
Very little intron loss/gain in Plasmodium: Intron loss/gain mutation rates and intron number
PlasmodiumAbstract
We compared intron positions in conserved regions of 3479 orthologous gene pairs from Plasmodium falciparum and Plasmodium yoelii, which likely diverged ≥100 million years ago (Mya). Only 27 out of 2212 positions were specific to one of the two species. Intron presence in related species shows that at least 19 and possibly 26 of the changes are due to intron loss, depending on phylogeny. The implied intron loss and gain rates are much lower than previously estimated for nematodes, arthropods, fungi, and plants, and are comparable only with the rates in vertebrates. That all observed changes were exact, occurring without loss or gain of flanking coding sequence, suggests intron loss via an mRNA intermediate, as does a nonsignificant trend toward loss of introns at adjacent positions. Many of the intron changes occurred in genes encoding proteins involved in nucleic acid-related processes, as previously found for intron gains in nematodes. Two changes occurred in the chloroquine resistance transporter, suggesting a role for positive selection in intron loss in Plasmodium. The dearth of intron loss and gain could be explained by the lack of known transposable elements in Plasmodium, since transposable elements and/or reverse transcriptase are thought to be necessary for both processes. The observed pattern suggests that the availability of stochastic intron loss and gain mutations can be a major determinant of changes in intron number.
Spliceosomal introns are largely quasi-random sequences that interrupt the coding regions of many eukaryotic genes. They are excised from mRNA transcripts by the spliceosome, an elaborate RNA–protein complex. The ultimate origins and evolutionary significance of spliceosomal introns have been hotly debated since their discovery 30 years ago (for recent reviews, see Rogozin et al. 2005; Jeffares et al. 2006; Roy and Gilbert 2006).
A central issue is the relative importance of intron loss and gain through eukaryotic history. Introns are often found at the exact same positions in orthologous genes of widely divergent eukaryotic species (Fedorov et al. 2002; Rogozin et al. 2003; Sverdlov et al. 2005) in a pattern suggesting intron-rich ancestors and massive recurrent intron loss along diverse lineages (Roy and Gilbert 2005b). This view is supported by the apparent presence of a complex spliceosome in the eukaryotic ancestor (Collins and Penny 2005) and by the two available genome-wide studies of more closely related species (Roy et al. 2003; Nielsen et al. 2004). However, other analyses suggest more moderate ancestral intron densities (Csuros 2005; Nguyen et al. 2005), with a more central role for intron gain (Babenko et al. 2004; Qiu et al. 2004).
The mechanisms of intron loss and gain also remain debated. New introns might arise either by (1) insertion of type II self-splicing introns laterally transferred from endosymbionts (Sharp 1985; Cavalier-Smith 1991; Stoltzfus 1999), (2) insertion of transposable elements into coding sequences (Crick 1979; Iwamoto et al. 1998, 1999; Roy 2004), or (3) reinsertion of a spliced RNA copy of an intron into a previously intron-less site of a transcript, followed by reverse transcription of this transcript and gene conversion (Cavalier-Smith 1985; Palmer and Logsdon Jr. 1991; Coghlan and Wolfe 2004; Logsdon Jr. 2004; Sverdlov et al. 2004). Intron loss might occur by recombination with a reverse-transcribed copy of a spliced mRNA transcript (Perler et al. 1980; Bernstein et al. 1983; Lewin 1983; Weiner et al. 1986; Fink 1987; Long and Langley 1993; Derr 1998; Sakurai et al. 2002; Wada et al. 2002; Lin et al. 2003; Mourier and Jeffares 2003; Sverdlov et al. 2004; Niu et al. 2005; Roy and Gilbert 2005a) or by simple genomic deletion of the intron sequence (Robertson 1998; Kent and Zahler 2000; Banyai and Patthy 2004; Cho et al. 2004).
Intron number varies massively across eukaryotes, from hundreds of thousands of introns per vertebrate genome to only two characterized introns in Giardia lamblia (Venter et al. 2001; Aparicio et al. 2002; Nixon et al. 2002), and often even within eukaryotic groups. Traditional selection-based explanations (Doolittle and Sapienza 1980; Gilbert 1987; Lynch 2002) do not predict recent findings of intron-rich early eukaryotes (Fedorov et al. 2002; Rogozin et al. 2003; Roy and Gilbert 2005b, 2006) or the diversity of intron-rich parasites and species with large population sizes.
