an example of a variant in which the proteins are different. Gene B is a nonshared sequence that is only present in B73 at this locus. Gene C is more highly expressed in Mo17 than in B73 because of differences in the repetitive elements surrounding this gene in B73 and Mo17. Gene D is expressed only in B73 and not in Mo17 because of altered sequence in nearby regulatory regions. Gene E shows no functional variation in B73 relative to Mo17.
Seven conserved genes were found in the bz region of chromosome 9 for both B73 and McC; however, an additional four gene-like fragments were found only in the McC haplotype (Fu and Dooner 2002). Initially these fragments were thought to be genes specific to the McC genotype. However, further investigation found that these fragments are actually partial gene segments that were likely the result of two transposition events mediated by Helitron transposable elements (Lai et al. 2005). The partial fragments present at the McC bz locus on chromosome 9 are derived from full-length gene sequences that are present on chromosome 5 in both B73 and McC (Lai et al. 2005). Helitron transposons, which use a rolling-circle method of replication, exhibit limited primary sequence signatures and can be difficult to identify using computational approaches (Kapitonov and Jurka 2001). The exact mechanism through which these elements can “capture” and transpose genic fragments is not well-understood (Brunner et al. 2005b; Lai et al. 2005).
The B73 and McC genotypes also demonstrated structural variation for repetitive elements present in this region (Fu and Dooner 2002). Only one short fragment from a repetitive Zeon1 element is conserved in both genotypes. Otherwise, the transposable elements present in McC and B73 are completely distinct in terms of their relative positions. The McC haplotype contains six full-length LTR retrotransposons and six partial elements that are not conserved in the B73 locus, while the B73 haplotype contains five full-length LTR retrotransposons and four partial elements that are not present in the McC locus (Fu and Dooner 2002). In many cases, the B73 and McC alleles of the same gene were surrounded by distinct repetitive elements that may contribute to differences in the chromatin neighborhood for the two alleles. A recent follow-up study compared the genomic sequence structure of the bz locus in eight different maize lines representing a wide range of geographic and genetic backgrounds (Wang and Dooner 2006). A tremendous degree of variation in genomic sequence structure was identified among these lines; the authors concluded that, on average, any two of the eight bz haplotypes share just 50% of their sequences (Wang and Dooner 2006).
Several other maize loci that were sequenced in multiple inbred lines also exhibit structural diversity for genic fragments and repetitive elements. The tandemly repeated zein1C gene family displays variation between maize inbred lines in terms of copy number and expression (Song and Messing 2003). The sequencing of four genomic loci in B73 and Mo17 identified 45 genes that were shared between genotypes, while another 27 genic fragments (which are likely pseudogenes) were only present in either one of the two inbred lines (Brunner et al. 2005a). These four loci also included a set of 27 LTR retroelements that are present at collinear locations in both B73 and Mo17, while another 62 LTR retroelements were only present in either one of the two inbred lines.
The genome-wide prevalence of nonshared genic fragments was investigated using BAC library hybridization methods (Morgante et al. 2005). Hybridization of short genic probes to fingerprinted BAC libraries from B73 and Mo17 resulted in mapping ∼20,500 genes, of which 80% were present in both inbreds, 11% were specific to B73, and 9% were specific to Mo17. Applying these frequencies to the anticipated genome size of maize suggests that the B73 and Mo17 genomes may contain ∼10,000 nonshared genic fragments; a subset of which may be functional.
There is evidence that mechanisms other than Helitron-mediated transposition can lead to the presence of nonshared genic fragments in different varieties of a species (Bennetzen 2005). For instance, other classes of transposons can modify genic content. Mutator-like transposons can “capture” genic sequences and move these sequences within the maize genomes (Bureau et al. 1994; Jin and Bennetzen 1994; Bennetzen 2005). The abundance of the Mutator-like elements that carry gene fragments, termed Pack-MULEs, is not known in maize; however, the completion of the rice genome has allowed for a comprehensive analysis of these elements in rice (Jiang et al. 2004). Fragments of >1000 genes have been captured and sometimes duplicated by Pack-MULEs, resulting in the presence of >3000 Pack-MULEs in rice. There is EST evidence for the expression of ∼5% of the Pack-MULEs, and some of these expressed sequences may encode functional peptides, although most contain numerous stop codons. There is also evidence for the presence of large numbers of tandem duplications in the maize genome, including some examples of tandem duplications that are present only in some inbred lines (Emrich et al. 2007). Duplicated genes and transposed gene fragments, like those discussed above, are likely to show variation within a species such that specific duplications or Pack-MULEs will be present within one inbred and absent in others.
The presence of nonshared genic sequences or retrotransposons might effect phenotypic variation through a variety of mechanisms. The majority of examples of the nonshared genic sequences that have been characterized to date are partial gene fragments. It remains possible that some of these nonshared genic sequences will include full-length genes or novel genes created by shuffling of exons from multiple genes (Brunner et al. 2005b). In fact, there is evidence for expression of some of these nonshared genes (Brunner et al. 2005b; Morgante et al. 2005). The expression level and pattern of these fragments are likely to be distinct from those of the original gene because the newly transposed gene copy often does not include the full promoter or regulatory sequences. In some cases, the transcription and translation of a transposed partial ORF may result in an aberrant protein that has a dominant-negative effect that inhibits the function of the original full-length protein.
The variation in maize repetitive elements may influence the expression level of nearby genes, and in some cases, this influence can extend over large regions. For example, most alleles of the maize transcription factor B have relatively small, closely linked, regulatory sequences. However, the transcription level of the B-I allele can be influenced in cis by a set of tandem repeats that are located >100 kb upstream of the coding region (Stam et al. 2002). Additionally, the tb1 locus, which influences maize architecture, is influenced in cis by a region of repetitive elements >50 kb upstream of the gene (Clark et al. 2006). These examples suggest that in some cases, repetitive sequences may affect the expression level of maize genes, ultimately resulting in altered phenotypes. However, it is unknown whether these examples represent rare cases of long-distance effects of repetitive sequences, or if this phenomenon is common in maize gene regulation.
Expression diversity in maize
Intraspecific allelic variation also includes variation in gene expression. Several recent studies have documented expression diversity in maize. Analysis of gene expression levels using an Agilent long oligonucleotide microarray platform found evidence for the differential expression of ∼5% of genes in the maize inbred lines A619 and W23 at several developmental stages (Ma et al. 2006). A cDNA microarray platform used by Swanson-Wagner et al. (2006) identified ∼10% of genes as being differentially expressed in B73 and Mo17 seedlings. Affymetrix microarray analysis also found prevalent differential expression between B73 and Mo17 in seedling, immature ear, and embryo tissues (Stupar and Springer 2006).
The Affymetrix microarray study suggested that ∼2.5% of the genes present on the array were detected in only one of the two transcriptomes, either B73 or Mo17 (Stupar and Springer 2006). Validation of these results provided evidence that many of these differences in B73–Mo17 transcriptome content are not the result of differences in genomic content. Instead, these presence–absence transcriptome differences appear to be the result of differentially expressed genes that are present in the genomes of both B73 and Mo17.
The expression diversity observed in maize could be the result of cis-acting variation at each of the differentially expressed genes or the result of variation at a small number of trans-acting loci that have downstream regulatory effects. Cis-acting variation can be the result of alterations in regulatory sequences (i.e., enhancers and promoters), sequence changes that affect the RNA stability, or heritable variation in chromatin structure. Trans-acting variation can be the result of quantitative or qualitative variation in a factor that influences the expression of the gene, such as a transcription factor. The prevalence of cis- and trans-regulatory variation can be assessed using expression quantitative trait loci (eQTL) (Cheung and Spielman, 2002) and allele-specific expression (ASE) (Wittkopp et al. 2004) analyses. Numerous studies in animal systems using both eQTL (Monks et al. 2004; Morley et al. 2004; Cheung et al. 2005; Doss et al. 2005; Pastinen et al. 2005; Stranger et al. 2005) and ASE (Cowles et al. 2002; Yan et al. 2002; Bray et al. 2003; Lo et al. 2003; Pastinen et al. 2004; Wittkopp et al. 2004) have found that a significant amount of expression diversity is the result of cis-acting variation.