We studied orthologous gene pairs from the human malaria parasite Plasmodium falciparum and the rodent parasite Plasmodium yoelii, which diverged ≥100 million years ago (Mya). Among 2212 intron positions in regions of good alignment, we found only 27 species-specific introns, 19 in P. falciparum and eight in P. yoelii. Sequence searches against several nearly complete apicomplexan genomes suggest that at least 19 and probably at least 26 are due to intron loss, while none is clearly due to intron gain. These values imply intron loss/gain rates much lower than those previously found in several other eukaryotic groups. Nine of the 16 genes with known functions that have experienced intron changes are involved in nucleic acid-related processes. Two other changes occurred in the chloroquine-resistance transporter gene, a gene likely under strong selection (Vennerstrom et al. 2004), perhaps suggesting a role for positive selection in intron loss. The dearth of intron gain and loss could reflect rarity of intron loss/gain mutations due to the lack of known transposable elements (TEs) and associated reverse transcriptases in Plasmodium, suggesting an important role for intron loss/gain mutations in determining intron number.
Results
Intron loss and gain between Plasmodium falciparum and Plasmodium yoelii
We found 2185 shared intron positions, eight P. yoelii-specific positions and 19 P. falciparum-specific positions in ∼3.5 Mb of conserved regions of 3479 ortholog pairs. We determined intron presence/absence of these 27 introns in other apicomplexans by BLAST searches against available genomic sequence and predicted genes structures. In the most likely Plasmodium phylogeny (Fig. 1A) (Qari et al. 1996; Perkins and Schall 2002), the presence of 26/27 introns in P. gallinaceum indicates intron loss. If, instead, P. yoelii is the outgroup (Fig. 1B) (Waters et al. 1991; Escalante and Ayala 1994, 1995; Escalante et al. 1995, 1997, 1998; McCutchan et al. 1996), at least 19 are due to loss. In no case is there clear evidence of intron gain.
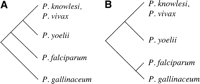
Relationship of Plasmodium species used in this study. (A) Most probable relationship, in which P. falciparum is sister to other mammalian malarias; (B) alternative relationship, in which P. falciparum is sister to the avian malaria P. gallinaceum.
The pattern of intron change
The six genes experiencing multiple intron changes is more than expected by random chance (P = 10−5). Introns experiencing changes were not more likely to fall in 3′ regions, nor in any particular phase. Among the five genes with multiple P. falciparumspecific introns, the introns were adjacent in four cases (P = 0.107). The average length of the 19 P. falciparum introns that are absent in P. yoelii is 174 bp, similar to the overall P. falciparum average (177 bp). The average length of the eight P. yoelii introns that are absent in P. falciparum is 154 bp, nonsignificantly shorter than the overall average length of P. yoelii introns (221 bp, P = 0.30). Table 1 lists the 16 intron losing/gaining genes for which putative or confirmed gene functions are available. Nine of the 16 genes encode proteins involved in nucleic acid-related processes.
Summary of observed intron gains and losses
All observed intron changes are exact
Twenty-six of 27 discordant introns fell in ungapped regions of the alignment; thus, no addition or deletion of flanking sequence appeared to have been associated with the change. The remaining intron (in P. falciparum) lies adjacent to a one-amino-acid alignment gap (valine in P. yoelii). The intron begins “GTAGTA.” If, in fact, the second GT was the true 5′ intron boundary, the change would be exact and the valine (encoded by GTA) restored. A BLASTN search of dbEST yielded no hits, thus this conjecture could not be confirmed.