There is evidence for prevalent cis-acting regulatory variation in maize. An eQTL study found that 80% of the eQTL with a LOD score >7.0 mapped to the same physical location as the differentially expressed gene, indicating that allelic cis-variation may be causing regulatory differences for many maize genes (Schadt et al. 2003). Guo et al. (2004) used ASE to study the relative expression of two alleles in maize F1 hybrids. Both alleles present in an F1 hybrid have access to an identical set of trans-acting factors, thus unequal, or biased, allelic expression suggests cis-acting variation between alleles. Frequent allelic expression bias in F1 hybrids was observed (11/15 genes), suggesting that cis-variation is present in many maize genes (Guo et al. 2004). These findings were supported by another ASE study that found allelic cis-variation contributing to inbred expression differences in 46 of 53 maize genes (Stupar and Springer 2006).
In addition to structural and expression diversity, there are additional sources of diversity within maize. Variation in sense–antisense transcription, allelic variation in DNA methylation patterns, and allelic variation in chromatin structure are all topics that have not been extensively addressed to date. One study of the targets of CpNpG methylation in maize found substantial variation for the epialleles present in B73 and Mo17 (I. Makarevitch and N.M. Springer, unpubl.). In addition, studies of polyploids have found evidence for widespread epigenetic alterations that can lead to phenotypic variation (Osborn et al. 2003). The epigenetic diversity within a species may be particularly important in contributing to overdominance. For example, one of the few examples documenting overdominant gene action for a single locus involves an epiallele at the Pl locus in maize (Hollick and Chandler, 1998). Phillips (1999) presented an excellent discussion of the evidence for de novo variation in inbred lines and how this variation may contribute to heterosis.
Interactions of alleles in hybrids
The allelic variation described above is brought together in the hybrid organism. The allelic combinations present in a hybrid may result in interactions that alter expression profiles, new protein–protein interactions, or epistatic interactions. The studies of gene expression in inbreds and hybrids can be divided according to whether they have found substantial evidence for frequent nonadditive expression patterns compared to the expression levels in the inbred parents (see Text Box 2 for a primer on the terminology and concepts associated with gene expression in inbreds relative to hybrids). The concept of transcriptional epistasis and the evidence for this phenomenon in hybrid maize are also discussed.
Evidence for allelic interactions that lead to novel hybrid expression patterns
Several studies have found evidence for novel expression patterns in hybrids relative to inbred parents such that the hybrid expression level is outside the range of the level of the parents. The first studies to provide evidence of this phenomenon focused on using 2D-PAGE to observe protein levels. Romagnoli et al. (1990) and Leonardi et al. (1991) used such methodologies to suggest that maize hybrids exhibit significant levels of non-additive expression.
Hollick and Chandler (1998) provided evidence for overdominant action by some alleles at the Pl locus of maize. The anthocyanin levels were significantly higher in heterozygotes containing one copy of an epigenetically modified Pl allele than in either of the homozygous parent plants. The higher levels of pigmentation likely result from over-high-parent expression of the Pl gene in these hybrids relative to inbreds (Hollick and Chandler 1998).
Song and Messing (2003) focused on characterizing the relative expression of zein1C genes in developing endosperm tissue using an RT-PCR/sequencing approach. BAC sequencing was used to show that the B73 and BSSS53 genotypes encode a different set of zein1C genes. By performing reverse-transcriptase-mediated PCR and then sequencing large numbers of the resulting zein1C cDNAs, it was possible to determine which genes were expressed and their relative expression levels. B73 expresses six different zein1C genes, while BSSS53 expresses seven zein1C genes, but only three of these genes are shared between the genotypes. Analysis of these genes in hybrid endosperm tissue found that all 10 zein1C genes were expressed, but the relative expression levels were often nonadditive relative to the parental levels (Song and Messing 2003).