Expression level
For each species, we determined the number of expressed sequence tags (ESTs) from that species matching each gene from each ortholog pair (Table 2). Direct comparisons between such numbers are not straightforward (see Discussion). However, investigation showed that there are roughly an equal number of cases among all 3479 pairs of orthologous genes in which the P. falciparum gene matched more or alternatively less than twice as many ESTs as the P. yoelii gene, thus identifying roughly equal numbers of pairs with “excess expression” in P. falciparum and P. yoelii. This approximate equality held when more stringent cutoffs based on this metric (e.g., that the difference between twice the number of P. falciparum matches and the number of P. yoelii matches be greater than some positive value) were used. Using this crude metric, we studied the data for the genes undergoing intron gain/loss. We first asked whether there was a directional trend of change in expression level (i.e., whether the intron-lacking gene tended to be more highly expressed, or alternatively the intron-containing gene tended to be more highly expressed). Using the simplest comparison of P. falciparum matches against twice the number P. yoelii matches, in nine cases the intron-lacking gene had “higher expression,” whereas in seven cases the intron-containing gene did. Using a more stringent criterion requiring that the difference between the number of P. falciparum EST matches and twice the P. yoelii matches is at least three, the numbers were six and four.
Numbers of P. falciparum or P. yoelii ESTs in dbEST matching each gene experiencing an intron change
We next asked whether, regardless of direction, there tended to be large changes in expression level in orthologous gene pairs with intron changes than in others. In particular, three cases in which there were apparently large changes appeared notable. For the strongest P. falciparum-biased gene pair, with 35 P. falciparum ESTs and only two P. yoelii ESTs, there are 115 pairs of orthologs out of 3479 (3.3%) with at least 35 P. falciparum ESTs and two or fewer P. yoelii ESTs. For the strongest apparently P. yoelii-biased gene pair, with 18 P. yoelii ESTs but no P. falciparum ESTs, there are 67 pairs (2.0%) with 18 or more P. yoelii ESTs but no P. falciparum ESTs. Given the number of genes with intron changes, such differences are not unexpected, thus we see no clear pattern of change in expression level between genes experiencing intron changes.
Discussion
Low rates of intron gain and loss in Plasmodium falciparum and Plasmodium yoelii
In 3.5 Mb of conserved regions of alignment of orthologous gene pairs, we found 27 intron positions that were specific to one of the two Plasmodium species, P. falciparum and P. yoelii, compared with 2185 clearly shared intron positions presumably retained from the common ancestor. Depending on phylogeny, at least either 19 or 26 of these differences are due to intron loss, supporting an excess of intron loss over gain in eukaryotic evolution. These numbers imply 0.5% per 100 My intron loss and less than one gain/Mb per 100 My, much less than previous estimates of 3%–30% per 100 My loss and 60–240 gains/Mb per 100 My for various fungi, plants, and animals (Nielsen et al. 2004; Roy and Gilbert 2005c), and comparable only to a mouse–human comparison that showed <0.1% loss and no gain in 1560 genes over 100 My (Roy et al. 2003).
These low rates in Plasmodium could reflect the lack of known retrotransposons and associated reverse transcriptase activity, since intron loss likely occurs via reverse transcription of spliced mRNAs (see below), and intron gain likely occurs either via reverse transcription of spliced mRNAs or via transposon insertion (Crick 1979; Cavalier-Smith 1985; Palmer and Logsdon Jr. 1991; Giroux et al. 1994; Iwamoto et al. 1998, 1999; Coghlan and Wolfe 2004; Logsdon Jr. 2004; Roy 2004; Sverdlov et al. 2004). Alternative models of intron loss by simple genomic deletion and of intron gain by genomic duplication do not predict low rates in Plasmodium (Rogers 1989; Robertson 1998; Venkatesh et al. 1998; Kent and Zahler 2000; Llopart et al. 2002; Banyai and Patthy 2004; Cho et al. 2004).
Patterns of intron loss
This is the first genome-wide study to assess whether intron loss is associated with coding sequence indels, as previous studies excluded introns near alignment gaps (Fedorov et al. 2002; Rogozin et al. 2003; Roy et al. 2003; Nielsen et al. 2004). Strikingly, no observed loss/gain is associated with an alignment gap. Such a pattern is expected by loss by an mRNA intermediate (Perler et al. 1980; Bernstein et al. 1983; Lewin 1983; Weiner et al. 1986; Fink 1987; Long and Langley 1993; Derr 1998; Sakurai et al. 2002; Wada et al. 2002; Mourier and Jeffares 2003; Sverdlov et al. 2004; Roy and Gilbert 2005a). mRNA-mediated loss further predicts loss of adjacent introns from the same gene, as observed here, and preferential loss of 3′ introns (Mourier and Jeffares 2003), a trend not observed here. Our data thus cautiously support mRNA-mediated intron loss. Phase zero introns, which are over-represented in eukaryotic genes (Fedorov et al. 1992; Long et al. 1995) were neither preferentially lost (Roy and Gilbert 2005a) nor retained (Lynch 2002).