Auger et al. (2005a) used quantitative Northern blot hybridization to study gene expression levels in mature leaf tissue for ∼30 genes, including nuclear-, mitochondrial-, and chloroplast-encoded genes. A surprisingly high level of nonadditive gene expression was documented in the reciprocal hybrids compared to their inbred parents, B73 and Mo17. More than three-quarters of the monitored genes (24/30) were found to display nonadditive expression patterns in at least one of the two reciprocal hybrids relative to the inbred parents. In addition, 16 of the 24 nonadditive genes displayed hybrid expression levels outside the range of the levels of the two parents (Auger et al. 2005a). This study suggested that maize hybrids may have different expression patterns from what would be predicted by the mid-parent values of the two inbred parents. Several specialized microarray approaches were used to document evidence for nonadditive gene expression in maize hybrids, although in most cases the nonadditive expression was within the range of the parental expression levels (Meyer et al. 2007; Użarowska et al. 2007).
A similar picture of prevalent nonadditive gene expression has been reported in microarray-based experiments designed to monitor the level of additive and nonadditive expression in Arabidopsis, Drosophila, and rice (Gibson et al. 2004; Ranz et al. 2004; Vuylsteke et al. 2005; Huang et al. 2006). A significant fraction (∼10%) of the genes monitored in Arabidopsis displayed evidence for hybrid gene expression outside of the parental range (Vuylsteke et al. 2005). A well-designed experiment in Drosophila melanogaster also found prevalent nonadditive expression in hybrids. In fact, the gene expression patterns of the two inbred parents were more similar to each other’s than to the hybrids’ (Gibson et al. 2004). Analysis of the differentially expressed genes in this experiment found that the number of genes with above high-parent or below low-parent expression patterns was significantly higher than the number of genes with additive, high-parent or low-parent expression patterns. Almost one-third of the differentially expressed genes identified in rice displayed expression levels that deviated from the mid-parent levels (Huang et al. 2006). However, there was no evidence that the expression level for these genes was outside the range of the parental expression values. In combination, these experiments suggest that hybrids may have significant levels of nonadditive gene expression including many cases of expression outside of the parental range.
Evidence for prevalent additive expression in hybrids
In contrast to the above studies, several groups have reported that maize hybrids tend to exhibit frequent additive expression patterns. These studies do find evidence for nonadditive expression, but these are primarily examples of expression within the range of the parents. Guo et al. (2003) used a cDNA-AFLP approach to analyze expression levels in maize endosperm tissue isolated from genotypes that display a range of heterosis for yield. The majority of genes were expressed at additive levels in the hybrids. By studying the reciprocal hybrids, they found that ∼10% of genes are expressed at nonadditive levels; these genes typically displayed maternal-like or paternal-like expression levels. There was no significant correlation between the amount of heterosis and the frequency of nonadditive patterns (Guo et al. 2003). More recently, this technique was applied to profiling expression in immature ear tissue derived from a set of 16 hybrids (Guo et al. 2006). In this study, it was found that the majority of differentially expressed genes display hybrid expression patterns within the range of the parents. This study included a series of hybrids with a range of heterotic response, and it was demonstrated that the proportion of genes displaying mid-parent expression is positively correlated with heterosis and yield (Guo et al. 2006).
Two recent microarray studies have also found evidence for low levels of novel expression in hybrids relative to inbreds. Swanson-Wagner et al. (2006) used a cDNA microarray to monitor the expression levels of ∼14,000 genes in 14-d seedlings of B73, Mo17, and the F1 hybrid. Approximately 10% of the genes were identified as differentially expressed in at least one genotype. The majority of these differentially expressed genes (1062/1367) displayed an additive pattern of gene expression in the hybrids relative to the inbred parents. While most of the remaining genes exhibited expression patterns similar to high-parent or low-parent levels, there was a subset of 44 genes that displayed nonadditive expression patterns outside of the parental range. Most of the genes (42/44) that displayed above high-parent or below low-parent expression were cases in which the expression of B73 was not significantly different from Mo17 but the hybrid displayed a nonequivalent expression pattern. Within the set of genes that displayed novel hybrid expression levels, there were only a few examples with large fold changes (33 of 44 were <1.4-fold different from the near parent) (Swanson-Wagner et al. 2006).
Gene expression in 11-d seedlings, immature ear, and embryo tissue was monitored in essentially the same genotypes (B73, Mo17, and reciprocal F1 hybrids) using Affymetrix microarrays (Stupar and Springer 2006). A series of statistical and clustering analyses indicated that most genes display additive expression patterns in the hybrids relative to the inbred parents. Nearly 80% of the genes that are differentially expressed in B73 relative to Mo17 demonstrate mid-parent expression in the hybrid. The vast majority of the genes that displayed nonadditive expression patterns were found to be expressed at levels within the range of the two parents. Additionally, this study found no evidence for novel hybrid expression patterns in genes that were not differentially expressed in the two inbred parents.
The results of these two microarray studies are similar in that the majority of genes that are differentially expressed in the inbred parents show an additive pattern of expression in the hybrids, and most of the genes with nonadditive expression are still expressed within the parental range. Another microarray experiment that compared maize hybrid and inbred expression levels using an Agilent platform supports the conclusion that novel hybrid expression patterns are rare (Ma et al. 2006).
A recent study from the mouse community also found evidence of prevalent additive expression levels in F1 hybrids. Cui et al. (2006) used an Affymetrix mouse GeneChip platform to compare the gene expression of liver tissues from two relatively closely related mice inbred strains and their reciprocal F1 hybrids. The authors found that the F1 hybrids exhibited additive expression levels much more frequently than nonadditive levels. The few observations of hybrid gene expression outside of the parental range were typically identified in genes with little expression difference between the inbred parents (Cui et al. 2006), similar to the results obtained by Swanson-Wagner et al. (2006) in maize.
Epistasis in gene expression
While the concept of epistasis is relatively straightforward (see Text Box 1), the specific application to heterosis has been varied. We discuss here the potential of transcriptional epistasis, defined here as gene interactions that affect the transcription rate of another gene or set of genes. The anthocyanin biosynthesis pathway of maize provides an excellent example of transcriptional epistasis. The maize B and Pl gene products are transcription factors that interact to up-regulate the synthesis of genes such as A1, A2, and Bz1, which control anthocyanin production (Dooner et al. 1991). An inbred with a nonfunctional b allele would likely display a green phenotype due to the low or absent expression of A1, A2, and Bz1. Similarly, a second inbred fixed for a nonfunctional pl allele would also be green. A hybrid produced between these two plants would be B/b Pl/pl and have high levels of expression for A1, A2, and Bz1 and a red pigmentation (Fig. 3). The expression phenotype of A1, A2, and Bz1 would be above high-parent in the hybrid relative to the inbred parents, while the expression phenotype of B and Pl would be additive. This overdominance would be the result of epistatic interactions between B and Pl, not the result of direct allelic interactions at A1, A2, or Bz1.
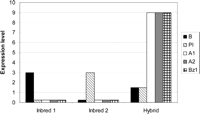
Example of potential transcriptional epistasis in maize hybrids. A potential example of transcriptional epistasis in maize is illustrated using genes from the well-characterized anthocyanin biosynthesis pathway. Inbred 1 (genotype B, pl, A1, A2, Bz1) and Inbred 2 (genotype b, Pl, A1, A2, Bz1) each contains a nonfunctional allele for one of the regulatory genes B or Pl, and both show negligible levels of expression for the structural genes A1, A2, and Bz1. The hybrid shows additive expression for B and Pl but nonadditive, over-high-parent expression for A1, A2, and Bz1.