Genes experiencing intron changes
Most intron losses occurred in genes encoding nucleic acid-related functions, as previously noted for intron gains in Caenorhabditis (Coghlan and Wolfe 2004). Perhaps relevantly, the only known Plasmodium reverse transcriptase targets telomeres and localizes to the nucleolus, where many nucleic acid-related processes occur (though why this should lead to association of nucleic acid-related transcripts, as opposed to proteins, with reverse transcriptase, is unclear). Coghlan and Wolfe (2004) identified introns specific to one of two Caenorhabditis species and absent in various outgroups as putative gains. However, as the outgroups used have very divergent intron–exon structures (Guiliano et al. 2002; Rogozin et al. 2003), some losses may have been incorrectly identified as gains, thus a nucleic acid-related functional bias among intron losses could conceivably explain their result.
Two changes occurred in the gene encoding chloroquine resistance transporter (crt), which is implicated in P. falciparum sensitivity to a wide variety of compounds (Fig. 2) (Vennerstrom et al. 2004) and is likely to be under very strong selection. One of the changes is apparently due to loss in P. yoelii, while the lack of homologous sequences from P. gallinaceum or distantly related apicomplexans prohibits determination of intron loss/gain for the other. The apparent loss did not pass our filter because of the high degree of sequence divergence in the flanking coding regions (Fig. 2). Chloroquine-sensitive (3D7) and chloroquine-resistant (Dd2, accession number AF030694) P. falciparum isolates show the same intron–exon structure, thus the introns are not directly associated with chloroquine resistance.

Alignment of P. falciparum CRT protein and putative orthologs with intron positions and phases. P.fal, P.yoe, P.cha, and P.viv indicate P. falciparum, P. yoelii, P. chabaudi (a rodent malaria most closely related to P. yoelii), and P. vivax, respectively. P. vivax and P. chabaudi sequences and intron–exon structures are from GenBank accession numbers AF314649 and AY304549, respectively. Gray boxes indicate intron positions that have undergone a change since the P. falciparum–P. yoelii divergence. The first position shows a loss in rodent malarias, the second either a loss in P. falciparum or a gain in the other species. The first position did not pass the filter because of the high degree of sequence divergence in the flanking coding region, although the presence of well-characterized homologs for several species in GenBank indicates that it is a bona fide change.
Alternative splicing and intron loss/gain
We explored the possibility of alternative splicing in genes experiencing intron loss/gain. For 20/21 orthologous gene pairs, no evidence was found for alternative splicing of either gene among available ESTs, although this is not conclusive because of incomplete sampling of transcripts in EST databases. In the final case, P. yoelii ESTs were found that both included and excluded a P. yoelii-specific intron. If this apparent intron retention event reflects inefficiency of splicing of this intron, positive selection for transcript fidelity could have driven the loss of this intron in P. falciparum. If, instead, this alternative splicing event is functional, it would be surprising for the intron to be lost in P. falciparum, unless the alternative splicing pattern has more recently evolved in P. yoelii or had been lost in the P. falciparum lineage prior to intron loss.
Expression level and intron gain/loss
Intron presence is well known to affect expression level, and the one known intraspecific intron presence/absence polymorphism is associated with a change in expression level (Llopart et al. 2002). Heritable changes in expression levels could drive selection for intron loss/gain. However, we found no clear change in EST number either in direction (increase/decrease) or magnitude of change between intron-lacking/containing ortholog pairs. However, the EST sequence libraries used are biased in many ways, including parasite life stage as well as timing and level of expression, and comparison of expression level between such widely diverged species is difficult. More nuanced studies are necessary.
The determinants of intron number
Intron number per gene varies across eukaryotes from several introns per gene down to only a few introns per genome and shows no simple phylogenetic pattern, with intron-rich and intron-poor species commingled across the eukaryotic tree (Logsdon Jr. 1998; Jeffares et al. 2006; Roy and Gilbert 2006). Such differences have been thought to largely reflect stronger genomic streamlining in unicellular than in multicellular species (Doolittle and Sapienza 1980; Gilbert 1987) or differential efficiency of selection in species with different population sizes (Lynch 2002; Lynch and Richardson 2002; Lynch and Conery 2003). However, neither proposal predicts the apparently high intron densities of early eukaryotes (Fedorov et al. 2002; Rogozin et al. 2003; Roy and Gilbert 2005a) or the large population size unicellular species Cryptococcus neoformans and Chlamydomonas reinhardtii (Lynch and Conery 2003; Lin and Zhang 2005; Loftus et al. 2005), nor the persistence of a large number of introns in otherwise reduced genomes (Gilson and McFadden 1996, 2002; Zagulski et al. 2004).
Importantly, simple general selection on intron number cannot explain the low rates of both intron loss and gain observed here. Selection against introns could explain the lack of Plasmodium intron gain, but not the lack of loss; selection for introns could explain the lack of loss, but not of gain. Instead, the rarity of both intron loss and gain in Plasmodium could simply reflect a dearth of stochastic intron loss and intron gain mutations. Low intron loss/gain mutation is predicted by the lack of known transposable elements and their encoded reverse transcriptases, which hold central roles in prominent models of intron loss and gain.
In this case, the dependence of both intron loss and gain on TE abundance would predict a direct correlation between rates of intron loss and gain. On the other hand, paralogous recombination, likely an important event in intron loss, could be suppressed in some TE-rich lineages because of selection against ectopic recombination between TEs, leading to high gain but low loss rates, and thus to high intron number. Alternatively, species whose spliceosomes are less efficient at removing new TE insertions from transcripts (or whose TEs are less recognizable) could have high intron loss rates but low gain rates, leading to low intron number.
Conclusions
We have shown a dearth of intron loss and gain in malaria parasites. These results suggest against intron number control by sensitive natural selection and suggest an important role for mutational mechanisms of intron gain and loss, underscoring the need for new models of the evolution of genome complexity.
Methods
We downloaded Plasmodium falciparum (10/3/2002 release, version 2) and Plasmodium yoelii (version 1) genome annotations from PlasmoDB (www.plasmodb.org). Reciprocal BLASTP searches between the two proteomes yielded 3479 putatively orthologous gene pairs. We used ClustalW with default parameters to align the protein sequences of each pair and mapped intron positions onto the alignments, yielding roughly 3.5 Mb of good alignment (>50% amino acid identity).
We next excluded large numbers of obvious and recurrent annotation errors as well as intron positions in nonhomologous regions of alignment, using permissive criteria in order to retain even questionable cases of intron difference, which were later analyzed by eye (see below). We first excluded introns that were found in regions of bad alignment (<50% amino acid-level identity in the 15 aligned amino acids on either side of the position). This filter retained introns that are present in regions with gaps, even cases including large and/or numerous gaps. We then excluded cases in which an intron in one sequence was opposite or within five residues of a 15-amino-acid or greater gap in the same sequence, as such cases are strongly suggestive of either an exonic stretch of sequence having been erroneously called an intron in the annotation (in the intron-containing sequence) or vice versa (in the other sequence). However, we retained cases in which an intron in one sequence fell adjacent to or within a 15-amino-acid or longer gap in the other sequence, as such cases are not explicable as simple annotation errors and are possible instances of an inexact intron loss or gain involving coding sequence loss or gain. We next excluded sequences that fell at the beginning or end of the alignment. Custom Perl programs were written to perform these filters. Every excluded intron position was analyzed by eye, yielding no additional bona fide intron position differences relative to the automated results.
The successive filtering left 192 ortholog pairs with an apparent intron discordance. Visual inspection showed that the vast majority of these cases involved a discordant intron position near another intron position with an intervening gap and no intervening region of clear homology, easily explained as errant prediction in which an intron–exon–intron had been called a single intron or vice versa. Many others involved differences in intron position of one to four bases between species. Multiple such cases with the same offset were often found in the same gene, strongly suggesting annotation error. We excluded one discordant position in repetitive coding sequence because of alignment uncertainty. We excluded four short introns (6, 15, 21, and 31 base pairs long) adjacent to short alignment gaps as unlikely.