One might expect to observe numerous instances of transcriptional epistasis in maize hybrids; the merger of two genomes may result in new interactions within gene regulation pathways that were not present in the inbred parents, as in the anthocyanin example described above. If such cases were common in hybrids, nonadditive expression profiles outside of the parental range would be frequently observed, as was the case in Drosophila (Gibson et al. 2004). Therefore, it was surprising that there are few examples of this type in the microarray studies of maize. Therefore, transcriptional epistasis, in which novel gene interactions affect the expression levels of downstream genes, is not common in hybrids of B73 and Mo17. Additionally, quantitative genetics analyses have suggested that epistasis does not play a major role in contributing to hybrid maize yield (Hinze and Lamkey 2003; Mihaljevic et al. 2005).
Despite multiple studies addressing the topic, a consensus view of gene expression in maize hybrids has not emerged. The prevalence of novel expression patterns in maize hybrids remains an open question. It is possible that the tissue sampling and experimental design differences have influenced the expression profiles observed in the studies discussed above.
Contribution of allelic variation to heterotic phenotypes
In the previous two sections, we have reviewed intraspecific variation in maize (allelic variation) and the relative expression patterns in inbreds relative to hybrids (allelic interactions). The subsequent question is to determine how allelic variation and interactions result in a superior hybrid phenotype. While outside the scope of this review, a complete model of heterosis undoubtedly involves contributions from quantitative genetics, physiology, and biochemistry.
Considerations of heterosis should take into account the following four factors.
-
The magnitude of heterosis varies in different species. For example, heterosis has much stronger and more ubiquitous effects in maize than in Arabidopsis (Zanoni and Dudley 1989; Meyer et al. 2004).
-
The level of heterosis for specific traits varies and is not correlated in different hybrids of the same species. This indicates that heterosis is not the result of action at a single locus, nor does heterosis simply reflect the overall extent of heterozygosity between parents.
-
As the genetic distance between the parental inbreds increases, there is generally an increase in heterosis; yet when the parental distance exceeds a threshold, heterosis decreases (Moll et al. 1965). Thus, there appears to be a relationship between genetic diversity and heterosis; however, the correlation is not strong enough to be used as an accurate predictive tool (Melchinger 1999).
-
The allelic variation that produces heterosis is not representative of all variation that occurs. Not all allelic variants that arise in a maize population will become fixed in inbred lines. Variants with strong deleterious phenotypes will be selected against by breeders. Therefore, the range of allelic variation that is captured in inbred lines that can affect heterosis is limited to the variation that has beneficial affects for a specific trait or has limited deleterious affects (Troyer 2006).
Collectively, these factors provide insight into several important features of heterosis. First, heterosis is the result of variation at multiple genomic locations. Second, many of the complex phenotypes that are often assessed for heterosis, such as yield, are likely influenced by many (hundreds of) genes. Third, heterosis for different phenotypes is determined by variation at a partially nonredundant set of loci. These features of heterosis are supported by QTL studies that have mapped QTLs controlling heterosis for yield, height, and flowering date in maize and rice (Stuber et al. 1992; Xiao et al. 1995; Li et al. 2001; Luo et al. 2001; Lu et al. 2003). Although there was evidence for overdominance in some of these studies, it is quite possible that this apparent overdominance could reflect pseudo-overdominance resulting from repulsion-phase linkages (see Text Box 1). A QTL fine-mapping approach confirmed that this was the case for one apparent overdominant locus (Graham et al. 1997).
Complementation of present–absent genes
The dominance hypothesis proposes that heterosis is the result of complementation of slightly deleterious alleles in the hybrid. The inbreds become fixed for these alleles, resulting in inbreeding depression. Fu and Dooner (2002) refined this model to include the potential of present–absent genes. There is growing evidence that the suite of active genes in heterotic hybrids is greater than that of either of the parental inbred lines.
Nonshared genic fragments in maize inbred lines (for reviews, see Buckler et al. 2006; Messing and Dooner 2006) provide instances of variation in which one inbred contains genic information that is absent in another inbred. The majority of these nonshared genic sequences appear to be novel sequence gain (duplication events) in one inbred relative to the other, as opposed to deletion events of previously shared genes (Lai et al. 2005). Novel functions may arise as gene fragments, or full-length genes, are copied to new chromosomal locations and potentially acquire novel expression patterns. In addition, the mechanism of transposition appears to have the capacity to perform exon shuffling and create new ORFs at a relatively high frequency (Brunner et al. 2005b; Lai et al. 2005).
In addition to the differences in genic content of the inbred genomes, there is also evidence for a significant number of genes that are expressed in one inbred line but not detected in another inbred (Song and Messing 2003; Stupar and Springer 2006). These genes with presence–absence inbred expression patterns are expressed at detectable levels in the hybrid. Both gain of transcripts (through altered regulation or new genic fragments) and loss of transcripts (through altered regulation or transposon insertions) would result in the presence–absence transcriptome differences between maize inbreds.
What are the specific mechanisms driving these differences in genomic and transcriptome content? There is evidence that a primary source of the variation in genome content, and potentially transcriptome content, is the action of transposable elements. Helitron elements, DNA transposons, and retrotransposons constitute two-thirds of the maize genome and are likely to be continuously generating variation. The “capture” and movement of genic fragments by DNA transposons, retrotransposons, and rolling circle transposons can increase the copy number for genic fragments (Bennetzen 2005). Furthermore, transposable element insertion events may cause a reduction in the expression level of genes through disruption of coding regions or regulatory regions. Alternatively, transposable elements may have more complex effects on gene regulation through the provision of novel regulatory sequences (Barkan and Martienssen 1991).
Complementation of novel beneficial alleles
One clue to the underlying causes of heterosis comes from the analysis of diversity limitations in heterotic crosses. Crosses between highly diverse maize germplasm often exhibit limited heterosis. Tropical germplasm has not been adapted to environmental conditions in the temperate United States, and this may cause the inferior heterotic performance of these stocks. Troyer (2006) points out the importance of adapted, or beneficial, alleles in heterosis. Any breeding program will include artificial selection for specific traits in addition to inadvertent selection dictated by environmental factors. During the domestication and spread of maize, different inbreds have evolved unique sets of adaptations specific to the selective pressures of growth and reproduction in different habitats. These adaptations might include altered flowering time, disease resistance, growth habits, or stress tolerances. In some cases, different sets of genes may have even been selected for the same traits. If we assume that these traits behave in a dominant or codominant fashion, then the improved phenotype of hybrids may be the result of an increase in the cumulative adaptedness of hybrids for specific traits. The hybrid will contain one complete set of alleles from each of the inbred parents, resulting in a combination of each of the beneficial alleles accrued by each parent. This concept is simply a minor revision of the dominance model. In the dominance model, hybridization essentially masks the accumulated deleterious alleles in the inbreds. In the beneficial allele model, hybridization merges the accumulated specific and divergent beneficial alleles from both inbreds. This model could also be applied to overdominance. If two different alleles of the same gene acquire unique beneficial adaptations, the heterozygote (containing both adaptations) may be more beneficial than either of the parental alleles alone.
A potential example is provided by a study of phosphate acquisition efficiency in Arabidopsis (Narang and Altmann 2001). The F1 of a cross of inbred ecotypes C24 and Col-0 displays heterosis for phosphate acquisition efficiency. Col-0 and C24 each have adaptations to improve nutrient uptake characteristics: longer overall root length for Col-0 and longer root hair length plus higher phosphate transporter expression for C24. The combined inheritance of these three dominant physiological adaptations results in heterosis for phosphate acquisition efficiency.
Potential advantages of mid-parent expression levels
As discussed above, there is a surprisingly high level of variation in the content and structure of the repetitive regions between maize genes. The repetitive regions may generate metastable epigenetic or chromatin modifications. There is evidence that these regions can influence the expression of adjacent genes (Stam et al. 2002; Clark et al. 2006) and may explain the high rate of cis-linked regulatory variation observed in maize (Guo et al. 2004; Stupar and Springer 2006). The high frequency of maize cis-acting regulatory variation may, in turn, lead to another potential mechanism of heterosis, mid-parent levels of gene expression.