Finally, for one P. yoelii intron next to a 34–amino-acid gap in P. falciparum, a BLASTN search against the PlasmoDB P. yoelii EST database (release date 3/12/2004) yielded a sole EST containing not only the gapped sequence, but also the supposedly intronic sequence. The supposed intron is a multiple of three bases and contains no in-frame stop codons. Thus, this sequence is likely a large coding sequence indel, not an intron.
For each of the 27 remaining intron positions, we used sequences flanking the discordant position as the query for a TBLASTN search against genomic P. gallinaceum sequence (www. sanger.ac.uk/Projects/P_gallinaceum, available shotgun reads on 6/10/2005). In 24 cases, a sizable gap in the alignment at the discordant position clearly suggests intron presence in P. gallinaceum. In two more cases, a hit to only one of the two flanking exons was found, with sequence similarity ending abruptly at the intron position. Here it is likely that the adjoining P. gallinaceum sequence is intronic and that the other exon has not yet been sequenced; thus such cases were scored as intron presence. In the remaining case no corresponding P. gallinaceum sequence was found. For each discordant intron from P. falciparum, we performed analogous searches against available genome sequence for the macaque parasite P. knowlesi (http://www.plasmodb.org, performed on 6/15/2005) and for Toxoplasma gondii (http://tigrblast.tigr.org/ufmg/index.cgi?database=t_gondii, performed on 6/15/2005), and against the genomic sequences and predicted proteomes of Eimeria tenella (http://www.genedb.org/genedb/etenella/, performed on 1/14/2006), Theileria parva (AAGK01000001.1), and T. annulata (version 1). The data are summarized in Table 1.
Simulating intron gain/loss
We used Monte Carlo simulation to simulate 27 intron changes among the 2212 positions in conserved regions and calculated mean intron length and counted genes with multiple changes. Ninety-two of 100,000 such sets had three or more genes with multiple changes. Only 1/100,000 had six or more (P = 10−5). Out of 10,000 random sets of eight P. yoelii introns, the average intron length was less than the observed average of 154 bp among the eight observed P. yoelii-specific introns in 3012 sets (P = 0.30).
Adjacent changes
The five genes with two P. falciparum-specific introns have 3, 4, 5, 6, and 7 intron positions in good regions of alignment. The probability of randomly selecting an adjacent pair of introns among n introns is 2/n, thus the probability of selecting adjacent pairs in all five genes is 2/3 × 2/4 × 2/5 × 2/6 × 2/7 = 0.012. The probability of selecting adjacent pairs in all genes except the three-intron gene is (3 – 2)/3 × 2/4 × 2/5 × 2/6 × 2/7; the chance of selecting adjacent pairs in all genes except the i-intron gene is (i – 2) × 24 / (3 × 4 × 5 × 6 × 7). Thus, the total probability of four adjacent pairs is this value summed over all five genes, or 0.095, and the total probability of four or five adjacent pairs is 0.107.
Gene representation in EST databases
We downloaded all available ESTs for both P. yoelii and P. falciparum from PlasmoDB (plasmodb.org, release date 3/12/2004 for both). For each gene from each pair of orthologous genes, we performed a BLASTN search against the ESTs from the same species and counted hits with at last 90% sequence identity over at least 60 base pairs.
Alternative splicing
For each of the 21 ortholog pairs with intron changes, we BLASTed the entire genomic gene region (intronic and exonic sequences) for each species against all ESTs. We compared each pair of ESTs with overlapping alignments and looked for evidence of alternative splicing. In 20 cases, no alternative splicing event was found. In the final case of PY05736, two EST forms were found, one in which the P. yoelii-specific intron in that gene had been removed and another in which the P. yoelii intron was included in the transcript.
Position and phases of intron changes
We determined whether each intron in each gene was 5′ or 3′ of the median position among intron positions in conserved regions of that gene. There were equal numbers of 5′ and 3′ introns experiencing changes (10 vs. 10, with seven introns in median positions). The fractions of all discordant introns falling in the three phases were not different.
Acknowledgments
This work was supported by the Ellison Medical Foundation.
Footnotes
- 1
↵1 Corresponding author
↵1 E-mail scottwroy{at}gmail.com; fax +64-6-350-5682.
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.4845406
-
- Received October 22, 2005.
- Accepted March 27, 2006.
- Cold Spring Harbor Laboratory Press