There is ample genetic evidence, such as the phenotypic alterations displayed by over- or underexpressed transgenes, that altered gene expression levels can affect phenotypes in a quantitative manner. For example, many of the genes involved in disease resistance or pathogen defense display altered fitness in response to very high or low levels of expression (Heidel et al. 2004). While altered expression can affect phenotype, there is also evidence that expression level variation within an acceptable range does not affect phenotype. The prevalence of recessive mutations relative to dominant or codominant mutations suggests that many genes have a range of acceptable gene expression levels in which equally good phenotypes are produced. It is only when expression varies above or below thresholds that phenotypic consequences will ensue.
We propose a model that includes the following assumptions: (1) Gene expression levels will show a high rate of change within a maize population; (2) the cause of many of these gene expression differences is cis-linked; (3) these expression level changes can affect the phenotype when they deviate outside of an optimal range of expression and may result in a minor loss of fitness; (4) a subset of genes that display excessively high or low expression levels will become fixed within different inbred lines; and (5) the combination of two alleles in a hybrid that vary because of cis-linked changes will result in mid-parent expression levels. Given these assumptions, a model can be developed that would predict increased fitness of the hybrid relative to one or both of the inbreds (Fig. 4). Inbred gene expression levels may frequently be altered and then subsequently fixed above or below the optimal range, which may result in subtle effects on the phenotype. Mid-parent expression levels in the hybrid may dilute the detrimental effects of over- or underexpression of these genes by causing the total expression level for a gene to be within the optimal range. The hybrid may exhibit dominant gain (genes A or B) or overdominant gain (gene C) due to the hybrid mid-parent expression level. This model proposes that there will be instances in which the hybrid mid-parent expression level is advantageous relative to one (examples of genes A and B) or both (example of gene C) of the inbred parents.
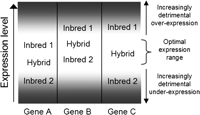
Potential model to explain how additive expression could be advantageous relative to one or both parents. Optimal gene expression levels (or patterns) occur in the white range at the middle of the graph. Increasing levels of shading indicate increasing deleterious effects on the phenotype due to low or high levels of expression. The exact range of optimal expression as well as the rate at which over- or underexpression becomes detrimental is likely to vary for different genes. We illustrate potential gene expression variation at three hypothetical genes. Gene A expression levels are within the optimal range for inbred 1 but under the optimal level for inbred 2. Gene B expression levels are within optimal range for inbred 2 but above the optimal level for inbred 1. Gene C exhibits suboptimal expression in both inbred 1 and inbred 2. The expression level in the hybrid is within the optimal range for all three genes.
Concluding remarks
There are several potential mechanisms through which genomic variation could combine to produce a heterotic phenotype. Each of these mechanisms could occur at a subset of genes, and the combination of effects will result in heterosis. In addition, there are other nonmutually exclusive mechanisms such as altered protein–protein interactions, novel epigenetics states, siRNAs, or altered hormone levels that can profoundly alter phenotypes (Phillips 1999; Birchler et al. 2003; Osborn et al. 2003). As additional genomic and genetic resources become available, it is anticipated that a better understanding of heterosis will emerge. Given the rate and types of allelic variation observed in maize inbreds, there is substantial evidence that allelic variants at a large number of loci act through partial to complete dominance to provide favorable complementation resulting in superior hybrid phenotypes. The contributions of epistatic interactions and overdominance to heterosis are more difficult to establish and remain enigmatic.
Acknowledgments
We apologize to all those researchers whose work we have overlooked or could not include because of page limitations. This review was greatly improved by the comments of Virginia Walbot and two anonymous reviewers. We thank Shawn Kaeppler, William Tracy, Ronald Phillips, Ruth Shaw, Irina Makarevitch, Karen McGinnis, William Haun, Wade Odland, Peter Tiffin, and Cynthia Weinig for helpful comments and discussions on the manuscript. N.M.S. is supported by the National Science Foundation (grant no. DBI-0227310).
Footnotes
-
↵1 Corresponding author.
↵1 E-mail springer{at}umn.edu; fax (612) 625-1738.
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.5347007
- Copyright © 2007, Cold Spring Harbor Laboratory Press








